

Abstract
The STAT3 transcription factor is well known to function as an anti-apoptotic factor, especially in numerous malignancies. Recently we showed that STAT3 is cytoprotective and that cells lacking STAT3 are more sensitive to oxidative stress. A key feature of oxidative stress involves activation of the DNA damage pathway. However, a role for STAT3 or its contribution in response to DNA damage has not been described. In the present study we show that cells lacking STAT3 are less efficient in repairing damaged DNA. Moreover, STAT3 deficient cells show reduced activity of the ATM-Chk2 and ATR-Chk1 pathways, both important pathways in sensing DNA damage. Finally we show that MDC1, a regulator of the ATM-Chk2 pathway and facilitator of the DNA damage response, is modulated by STAT3 at the transcriptional level. These findings demonstrate that STAT3 is necessary for efficient repair of damaged DNA, partly by modulating the ATM-Chk2 and ATR-Chk1 pathways.
Keywords: ATM, ATR, Chk1, Chk2, DNA damage, DNA repair, STAT3
To ensure that cells pass accurate copies of their genomes on to the next generation, a series of surveillance pathways, the so-called cell-cycle checkpoint protein kinases are activated following DNA damage to allow appropriate time for DNA repair to take place.
The DNA-damage response pathway controls cell-cycle transitions, DNA replication, DNA repair and apoptosis. The major regulators of the DNA-damage response are the phosphoinositide 3-kinase (PI3K)-related protein kinases (PIKKs), including ataxia-telangiectasia mutated (ATM) and ATM and RAD3-related (ATR). ATM and ATR share many functional similarities including targeting an overlapping set of substrates that promote cell-cycle arrest and DNA repair (Cimprich & Cortez 2008). ATM is mainly activated following double-strand breaks (DSBs). By contrast, ATR is essential and is activated following replication fork damage on single stranded DNA during S phase to ensure normal firing of replication origins (Cimprich & Cortez 2008).
ATM kinase together with specific adaptor proteins (mediators), play a pivotal role in initiating the checkpoint cascade pathway following DSBs by phosphorylating and activating a subset of ATM substrates including Chk2 and γH2AX (Kastan & Lim 2000; Shiloh 2003). In mammalian cells, the BRCA1 carboxy-terminal domain (BRCT) containing proteins 53BP1 and MDC1, are key mediators in response to DNA damage and they play a central role in facilitating the downstream effector kinase, Chk2 (Stucki & Jackson 2003). ATR kinase with its specific adaptor proteins plays an important role in coordinating DNA replication and also acts as a sensor following exposure to agents which cause DNA damage. Activated ATR also targets specific substrates including Chk1 (Cimprich & Cortez 2008).
As well as genotoxic agents, reactive oxygen species (ROS), generated as a by-product of normal mitochondrial activity can cause severe damage to cellular macromolecules, including DNA. Oxidative stress is known to generate ROS and to be an inducer of the ATM-Chk2 pathway in response to DNA damage (Tanaka et al. 2006). Thus, pathways that are known to be activated by oxidative stress may also be coupled to the DNA damage cell cycle checkpoint pathway. One such pathway includes the JAK- signal transducers and activators of transcription (STATs) pathway (Levy & Darnell 2002). The STAT family of transcription factors were originally identified on the basis of their ability to transduce a signal from a cellular receptor into the nucleus and modulate the transcription of specific genes. STAT transcription factors have been reported to play a prominent role in cell fate following various stresses including ischaemia reperfusion (I/R) (Stephanou & latchman 2005; Barry et al. 2007). Interestingly, although STAT1 is known to promote apoptosis in a variety of cell types, STAT3 has an opposing anti-apoptotic role (Stephanou & Latchman 2005).
Phosphorylated STAT1 and STAT3 are significantly activated in the infarcted area in the in vivo rat myocardium exposed to I/R injury, known also to generate ROS (Mccormick et al. 2006). A hallmark of the cellular response to DNA double-strand breaks (DSBs) is histone γH2AX phosphorylation in chromatin mediated by ATM (Kastan & Lim 2000; Shiloh 2003). This serves as a molecular beacon for accumulation of DNA repair factors around the site of the DSBs. Recently we showed enhanced levels of phosphorylated γH2AX in the infarcted myocardium following I/R injury (Mccormick et al. 2006). These data strongly suggest a link between the DNA damage pathway and the JAK-STAT pathway. Evidence from this comes from the finding that STAT1 deficient cells have defects both in intra-S-phase and G2-M checkpoints in response to DNA damage (Townsend et al. 2005). However, a role for STAT3 in the DNA damage response pathway has not yet been investigated.
We now report that STAT3 is required for efficient repair of damaged DNA following UVB irradiation. Moreover, STAT3 deficient cells show reduced activity of the ATM-Chk1 pathway. Furthermore, MDC1 a key regulator of the ATM-Chk1 pathway and facilitator of the DNA damage pathway is modulated by STAT3 at the transcriptional level. These data strongly support a role for STAT3 in modulating the DNA damage response pathway.
Methods
Reagents and cell lines
All reagents were obtained from Sigma-Aldrich unless otherwise stated. STAT3 deficient mouse embryonic fibroblasts were a kind gift from Prof. Valeria Poli (University of Turin, Italy) (Celeste et al. 2002), the human fibrosarcoma cell 2FTGH were kindly provided by Ian Kerr (Cancer Research UK, London, UK) and Chinese hamster ovary cells (CHO) were purchased from American Type Culture Collection (ATCC). All cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal bovine serum (FBS) (Gibco, Invitrogen, CA, USA) with 100 U/ml penicillin and 100 μg/ml streptomycin. The STAT3-pcDNA3 expression plasmid was a kind gift from James Darnell (Rockefeller University, New York, NY, USA). Human Flag-tagged Chk2 was a kind gift from Thanos Halazonetis (Wistar Institute, Philadelphia, PA, USA).
Luciferase reporter assays
Cells were seeded in 96 well plates and transfected with 50 ng luciferase reporter construct, 20 ng CMV-renilla plasmid and 50 ng expression plasmid per well. Transfections were carried out using GeneJuice transfection reagent according to the manufacturer's instructions (Merck, Biosciences, NJ, USA). After 24 h, cells were lysed in passive lysis buffer (Promega, Madison, WI, USA) and luciferase activity was measured using the dual luciferase reporter system according to the manufacturer's instructions. Relative luciferase activity was calculated as luciferase activity/renilla activity and normalised to controls.
Host cell reactivation assay
The pCMVluc plasmid was a kind gift from Kenneth Kraemer (National Cancer Institute, Bethesda). The host cell reactivation (HCR) assay involves repair of a transcriptionally active gene and therefore measures the capacity of the host cells to perform transcription-coupled repair (TCR), which is a subset of the overall nucleotide excision repair (NER) pathway and this assay was carried out as described (Qiao et al. 2002). Briefly, pCMVluc was irradiated with 25–1000 J/m2 UVB and 50 ng plasmid was transfected into MEF cells along with 20 ng of CMV-renilla plasmid. After the indicated time points, luciferase activity was measured and the % HCR was determined by comparing luciferase values to a non-irradiated control which was set to 100%.
Western blot
Cells were seeded on 6-well plates and after the indicated treatment times were lysed in 200 μl of radio-immuno-precipitation assay (RIPA) buffer (0.75 M NaCl, 5% (v/v) Nonidet P-40, 2.5% (w/v) deoxycholate, 0.5% (w/v) SDS, 0.25 M Tris–HCl pH 8.0, 10 mM DTT containing protease inhibitor cocktail). Samples were then boiled in SDS sample buffer for 5 min and separated on a 10% SDS PAGE gel. Samples were transferred to nitro-cellulose filters and subjected to western blotting. Antibodies against Chk1, phosphor-Chk1S345, Chk2 and phosphor-Chk2-T68 pATRS428 (2853) and ATR (2790) were from Cell Signalling Technology (Beverly, MA, USA), ATM, pATM1981 and pH2AXS139 (γ-H2AX) antibodies were from Upstate, Anti-Flag was form Sigma (St Louis, MO, USA), STAT3 antibody was from Santa Cruz Biotechnology (Santa Cruz, CA, USA) and the GAPDH antibody was from Chemicon (Watford, UK). Horseradish peroxidase-conjugated secondary antibodies were from Dako (Stockport, UK).
Cell survival and flow cytometry
For survival assays, cells (2.0 × 106) were exposed to 10 Gy γ-IR and incubated for 24 h. Cells were then washed with 1x PBS before staining with Crystal Violet (0.2% Crystal Violet 2% EtOH).Viable cells were calculated as a percentage to control cells that were not exposed to γ-IR.
To assess levels of apoptotic cell death we measured the number of cells in sub-G1 fraction after 24 h exposure to cisplatin (Sigma) or doxorubicin (Sigma) and cells were stained with propidium iodide (Sigma) analysed FACScan flow cytometer (Becton Dickinson, San Jose, CA, USA)
Quantitative real-time PCR (qPCR)
RNA was extracted from cells using Trizol reagent (Invitrogen) according to the manufacturer's instructions and 1 μg was used to prepare cDNA using Superscript II (Invitrogen, Paisley, UK) and random hexamers (Promega, UK). qPCR was carried out using Platinum SYBR Green (Invitrogen) on the DNA Engine Opticon system (MJ Research, East Lyme, CT, USA). For PCR reactions, 5 μl SYBR Green was added to 5 μl cDNA with 500 nM primers in a 20 μl reaction and the PCR conditions were 95 °C for 3 min, followed by 45 cycles of 95 °C for 30 sec, 60 °C for 30 sec and 72 °C for 30 sec. A melt-curve was performed from 65 °C to 95 °C, reading every 0.3 °C with a 1 sec hold between reads. Specific primers were designed with the aid of CloneWorks and the Ensembl database. A control cDNA reaction without reverse transcriptase was included to confirm the absence of genomic DNA and all PCR reactions were visualised on agarose gels to ensure the presence of a single product. β2-microglobulin, Actin and GAPDH were used together as normalising genes and for each experiment both target and normalising gene PCR efficiency was firstly determined to ensure normalising genes were acceptable. To test primer efficiency, qPCR was carried out on a 2-fold dilution series from a pooled set of cDNA and the threshold Ct value was plotted against the log cDNA dilution. Efficiency was then calculated using the equation m = (−1/logE), where m is the slope of the line and E is the efficiency and primer pairs were used only if the PCR efficiency of the normalising and control genes were found to be within 10% of each other. Expression changes were calculated using the 2−ΔΔCt method and expressed as fold change over control. The primers used were as follows; MDC1 forward 5′-AGGTGATTGACTGGGATGCT-3′ MDC1 reverse 5′-GATGGTACTGGCAGGGAAA-3′, β2-microglobulin forward 5′-GTCTTTCTGGTG-CTTGTCTCA-3′, β2-microglobulin reverse 5′-GTGAGCCA-GGATATAGAAAGA-3′, Actin forward 5′-ATGCTCCCC-GGGCTGTATT-3′, Actin reverse 5′-TACGACCAGAGGC-ATACAG-3′, Gapdh forward 5′-GTGTGAACGGATTTG-GCCG-3′, Gapdh reverse 5′-CCAGTAGACTCCACGACATA-3′.
Statistical analysis
Data are expressed as mean ± SEM, all experiments were repeated in triplicate. Statistical analysis was carried out using a Student's t-test, one-way anova with Dunnet's post or two-way anova with Bonferroni post test, P-values of less than 0.05 were considered significant.
Results
MEFs lacking STAT3 repair DNA less efficiently and are more sensitive to DNA-damage-induced cell death than wild type cells
In order to ascertain what effect STAT3 has on the DNA repair pathway, the host-cell reactivation assay (HCR) was used (Qiao et al. 2002). The HCR assay (or repaired UV-damage DNA), involves introducing UV-induced DNA damage in a luciferase plasmid followed by transfection into cells. The damaged plasmids are repaired by the cellular DNA repair machinery and only fully repaired plasmids will be transcribed correctly to generate active luciferase. The resulting luciferase activity is then compared against an undamaged control plasmid (set to 100%) to calculate a % of repair (known as % HCR or repaired UV-damage DNA). Initially a dose response of UVB irradiation was carried out; plasmids were irradiated with 25–1000 J/m2 UVB, transfected into wild type MEFs for 24 h and the % HCR was calculated. Figure 1a shows that there was a dose dependent decrease in HCR with increasing UV irradiation; at the highest dose, 1000 J/m2 reduced the HCR or repaired UV-damage DNA to 3.4%.
Figure 1.
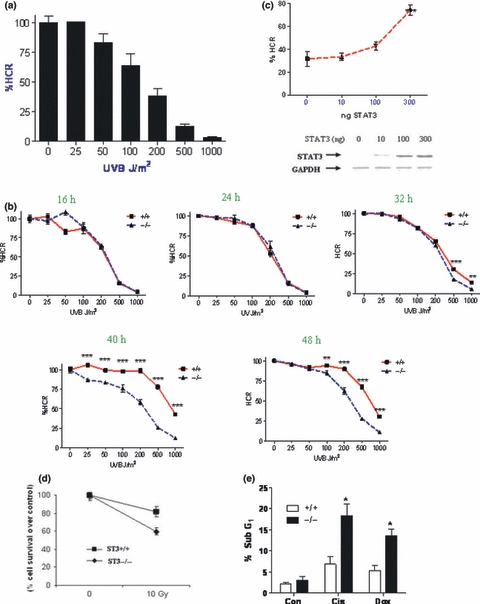
STAT3−/− MEFs have reduced capacity to repair damaged DNA. (a) CMV-luciferase was irradiated with the indicated dose of UV and transfected into MEF cells for 24 h, luciferase values were normalised to renilla. All values are relative to the non-irradiated control which was set to 100%. (b) CMV-luciferase was irradiated with the indicated doses of UV and transfected into STAT3+/+ and STAT3−/− MEFs for 16–48 h. Luciferase values were normalised to renilla and expressed as a percentage of un-irradiated control. A representative experiment is shown, n = 6 per experiment, repeated in triplicate (c) CMV-luciferase was irradiated with 500 J/m2 UV and transfected into STAT3−/− MEFS with 10, 100 or 300 ng STAT3-pcDNA3 or pcDNA3 alone (0) and luciferase activity was measured after 48 h, normalised to renilla and expressed as a percentage of non-irradiated control plasmid. The values were further normalised against pcDNA3 control values. The levels of STAT3 following transfection were also measured by western blot with GAPDH used as a loading control. (d) Cell survival was assessed following exposure to 10 Gy γ-irradiation in wild type (ST3+/+) and STAT3 deficient (ST3−/−) MEF cells for 24 h. After Crystal Violet staining, the percentage of cell survival was determined. (e) STAT3+/+ and STAT3−/− cells were treated with cisplatin or doxorubicin for 24 h and cell death was measured by flow cytometry as the sub-G1 fraction following propidium iodide staining. Error bars represent mean ± SEM, statistical analysis was carried out using a two-way anova with Bonfferoni post test (b) a one-way anova with Dunnett's post test (c) or Student's t-test (e) n = 3 per experiment, repeated in duplicate. *P < 0.05, **P < 0.01, ***P < 0.001.
Next the dose response was repeated and plasmids were transfected into both STAT3+/+ and STAT3−/− MEFs for 16–48 h. Figure 1b shows that at 16 and 24 h there are no differences between cell types. However as the time for DNA repair increased between 32 and 48 h, it was apparent that STAT3−/− MEFs had a lower % HCR or repaired UV-damage DNA, indicating a reduced efficiency of DNA repair. This data shows that STAT3 deficient MEFs, although they are capable of repairing damaged DNA, do so at a slower rate and may be defective in the late phase of DNA repair. Accumulation of damaged DNA in STAT3 deficient cells may therefore have effects on the rate of apoptosis in these cells.
To show that reduced DNA repair in the STAT3−/− cells was directly attributable to reduced STAT3 levels, STAT3−/− MEFs were transfected with increasing amounts of STAT3 and a luciferase plasmid irradiated with 500 J/m2 UV; the HCR or repaired UV-damage DNA assay was carried out after 48 h. Transfection of 100 ng STAT3 increased the % HCR from 31.8% to 43.5% and 300 ng STAT3 significantly increased the HCR to 74.4%, showing that STAT3 directly contributes to DNA repair (Figure 1c). These results are consistent with the previous data (Figure 1b) which showed that at 500 J/m2 after 48 h, the % repaired UV-damage DNA in STAT3+/+ MEFs was 67.1% and in STAT3−/− MEFs was 27.7%. Importantly this experiment shows for the first time that the level of STAT3 directly correlates with the level of DNA repair. This data thus predicts that cells which increase their expression of STAT3 (such as certain cancer cells) should repair damaged DNA more efficiently.
Since STAT3 has been shown to have anti-apoptotic effects, we examined whether STAT3 deficient cells show differences in sensitivity to DNA damage induced cell death following γ-irradiation or exposure to DNA damaging agents assessed by flow cytometry (sub-G1 fraction). As shown in Figure 1d, STAT3 deficient MEFs were more sensitive γ-irradiation-induced cell death and likewise had increased levels of cell death following exposure to DNA damaging agents compared to wild type MEFs (Figure 1e). These studies demonstrate that STAT3 is cytoportective against genotoxic stress and are in line with our previous work demonstrating reduced viability of STAT3−/− MEFs following oxidative stress (Barry et al. 2009) and provide further evidence that STAT3 has anti-apoptotic properties.
STAT3−/− MEFs show reduced activity of ATM-Chk2 and ATR-Chk1 pathways
Following DNA strand breaks, the DNA damage response pathway is quickly activated. The apical kinase in this pathway is ATM which phosphorylates several key proteins involved in DNA repair and apoptosis. Since STAT3 deficient cells repair DNA at a slower rate, the levels of activated ATM were examined in STAT3−/− MEFs to ascertain whether this key DNA damage kinase is altered in these cells. STAT3+/+ and STAT3−/− MEFs were treated for up to 4 h with 10 μM etoposide, a topoisomerase II inhibitor which induces DNA strand breaks. Etoposide treatment resulted in a time dependent increase in ATM phosphorylation in both STAT3 WT and knock-out cells; however the levels of phosphorylated ATM (pATM) were far lower at each time point in the STAT3−/− cells (Figure 2).
Figure 2.
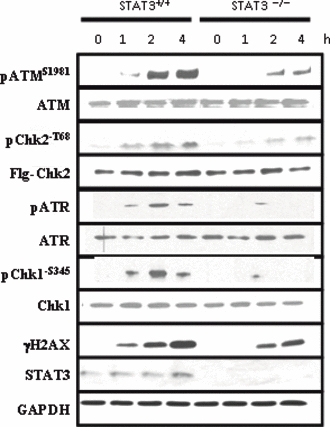
Phosphorylation of ATM and H2AX are reduced in STAT3−/− MEFs. STAT3+/+ and STAT3−/− MEFs were treated with DMSO (0 h) or 10 μM etoposide for 1, 2 or 4 h and western blots were carried out on cell lysates using the indicated antibodies for ATM, phospo-ATM (pATMS1981), Chk2, phospho-Chk2 (pChk2-T68), ATR, phosphor-ATR (pATR), Chk1, phosphor-Chk1 (pChk1-S345), γH2AX, STAT3 and GAPDH. For Chk2 and pChk2-T68 immunoblots, cells were transfected with human Flag-tagged Chk2 (1μg/time point) and Western blotted with the specific anti-Flag antibody to measure the levels of transfected Chk2 (Flg-Chk2) or an anti-pChk2-T68 following etoposide treatment.
Chk2 and γH2AX are known downstream ATM substrates which become phosphorylated following a DNA damage response. (Kastan & Lim 2000; Lowndes & Toh 2005). Since there is no available murine specific anti-phospho-Chk2-T68, we therefore overexpressed a human Flag-tagged Chk2 expression construct into MEFs. Similarly to ATM phosphorylation, both Chk2-T68, and γH2AX were phosphorylated by etoposide in a time dependent manner, and again the levels were reduced in STAT3−/− cells (Figure 2a).
Next we also examined whether another apical upstream kinase ATR, may also be activated after etoposide-induced DNA damage. As shown in Figure 2, ATR phosphorylation was induced following etoposide in wild type MEFs but not in STAT3 deficient MEFs. Moreover, Chk1 a downstream target substrate of ATR was also activated and phosphorylated (pChk1S35) following etoposide treatment in wild type MEFs but not in STAT3 deficient MEFs. Interesting, another DNA damage marker, phosphorylated p53S15 levels which can also be phosphorylated ATM but is also a target by other kinases was not affected in STAT3 deficient MEFs exposed to etoposide (data not shown). Taken together, this shows that activity of the ATM-Chk2 and the ATR-Chk1 pathway is reduced in the absence of STAT3.
STAT3 facilitates DNA damage mediated upregulation of MDC1
The previous sections have demonstrated that activation of ATM and its downstream substrates are reduced in STAT3 deficient MEFs. MDC1 functions to amplify DNA damage signals by binding to γH2AX through its BRCT domain and to ATM via its FHA domain (Lou et al. 2006). This allows further recruitment of ATM to the sites of strand breaks and enhances the ATM mediated DNA damage response by facilitating ATM dependent phosphorylation of Nbs1, 53BP1 and BRCA1 (Stucki et al. 2005). Although MDC1 has no effect on the initial activation of ATM and H2AX, it is required to maintain the DNA damage response past the initial activation phase (Lou et al. 2006). Since several other mediators of the DNA damage response were dysregulated in STAT3−/− MEFs, it was of interest to examine the regulation of MDC1 in these cells.
The mRNA level of MDC1 was measured in STAT3+/+ and STAT3−/− MEFs by qPCR and no difference was seen between cell types (Figure 3a). Next, the regulation of MDC1 levels by STAT3 following DNA damage was assessed. It is unknown whether the expression of MDC1 is altered by DNA damage, therefore WT MEFs were treated with 10 μM etoposide for up to 24 h and MDC1 mRNA levels were measured. Figure 3b shows that MDC1 levels increased over time following DNA damage with etoposide, and a significant increase could be seen by 12 h. STAT3−/− MEFs were then treated with 10 μM etoposide for 12 h and compared to wild type cells (Figure 3c). In contrast to the 2-fold increase in MDC1 levels in WT cells, STAT3−/− MEFs did not show any increase in MDC1 levels at 12 h.
Figure 3.
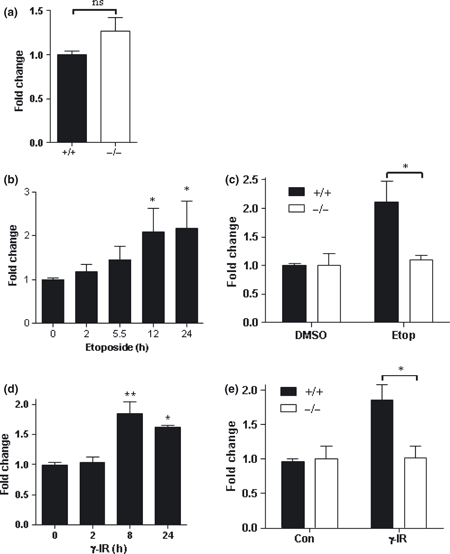
DNA damage mediated increase in MDC1 expression is compromised in STAT3−/− MEFs. (a) Basal MDC1 expression was measured in STAT3+/+ and STAT3−/− MEFs by qPCR. (b) Wild type MEFs were treated with 10μM etoposide for the indicated times and MDC1 levels were measured by qPCR. (c) STAT3+/+ and STAT3−/− MEFs were treated for 12 h with 10μM etoposide and MDC1 levels were measured by qPCR. (d) Wild type MEFs were irradiated with 10 Gy γ and MDC1 levels were measured at indicated time points after treatment. (e) STAT3+/+ and STAT3−/− MEFs were treated for 8 h with 10 Gy γ-irradiation and MDC1 levels were measured by qPCR. Error bars represent mean ± SEM, Statistical analysis was carried out using a student's t-test (a), one-way anova with Dunnett's post test (b, d) or a two-way anova with Bonfferoni post test (c, e), n = 2 per experiment, repeated in triplicate. *P < 0.05, **P < 0.01.
To ascertain if increased MDC1 levels is a general feature of DNA damage, WT MEFs were irradiated with 10 Gy γ-IR and allowed to recover for 2, 8 and 24 h (Figure 3d). At 8 h and 24 h after irradiation, MDC1 levels were significantly increased. Comparison of γ-IR mediated MDC1 upregulation showed that MDC1 failed to be upregulated in STAT3−/− MEFs which supports the findings with etoposide (Figure 3e).
The STAT3 dependent regulation of MDC1 expression was examined in greater detail using an MDC1 promoter luciferase construct (Townsend et al. 2005). The MDC1 luciferase reporter was co-transfected with pcDNA3 or STAT3 into three separate cell lines; Chinese hamster ovary cells (CHOs), MEFs and the fibrosarcoma cell line 2fTGH. Luciferase activity was measured after 24 h and in each cell line tested STAT3 enhanced MDC1 promoter activity (Figure 4a).
Figure 4.
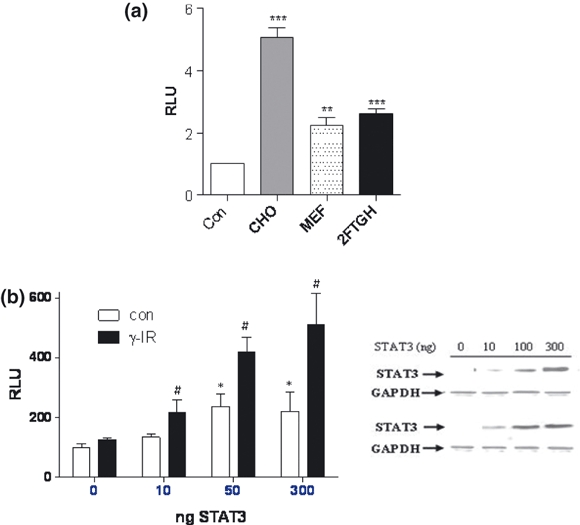
STAT3 regulates the MDC1 promoter. (a) CHO, MEF or 2FTGH cells were transfected with 50 ng pcDNA3 (Con), or 50 ng STAT3 expression plasmid together with 50 ng of MDC1 reporter and 20 ng of renilla. Luciferase activity was measured after 24 h and is given as the relative increase in STAT3 mediated MDC1 promoter activity in each cell type. (b) STAT3−/− MEFs were transfected with 50 ng MDC1, 20 ng renilla and 10, 100 or 300 ng STAT3 or the corresponding amount of pcDNA3 for 24 h and then left untreated (con) or treated with 10 Gy γ-IR, luciferase activity was measured after 48 h. Experiments were repeated in triplicate, n = 6 per experiment, luciferase values were normalised to renilla levels and then re-normalised to appropriate pcDNA3 controls and expressed as relative luciferase units (RLU). Cell lysates were also analysed by western blotting to reveal the expression of STAT3 and GAPDH. Statistical analysis was carried out using a student's t–test, **P < 0.01, ***P < 0.0001 (a) or a Two-way anova with Bonfferoni post correction (b), where *P < 0.05 relative to pcDNA3 control and # P < 0.05 relative to pcDNA3 + γ-IR.
Next, STAT3−/− MEFs were co-transfected with 10, 50 or 300 ng of STAT3 expression plasmid and the MDC1 reporter, treated with 10 Gy of γ-IR and assessed for luciferase activity after 24 h (Figure 4b). STAT3 dose dependently increased MDC1 promoter activity and this effect was enhanced by γ-IR, reaching a maximum level of 5 fold over control levels when transfected with 300 ng STAT3. Overall these data show that STAT3 regulates the MDC1 at the transcriptional level and that this is further modulated following DNA damage induced by γ-IR.
Discussion
Efficient repair of damaged DNA is essential to maintain genomic stability and cell survival. Although STAT3 has been shown to function as an anti-apoptotic transcription factor, very little is known regarding its role in the DNA damage response. STAT3 deficient MEF cells were found to be less efficient at repairing damaged DNA using the HCR assay or repaired UV-damage DNA, suggesting that STAT3 is necessary for optimum DNA repair. Since STAT3−/− MEFS are less efficient at repairing damaged DNA, it is possible that DNA breaks may persist for longer in STAT3 deficient cells. In addition, artificially increasing the levels of STAT3 correlated with increased ability to repair DNA and restored the repair defect phenotype of knock-out cells.
Several cancers are associated with elevated STAT3 levels, including multiple myelomas, lymphomas, breast, head and neck, lung, pancreatic and prostate cancers (Yu and Jove, 2004). Moreover, elevated STAT3 activity is thought to contribute to chemoresistance in these malignancies (Barréet al. 2007) and targeting of STAT3 has been shown to restore sensitivity to chemotherapeutic drugs (Alas & Bonavida 2003; Gariboldi et al. 2007). Thus over the past 5 years, STAT3 has emerged as a major new target for anti-cancer therapy (Yu and Jove, 2004). STAT3 mediated chemonresistance has been attributed to upregulation of anti-apoptotic STAT3 target genes such as cyclin D1, Bcl-XL and survivin (Turkson et al. 2005, Gritsko et al. 2006) and the data presented here now add to the understanding of how STAT3 may contribute to chemoresistance. Although the experiments were done in mouse embryonic fibroblasts, speculation can be made as to the contribution of STAT3 to chemoresistance. Expression of STAT3 was associated with an enhanced ability to repair damaged DNA. By inference this suggests that cancer cells which overexpress STAT3 may be resistant to chemotherapeutics through their increased efficiency of DNA repair and greater resistance to genotoxic stress. It would therefore be of interest to examine the rate of DNA repair following treatment with chemotherapeutic drugs in tumour cells with elevated STAT3 expression. Thus the HCR assay or repaired UV-damage DNA could be conducted on cancer cell lines which have varying expression of STAT3 to examine the correlation between STAT3 expression and DNA repair. Based on our results above, we would also predict that the STAT3−/− MEFs cells would also be more hypersensitive to exposure to UVB irradiation. If the conjecture that elevated STAT3 levels in cancer contribute to increased efficiency of DNA repair, it would offer an alternative explanation for chemonresistance in these tumours.
Molecular analysis of the DNA damage response pathway in STAT3−/− MEFS revealed several possible explanations for the reduced repair efficiency. The apical kinase in the DNA damage response is ATM which undergoes autophosphorylation after DNA damage, followed by dimer dissociation and activation (Bakkenist & Kastan 2003). ATM is responsible for phosphorylation of a host of proteins involved in DNA repair and cell cycle arrest and skin fibroblasts from Ataxia-telangiectasia patients (lacking active ATM) have a reduced capacity to repair damaged DNA in conjunction with decreased HCR levels (Hannan et al. 2002). The reduced activity of ATM in STAT3−/− MEFs may therefore lead to dysregulation of repair proteins resulting in sub-optimal efficiency of DNA repair.
Phosphorylation of H2AX by ATM is a key event in the DNA damage response; it leads to the recruitment of repair factors directly to the site of strand breakage (Celeste et al. 2002). Thus H2AX deficient cells are sensitive to radiation and have a reduced capacity to repair double strand breaks (Bassing et al. 2000). The reduced phosphorylation of H2AX in STAT3−/− deficient MEFs following etoposide treatment in conjunction with reduced active ATM might in part account for the slower rate of DNA repair in these cells. Reduced activity of H2AX would be expected to slow the rate of accumulation of DNA repair factors around the site of strand breakage, thereby hampering the DNA repair process.
Chk1 phosphorylation at seine 345, a known substrate of ATR was also shown to be reduced in STAT3−/− defieicent MEFs. Chk1 phosphorylation leads to its release from chromatin where it in turn phosphorylates the CDC25 phosphatases and targets them for destruction by ubiquitin-mediated proteolysis (Cimprich & Cortez 2008). This promotes CDK activation and cell cycle checkpoint block (Cimprich & Cortez 2008). The importance of serine 345 phosphorylation in Chk1 is underscored by the finding that mutation of this site on Chk1 dysregulates checkpoint control and leads to mitotic catastrophe (Niida et al. 2007). Reduced phosphorylation of Chk1 in cells lacking STAT3 cells would be expected to abrogate cell cycle arrest through the checkpoint system and examination of cell cycle profiles from STAT3-deficient cells could be used to address this.
Our data also demonstrated that STAT3 is able to modulate the expression of MDC1, which is a key player in the ATM mediated DNA damage response pathway. Thus, cells lacking STAT3 failed to induce mRNA expression of MDC1 in response to etoposide or γ-IR. Furthermore, increasing STAT3 levels enhanced γ-IR mediated MDC1 upregulation. Taken together, this shows that STAT3 is necessary for the upregulation of MDC1 levels following DNA damage and impaired MDC1 expression may contribute to the reduced efficiency of DNA repair in STAT3−/− MEFs. In addition, it can be speculated that cancer cells overexpressing STAT3 might also have elevated MDC1 levels, allowing more efficient DNA repair and thus contributing to chemo-resistance.
We have also previously reported that STAT1 is also able to upregulate the expression of MDC1 (Townsend et al. 2005). The significance of our present data and whether both STAT3 and STAT1 are required for modulating the expression of MDC1 is unclear. Since the DNA binding sites for STAT1 and STAT3 share similar DNA sequences it is not surprising that other genes are known to be regulating by both STAT1 and STAT3, such as acute phase proteins via IL-6 signalling (May et al., 2003). However, STAT3 is a stronger transactivator than STAT1 (May et al., 2003). We have also noticed that STAT3 is also more effective than STAT1 in transactivating MDC1 promoter activity (data not shown). It is also well know that STAT1 and STAT3 may regulate specific target genes via interaction with specific STAT1 or STAT3 transcriptional co-activators (Stephanou & Latchman 2005).
Although these studies were limited to mouse embryonic fibroblasts and DNA damage was induced predominantly with etoposide; some general conclusions can still be made. These studies have demonstrated that STAT3 is necessary for efficient repair of damaged DNA. Several candidate proteins which could mediate this affect have been highlighted, including ATM, H2AX, Chk2, ATR, Chk1 and MDC1. Increased expression and/or activity of STAT3 in tumours is suggested to contribute to chemoresistance through the possibility of increasing the activity of ATM and its downstream targets and upregulating MDC1 expression, allowing more efficient localisation of repair proteins to sites of damaged DNA. In future studies, direct imaging of DNA damage response factors at the sites of DNA breaks in tumour cells with varying levels of STAT3 will highlight any differences in duration of chromatin occupancy which might be influenced by STAT3.
Acknowledgments
This work was funded by the British Heart Foundation, London, UK.
References
- Alas S, Bonavida B. Inhibition of constitutive STAT3 activity sensitizes resistant non-Hodgkin's lymphoma and multiple myeloma to chemotherapeutic drug-mediated apoptosis. Clin. Cancer Res. 2003;9:316–326. [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Barré B, Vigneron A, Perkins N, Roninson IB, Gamelin E, Coqueret O. The STAT3 oncogene as a predictive marker of drug resistance. Trends Mol. Med. 2007;13:4–11. doi: 10.1016/j.molmed.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Barry SP, Townsend PA, Latchman DS, Stephanou A. Role of the JAK-STAT pathway in myocardial injury. Trends Mol. Med. 2007;13:82–89. doi: 10.1016/j.molmed.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Barry SP, Townsend PA, Mccormick J, et al. STAT3 deletion sensitizes cells to oxidative stress. Biochem. Biophys. Res. Commun. 2009;385:324–329. doi: 10.1016/j.bbrc.2009.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassing CH, Chua KF, Sekiguchi J, et al. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc. Natl. Acad. Sci. U S A. 2000;99:8173–8178. doi: 10.1073/pnas.122228699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste A, Petersen S, Romanienko PJ, et al. Genomic instability in mice lacking histone H2AX. Science. 2002;296:922–927. doi: 10.1126/science.1069398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gariboldi MB, Ravizza R, Molteni R, Osella D, Gabano E, Monti E. Inhibition of Stat3 increases doxorubicin sensitivity in a human metastatic breast cancer cell line. Cancer Lett. 2007;258:181–188. doi: 10.1016/j.canlet.2007.08.019. [DOI] [PubMed] [Google Scholar]
- Gritsko T, Williams A, Turkson J, et al. Activation of stat3 in primary tumors from high-risk breast cancer patients is associated with elevated levels of activated SRC and survivin expression. Clin. Cancer Res. 2006;12:20–28. doi: 10.1158/1078-0432.CCR-04-1749. [DOI] [PubMed] [Google Scholar]
- Hannan MA, Hellani A, Al-Khodairy FM, et al. Deficiency in the repair of UV-induced DNA damage in human skin fibroblasts compromised for the ATM gene. Carcinogenesis. 2002;23:1617–1624. doi: 10.1093/carcin/23.10.1617. [DOI] [PubMed] [Google Scholar]
- Kastan MB, Lim DS. The many substrates and functions of ATM. Nat. Rev. Mol. Cell Biol. 2000;1:179–186. doi: 10.1038/35043058. [DOI] [PubMed] [Google Scholar]
- Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Lou Z, Minter-Dykhouse K, Franco S, et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol. Cell. 2006;21:187–200. doi: 10.1016/j.molcel.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Lowndes NF, Toh GW. DNA repair: the importance of phosphorylating histone H2AX. Curr. Biol. 2005;15:99–102. doi: 10.1016/j.cub.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Mccormick JM, Barry SP, Sivarajah A, et al. Free radical scavenging inhibits STAT phosphorylation following in vivo ischemia/reperfusion injury. FASEB J. 2006;20:2115–2117. doi: 10.1096/fj.06-6188fje. [DOI] [PubMed] [Google Scholar]
- Niida H, Katsuno Y, Banerjee B, Hande MP, Nakanishi M. Specific role of Chk1 phosphorylations in cell survival and checkpoint activation. Mol. Cell. Biol. 2007;27:2572–2581. doi: 10.1128/MCB.01611-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y, Spitz MR, Guo Z, et al. Rapid assessment of repair of ultraviolet DNA damage with a modified host-cell reactivation assay using a luciferase reporter gene and correlation with polymorphisms of DNA repair genes in normal human lymphocytes. Mutat. Res. 2002;509:165–174. doi: 10.1016/s0027-5107(02)00219-1. [DOI] [PubMed] [Google Scholar]
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer. 2003;3:155–168. doi: 10.1038/nrc1011. [DOI] [PubMed] [Google Scholar]
- Stephanou A, Latchman DS. Opposing actions of STAT1 and STAT-3. Growth Factor. 2005;23:177–182. doi: 10.1080/08977190500178745. [DOI] [PubMed] [Google Scholar]
- Stephanou A, Brar BK, Knight RA, Marber MS, Yellon D, Latchman DS. Ischaemia-induced STAT-1 expression and activation plays a critical role in cardiac myocyte apoptosis. J. Biol. Chem. 2000;275:10002–10008. doi: 10.1074/jbc.275.14.10002. [DOI] [PubMed] [Google Scholar]
- Stucki M, Jackson SP. MDC1/NFBD1: a key regulator of the DNA damage response in higher eukaryotes. DNA Repair. 2003;3:953–957. doi: 10.1016/j.dnarep.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Halicka HD, Huang X, Traganos F, Darzynkiewicz Z. Constitutive histone H2AX phosphorylation and ATM activation, the reporters of DNA damage by endogenous oxidants. Cell Cycle. 2006;5:1940–1945. doi: 10.4161/cc.5.17.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend PA, Cragg MS, Davidson SM, et al. STAT-1 facilitates the ATM activated checkpoint pathway following DNA damage. J. Cell Sci. 2005;118:1629–1639. doi: 10.1242/jcs.01728. [DOI] [PubMed] [Google Scholar]
- Turkson J. STAT proteins as novel targets for cancer drug discovery. Expert Opin. Ther. Targets. 2004;8:409–422. doi: 10.1517/14728222.8.5.409. [DOI] [PubMed] [Google Scholar]
- Turkson J, Jove R. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene. 2000;19:6613–6626. doi: 10.1038/sj.onc.1204086. [DOI] [PubMed] [Google Scholar]
- Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer. 2009;9:798–809. doi: 10.1038/nrc2734. [DOI] [PMC free article] [PubMed] [Google Scholar]