

Abstract
BACKGROUND AND PURPOSE
In cardiovascular pharmacology, electrical and mechanical events can be distinguished, and the phrase ‘electro-mechanical window’ (EMw) describes the temporal difference between these events. We studied whether changes in EMw have potential predictive value for the occurrence of arrhythmias in fentanyl/etomidate-anaesthetized beagle (FEAB) dogs.
EXPERIMENTAL APPROACH
The EMw was calculated as differences between the QT interval and QLVPend in FEAB dogs during atrial pacing, treatment with isoprenaline or atropine, body temperature changes and induction of Torsade de Pointes (TdP) in an LQT1 model.
KEY RESULTS
The electrical systole (QT interval) was shorter than the duration of the mechanical event (QLVPend), providing a positive EMw. Atrial pacing, atropine or body temperature changes had no major effects on EMw, despite large changes in QT duration. However, β-adrenoceptor stimulation (with isoprenaline) decreased the EMw (from 90 to 5 ms) and in combination with HMR1556, a blocker of the slowly activating potassium current (IKs), induced a large negative EMw (−109 ms) and TdP. Prevention of TdP by atenolol or verapamil was associated with a less negative EMw (−23 to −16 ms). Mexiletine, a poorly effective long QT treatment, did not affect the EMw or prevent TdP induction.
CONCLUSIONS AND IMPLICATIONS
The EMw is a marker, other than QT prolongation, of TdP risk in the FEAB model. Therefore, we suggest examining the EMw as a risk marker in cardiovascular safety studies and as a potential biomarker to improve clinical management of long QT syndrome patients, especially in patients with borderline QT prolongation.
LINKED ARTICLE
This article is commented on by Vargas, pp. 1441–1443 of this issue. To view this commentary visit http://dx.doi.org/10.1111/j.1476-5381.2010.00980.x
Keywords: FEAB, Torsade de Pointes, electro-mechanical window, LQT1
Introduction
Prolongation of the repolarization of cardiac muscle – whether by congenital defects such as the long QT syndrome (LQTS) or by the effects of potassium channel blocking drugs such as terfenadine or dofetilide – is associated with a high incidence of ventricular arrhythmia and sudden cardiac death (Algra et al., 1991). As such, QT-interval prolongation has been commonly used as a biomarker for the pro-arrhythmic activity of non-cardiovascular drugs (Crumb and Cavero, 1999). The duration of the electrical systole (QT interval) is dependent on and controlled by several factors, especially heart rate (HR; Fridericia, 1920), but also by circadian rhythms (Molnar et al., 1996), autonomic influences (Magnano et al., 2002) and body temperature (Van der Linde et al., 2008). However, one must remember that this electrical event is not the end result but has the specific purpose of initiating mechanical contraction. Much work in the 1980s studied the so-called electro-mechanical (EM) coupling, which is the relationship between the duration of electrical systole (measured indirectly by the time between the Q wave and the end of the T wave; QT interval), and that of the mechanical systole (measured indirectly by the time between the Q wave and the second heart sound; QS2). In healthy individuals, the duration of the QT interval is shorter than, but closely parallels the duration of the QS2, throughout the normal range of resting HR (Boudoulas et al., 1981a). Changes in autonomic tone (De Caprio et al., 1984) or high circulating catecholamine levels (Boudoulas et al., 1981b) are associated with an inversion of this normal EM ratio, and this ratio has been singled out as a useful indicator of several cardiovascular diseases, such as mitral leaflet prolapse (Chambers and Ward, 1987), coronary artery disease (Boudoulas et al., 1982) and diabetes (Airaksinen et al., 1984). Indeed, Boudoulas et al. (1982) have described this inversion of the EM ratio as ‘the QT>QS2 Syndrome’. The EM ratio has been shown to be altered in patients with the Romano-Ward inherited LQTS (Vincent et al., 1991). Recent investigations of left ventricular contraction duration by tissue Doppler imaging in LQT patients showed that this syndrome is probably not a purely electrical condition (De Ferrari and Schwartz, 2009; Haugaa et al., 2009). We hypothesized that examining the relationship of the electrical systole to the mechanical systole might provide a more sensitive and reliable index of pro-arrhythmic danger than the now ‘traditional’ QT interval prolongation. We investigated the ‘electro-mechanical window’ (EMw) in the fentanyl-etomidate anaesthetized beagle (FEAB) dog model (Van Deuren et al., 2009), and studied the behaviour of this ‘window’ during changes in HR and body temperature, both of which are known to affect the QT-interval duration. We then examined the changes in EMw in an acquired-LQT1 model in anaesthetized beagle dogs (Gallacher et al., 2007), to evaluate its usefulness as a pro-arrhythmic biomarker.
Methods
All animal care and experimental procedures in this investigation were in accordance with ‘The provision of the European Convention’ on the protection of vertebrate animals, which are used for experimental and other scientific purposes, and with ‘the Appendices A and B’, made at Strasbourg on 18 March 1986 (Belgian Act of October 18, 1991). Furthermore, all dogs were examined prior to the experiments and found to be healthy and active. Food (but not water) was withheld for at least 12 h prior to anaesthesia and cardiovascular experimentation.
Anaesthesia and measured parameters
In this study, anaesthesia was induced by intravenous administration of 0.07 mg·kg−1 lofentanil (Janssen Pharmaceutica NV, Beerse Belgium), 0.0015 mg·kg−1 scopolamine (Alcon Laboratories Inc., Fort Worth, TX, USA) and 1.0 mg·kg−1 succinylcholine (Lysthenon 5%, Nycomed, Konstanz, Germany). This mixture was slowly injected into the saphenous vein, without any pre-medication. Dogs were quickly intubated with a cuffed endotracheal tube (FR34, ID8.5, Kruuse, Langeskov, Denmark), immediately (within 30 s) connected to a respirator (Servo ventilator 900C, Siemens, Munich, Germany) and ventilated with 30% oxygen in pressurized air to normocapnia (PaCO2 between 30 and 50 mm Hg). During the experiment, the anaesthesia was maintained with a continuous infusion of etomidate (Janssen Pharmaceutica NV): infusion was started at 1.5 mg·kg−1·h−1, but was slightly adapted according to the individual needs of each dog. Hourly slow bolus injections of 0.025 mg·kg−1 fentanyl were given to ensure continuous pain-free conditions.
All blood vessels needed for catheter insertion were exposed with an electro-cutting and coagulating equipment (Erbotom ACC450, Erbe, Tuebingen, Germany), to minimize bleeding. The surface ECG needles were then placed and connected to an amplifier (ECG lead II limb leads; Emka, Bourre, France). A catheter tip micromanometer with pigtail (Gaeltec, Dunvegan, Scotland) was inserted into the left carotid artery and positioned in the left ventricle for measuring left ventricular pressure (LVP). An open lumen catheter was placed into the femoral artery and positioned close to the heart to obtain blood samples for the measurement of arterial blood gases and metabolic blood parameters (ABL700; Radiometer, Bronshoj, Denmark). These blood parameters were used as additional checks on the physiological condition and stability of the anaesthetized dog over the course of the experiment.
HR (calculated from the RR interval; in b.p.m.) and the QT interval (measured from the onset of the QRS complex to the end of the T wave; in ms) were taken from the surface ECG (lead II). The duration of the mechanical event (QLVPend; measured from onset of the QRS complex to the end of the LVP signal; in ms) was taken from the surface ECG (lead II) and the LVP signal. The EMw was calculated as the difference between QLVPend and QT. Furthermore, the QT intervals were corrected for changes in HR according to the Van de Water formula (QTcV; in ms, Van de Water et al., 1989). One group of dogs was paced by a catheter (Boston Scientific-EP Technologies, San Jose, CA, USA) positioned in the lumen of the right atrium. In another group of dogs, core body temperature (Tc; in °C) was measured continuously within the right ventricle of the heart (Swan-Ganz, Edward Lifesciences LLC, Irvine, CA, USA). All signals and parameters were automatically analyzed (Notocord-Hem 3.3, Croissy-sur-Seine, Paris, France), checked and if necessary recalculated by hand at crucial time points or beats and represented in an Excel file.
Study protocols
The effect of changes in heart rate, autonomic tone and body temperature on the EMw
Twenty-six adult beagle dogs (11 male and 15 female; body weight between 9.6 and 14.7 kg) were used in this study. HR, QT, QTc, QLVPend and EMw were continuously measured in all dogs during baseline and after treatments. In eight dogs, a pacing catheter was placed into the right atrium and after stabilization; the heart was paced at frequencies of 100, 110 and 120 b.p.m. The β-adrenoceptor agonist, isoprenaline, was administered intravenously as a bolus at doses of 1.25, 2.5 and 5 µg·kg−1 to an additional five dogs, and another group of five dogs received atropine as a bolus at doses of 5, 10 and 20 µg·kg−1 i.v. Eight dogs were divided into two groups: four dogs were cooled to 35°C by a blanket of ice and a cold-air fan, and the other four dogs were warmed to 40°C using a heating plate in combination with a heating lamp.
IKs blockade and ‘adrenergic dependent’ Torsade de Pointes
Twenty adult beagle dogs (9 male and 11 female; body weight between 9.0 and 12.8 kg) were used in this study. All dogs were given HMR1556, a blocker of the slowly activating potassium membrane current (IKs: channel nomenclature follows Alexander et al., 2009), starting with an infusion of 0.025 mg·kg−1·min−1 over 30 min and followed by an infusion of 0.05 mg·kg−1·min−1 over 15 min, to achieve a maximum total dose of 1.5 mg·kg−1 i.v.) to mimic the LQT1 syndrome. Bolus injections of isoprenaline (2.5 µg·kg−1 i.v.) were used to simulate exercise and emotional stress, which are known triggers of ‘adrenergic dependent’ Torsade de Pointes (TdP) in LQT1. The first bolus of isoprenaline was given 15 min after starting the HMR1556 infusion and repeated every 5 min until TdP occurred. Some dogs showed TdP after the first isoprenaline challenge, but all 20 dogs showed TdP before or at the total HMR1556 dose of 1.5 mg·kg−1 i.v. (for more details, see Gallacher et al., 2007). Furthermore, the effects of drugs used in the clinic were examined on the inducibility of TdP in the dogs: a sodium channel blocker (mexiletine; 5 mg·kg−1 i.v.), a long acting β-adrenoceptor blocker (atenolol; 0.5 mg·kg−1 i.v.), an L-type Ca2+ channel blocker (verapamil; 0.4 mg·kg−1 i.v.) and saline (0.9% NaCl; 1 mL·kg−1 i.v.). HR, QT, QTc, QLVPend and EMw were continuously measured at baseline, during HMR1556 infusion, and after isoprenaline administration, in the presence or absence of the anti-arrhythmic drugs (Khan, 2004).
Solutions and drugs
Two stock solutions were used; HMR1556 (JNJ-27448538-AAA, both with the same batch no. 23106846) was dissolved at 0.25 mg·mL−1 in hydroxypropyl-β-cyclodextrin (Roquette frères, Lestrem Cedex, France), 20% in pyrogen-free water with added mannitol (pH = 8.62 and 8.25 with an osmolarity of 294 and 314 mosmol·kg−1 respectively). Isoprenaline (Cilag AG, Schaffhausen, Switzerland) was dissolved at a concentration of 0.015 mg·mL−1 in 0.9% NaCl with L(+)-ascorbic acid (pH = 4.2 with an osmolarity of 277 mosmol·kg−1). Mexiletine (Boehringer Ingelheim GmbH, Ingelheim am Rhein, Germany) was diluted to a concentration of 3 mg·mL−1 in pyrogen-free water with added mannitol (pH = 5.2 with an osmolarity of 272 mosmol·kg−1). Atenolol (AkzoNobel N.V., Amsterdam, The Netherlands) was diluted to a concentration of 1 mg·mL−1 in pyrogen-free water with added tartaric acid and mannitol (pH = 4.1 with an osmolarity of 293 mosmol·kg−1). Verapamil (Knoll AG, Ludwigshafen, Germany) was diluted to a concentration of 0.5 mg·mL−1 in pyrogen-free water with added mannitol (pH = 4.3 with an osmolarity of 280 mosmol·kg−1). In the saline group, infusions were given of 0.9% NaCl (Baxter S.A., Lessines, Belgium) with pH = 5.5 and an osmolarity of 308 mosmol·L−1.
Data analysis
Pooled data are expressed as mean ± SEM. Intergroup comparisons were made with ANOVA Dunnett's test on repeated measures (WINKS SDA Software, Cedar Hill, TX, USA). Comparisons within a group were made with a paired Student's t-test (Microsoft Excel 2000). A two-tailed P < 0.05 was considered statistically significant.
Results
The effect of changes in HR, autonomic tone and body temperature on the EMw
Figure 1 shows a representative tracing of the ECG (lead II) and of the LVP signal during a single beat in a FEAB dog. This figure indicates how the different parameters in this study were measured. In eight atrial-paced anaesthetized dogs, the increase in HR (from 74 ± 4 to 120 ± 0 b.p.m.) was associated with similar decreases in QT (–22 ms) and QLVPend (–26 ms), and linear relationships between HR and QT, and between HR and QLVPend. The EMw did not significantly change (Table 1A and Figure 2A).
Figure 1.
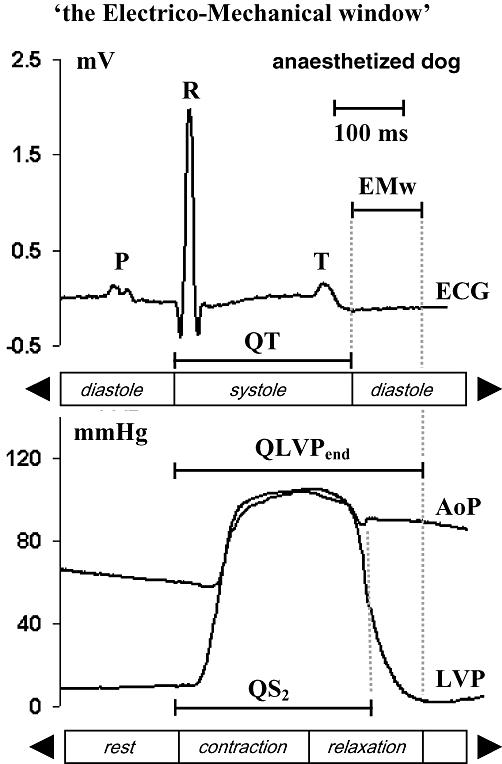
Method for calculating the QT, QLVPend, EMw and QS2, of ECG, AoP (aortic blood pressure) and left ventricular pressure signals of an anaesthetized dog. P, P-wave, R, R-wave, T, T-wave and Q, Q-wave of the ECG (lead II), S2, second heart sound, EMw, electro-mechanical window.
Table 1.
Heart rate (HR), QT interval (QT), mechanical systole (QLVPend) and electro-mechanical window (EMw) values in anaesthetized beagle dogs before and after pacing (A), isoprenaline (B), atropine (C) and during hypo-, normo- and hyperthermia (D)
| Treatment | n | HR (b.p.m.) | QT (ms) | QLVPend (ms) | EMw (ms) | ||
|---|---|---|---|---|---|---|---|
| A | Pacing | Baseline | 8 | 74 ± 10 | 231 ± 11 | 311 ± 12 | 80 ± 7 |
| 120 b.p.m. | 8 | 120 ± 0† | 209 ± 10† | 285 ± 14† | 76 ± 9 | ||
| B | Isoprenaline | Baseline | 5 | 97 ± 10 | 229 ± 7 | 320 ± 10 | 90 ± 7 |
| 2.5 µg·kg−1 | 5 | 191 ± 31† | 188 ± 13† | 193 ± 29† | 5 ± 28† | ||
| C | Atropine | Baseline | 5 | 71 ± 19 | 253 ± 10 | 356 ± 28 | 104 ± 31 |
| 10 µg·kg−1 | 5 | 205 ± 19† | 184 ± 6† | 251 ± 10† | 67 ± 16 | ||
| D | Body | 35.0°C | 4 | 60 ± 9 | 311 ± 34† | 409 ± 24† | 98 ± 33 |
| Temperature | 37.5°C | 8 | 65 ± 16 | 259 ± 23 | 347 ± 21 | 88 ± 18 | |
| 40.0°C | 8 | 81 ± 35 | 213 ± 23† | 296 ± 23† | 83 ± 12 |
Values are expressed as mean ± SEM; data were statistically tested against baseline (A, B and C), and in group D hypothermic (35.0°C) and hyperthermic (40.0°C) were tested against normothermic (37.5°C) dogs.
Intergroup comparisons:
P < 0.05; ANOVA/Dunnett's test (two-sided, unpaired).
Figure 2.
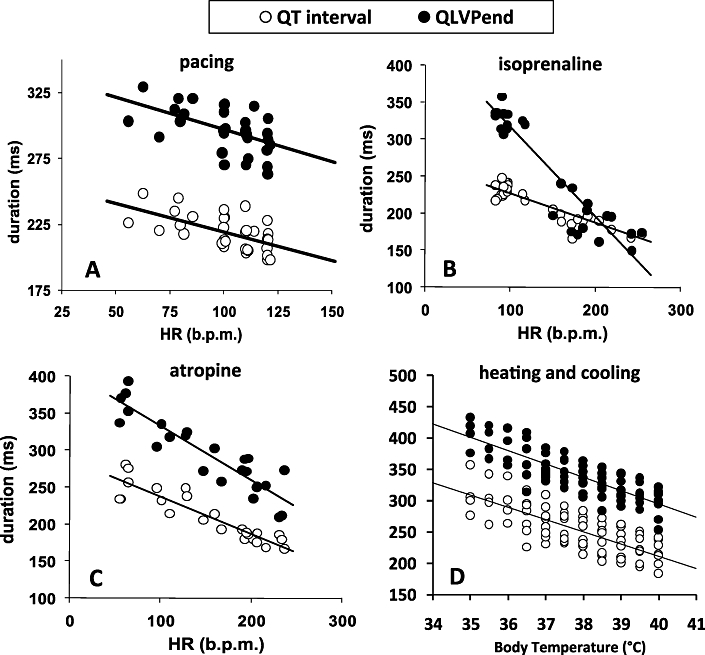
Relationship between mechanical systole (QLVPend) and electrical systole (QT) in after (A) atrial pacing (100, 110 and 120 b.p.m.; n = 6), after (B) isoprenaline (1.25, 2.5 and 5 µg·kg−1; n = 5), after (C) atropine (5, 10 and 20 µg·kg−1; n = 5) and after (D) heating and cooling (n = 8). The straight lines represent the linear regression lines.
Table 1B,C shows that both isoprenaline (2.5 µg·kg−1 iv) and atropine (10 µg·kg−1 iv) induced a comparable increase in HR and a comparable decrease in QT-interval duration. However, isoprenaline shortened QLVPend to a greater extent than atropine, almost abolishing the EMw, whereas atropine did not significantly affect EMw (Table 1B,C). Both compounds showed a linear relationship between HR and QLVPend (Figure 2B,C).
Alterations in body temperature between 35.0 and 40.0°C caused minor changes in HR, but notable changes in the duration of the QT interval and QLVPend (Table 1D and Figure 2D). However, the EMw was not significantly altered.
Effects after IKs blockade and ‘adrenergic-dependent’ TdP
Infusion of HMR1556 (0.05 mg·kg−1·min−1 i.v.) induced a clear increase after maximally 45 min in the duration of the QT interval (P < 0.05; Table 2) and in QTcV-interval duration (P < 0.05), but no notable effect on QLVPend (P = 0.48) and a significant decrease in the EMw (P < 0.05). At this point, no TdPs were noted (Table 2). Bolus injections of isoprenaline (2.5 µg·kg−1 i.v.), in addition to HMR1556, caused an increase in HR (P < 0.05), no effect on the duration of the QT interval (P = 0.51), a slight prolongation of the QTcV interval (P < 0.05) and a marked decrease in QLVPend (P < 0.05), resulting in a large negative EMw (Table 2). During this short period of complete mechanical systole and incomplete electrical systole, aftercontractions were noted on the LVP signal (Figure 3A) and after several beats, an ‘adrenergic-dependent’ TdP appeared (Figure 4). After the induction of TdP, the HMR1556 infusion was stopped and the dogs were immediately defibrillated within 10–20 s after the onset of TdP (as many times as necessary), and stabilized until normal sinus rhythm was achieved.
Table 2.
Heart rate (HR), QT interval (QT), QTcV interval (QTcV), mechanical systole (QLVPend) and electro-mechanical window (EMw) before and after HMR1556 (maximal 1.5 mg·kg−1 i.v.) and isoprenaline (2.5 µg·kg−1 i.v) in anaesthetized dogs (n = 20)
| HR (b.p.m.) | QT (ms) | QTcV (ms) | QLVPend (ms) | EMw (ms) | TdP (%) | |
|---|---|---|---|---|---|---|
| Baseline | 73 ± 3 | 252 ± 6 | 265 ± 5 | 339 ± 8 | 87 ± 7 | 0 |
| HMR1556 | 75 ± 5 | 319 ± 11† | 330 ± 10† | 348 ± 10 | 28 ± 12† | 0 |
| Isoprenaline | 160 ± 9† | 310 ± 8 | 362 ± 8† | 200 ± 6† | −109 ± 6† | 100 |
| Saline | 153 ± 12 | 369 ± 13 | 422 ± 13 | 223 ± 8 | −147 ± 20 | 100 |
| Mexiletine | 152 ± 15 | 301 ± 29 | 352 ± 26† | 175 ± 5† | −126 ± 27 | 100 |
| Atenolol | 108 ± 12† | 316 ± 21 | 353 ± 16† | 293 ± 11† | −23 ± 19† | 0 |
| Verapamil | 159 ± 26 | 255 ± 9† | 304 ± 4† | 240 ± 24 | −16 ± 17† | 0 |
After induction of Torsade de Pointes (TdP), dogs were defibrillated and treated with saline (1 mL·kg−1 i.v.; n = 5), mexiletine (5 mg·kg−1 i.v. n = 5), atenolol (0.5 mg·kg−1 i.v.; n = 5) or verapamil (0.4 mg·kg−1 i.v. n = 5).
Values are expressed as mean ± SEM.
Intergroup comparisons:
P < 0.05; ANOVA/Dunnett's test (two-sided, unpaired).
Figure 3.
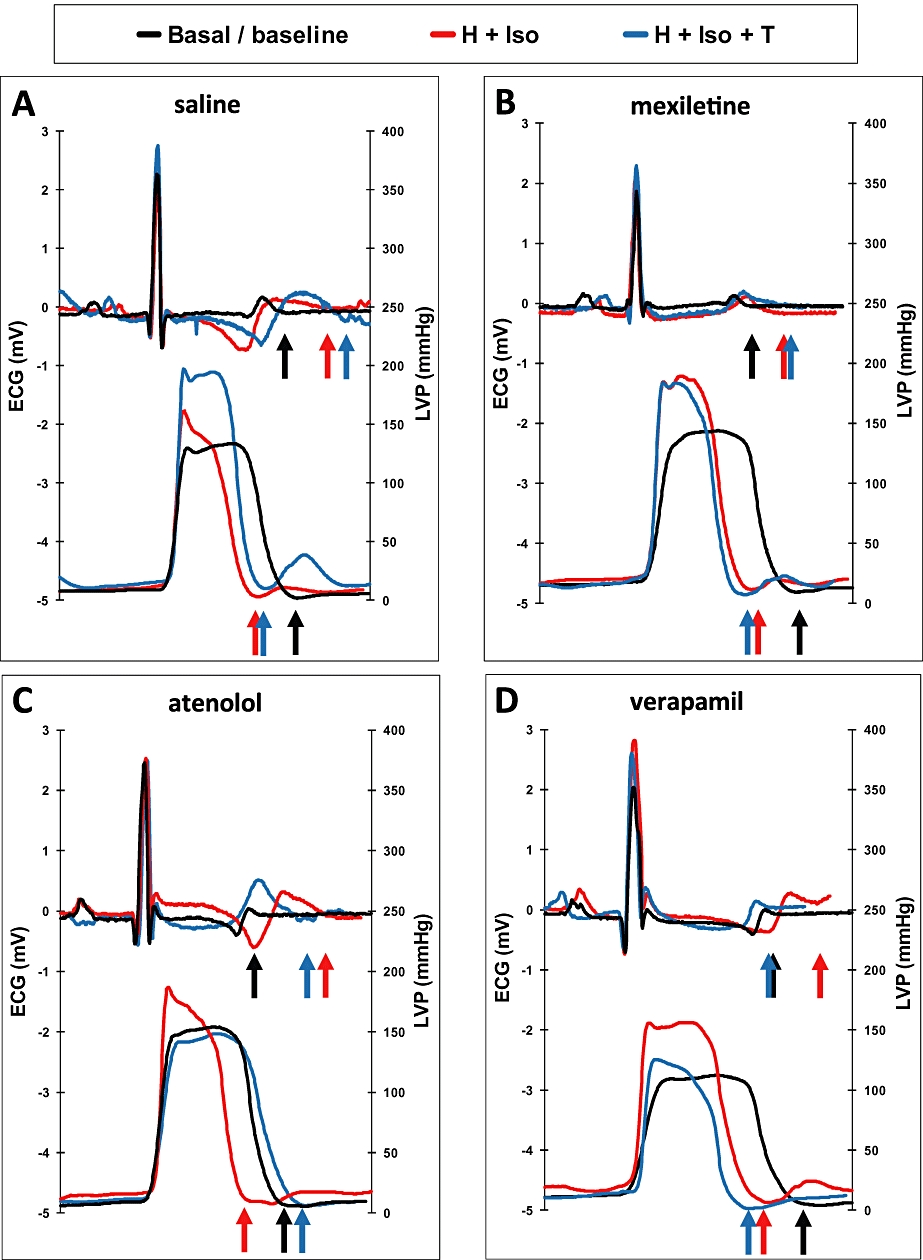
ECG and left ventricular pressure (LVP) signals at baseline (basal/baseline), after HMR1556 + isoprenaline (H + Iso) and after HMR1556 + isoprenaline + treatment (H + Iso + T). Treatments are saline (A), mexiletine (B), atenolol (C) and verapamil (D), and arrows show the end of T-wave or the end of LVP signal, for each of the three conditions.
Figure 4.
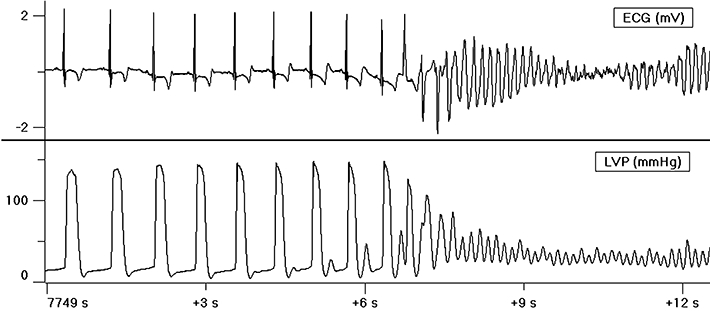
Example of an adrenergic dependent Torsade de Pointes (TdP), with an increased rate pattern and R on T phenomenon. Note the ‘aftercontractions’ on the left ventricular pressure (LVP) signal, just before the induction of TdP.
The protocol [infusion of HMR1556 (0.05 mg·kg−1·min−1 i.v.) and bolus injections of isoprenaline (2.5 µg·kg−1 i.v.) after 15 min] was then repeated after pre-treatment with saline (n = 5), mexiletine (n = 5), atenolol (n = 5) or verapamil (n = 5), administered by infusions at 5 min before the isoprenaline challenges. Vehicle pre-treatment (saline) before the second induction of ‘adrenergic-dependent’ TdP could not prevent a further prolongation of the QT and QTcV intervals and of QLVPend and a more negative EMw (Table 2, lower half). Mexiletine (Figure 3B; Table 2 lower half) pre-treatment produced several changes, relative to those induced by saline pre-treatment. Thus, mexiletine resulted in a tendency for shortening of the QT interval (P = 0.34), a significant shortening of the QTcV interval (P < 0.05) and of the QLVPend (P < 0.05), but the EMw remained large and negative. Neither of these pre-treatments (saline or mexiletine) blocked the development of ‘after contractions’ or the induction of TdP.
In contrast, the induction of TdP was prevented by pretreatment with atenolol or verapamil (Table 2, lower half). The long-acting β-blocker, atenolol (Figure 3C), increased QLVPend, relative to saline pre-treatment (P < 0.05) and the Ca2+ channel blocker, verapamil (Figure 3D), decreased the duration of the QT interval (P < 0.05). In both cases, the EMw increased, that is, was less negative than the values for the saline and mexiletine groups.
Discussion
Our interest in the EM relationship stems from in-depth observations made in an aLQT1 model (blockade of the IKs = KCNQ1/KCNE1) in anaesthetized beagle dogs (Fabritz, 2007; Gallacher et al., 2007). However, our preliminary studies suggest that the same phenomenon can be observed in aLQT2 (IKr+ IKs blockade), and (using heart sounds) also in conscious animals (H.J. van der Linde, unpubl. obs.). LQT1 syndrome is a congenital disease associated with TdP and sudden cardiac death (Shimizu and Antzelevitch, 1998), and is caused by several ‘loss of function’ mutations in the IKs channel (Jost et al., 2005). When we mimicked this pathology in our anaesthetized dog model by blocking the IKs channel with a selective IKs blocker (HMR1556) and adding isoprenaline as a β-adrenoceptor stimulus, we observed no shortening of the QT interval by isoprenaline, whereas the duration of the mechanical systole notably decreased. Shortly before the onset of a reproducible TdP, there was a ‘window’ of up to or more than 100 ms between the end of the short mechanical systole and the end of the relatively long electrical systole.
The existence of this ‘window’ at first appeared paradoxical, because the ventricular muscle was now relaxed, but still electrically depolarized. However, this situation provides an ideal pro-arrhythmic condition, made possible by potential continued activation of voltage-sensitive Ca2+ channels, but more likely by ‘voltage-dependent’ intracellular Ca2+ release (Ferrier and Howlett, 2001; Pasquié & Richard, 2009), and indeed, TdP occurred several beats later. Also noticeable were ‘after contractions’, which consistently occurred during this ‘window’, and generally increased beat-by-beat in magnitude within the last few beats preceding the initiation of the TdP (Gallacher et al., 2007).
Because of the lack of published information about the relationship between the electrical and mechanical systole in pre-clinical dog models, and to achieve more insight in the behaviour of this observed EMw, we measured the QT and QLVPend intervals under different conditions in our FEAB model. We found that under baseline conditions, the mechanical systole (QLVPend) was longer than the electrical systole (QT), as is also the case in healthy human subjects. Furthermore, both intervals (QT and QLVPend) were linearly related to HR with a similar slope, resulting in a fairly constant EMw over a range of HR between 74 and 120 b.p.m. Thus, the EMw seemed to be HR-independent, under normal conditions.
In healthy subjects, the electro-mechanical index (QT/QS2), is affected by changes in autonomic tone (De Caprio et al., 1984) or high circulating catecholamine levels (Boudoulas et al., 1981a). Adrenergic stimulation in humans resulted in an increase in the QT/QS2 ratio, by QS2 shortening. Atropine, in contrast, induced less shortening of the QS2 than expected considering the large increase in HR (Conrad, 1981). Similarly, in our dogs, isoprenaline greatly reduced QLVPend resulting in a significant decrease in the EMw, while atropine reduced EMw to a lesser extent. Our study demonstrates that effects on our proposed pre-clinical biomarker, the EMw, are similar to effects published on the EM index (QT/QS2) in humans.
We previously reported that changes in body temperature have a notable influence on the QT-interval duration (Van der Linde et al., 2008). Lowering the body temperature of the FEAB dog increased the QT-interval duration to potentially pro-arrhythmic lengths (+20%), and increasing its body temperature shortened the QT interval to potentially pro-fibrillatory lengths (–18%). In spite of these extreme changes, no arrhythmias were noted. Interestingly, the EMw did not seem to be affected by body temperature changes and the concomitant alterations in the duration of the QT interval.
Our first observation of the existence of negative values for the ‘window’, prior to the onset of TdP in our aLQT1 model, led to the hypothesis that a negative EMw might predict the occurrence of arrhythmias.
IKs channel blockade, by infusion of HMR1556 to anaesthetized dogs (aLQT1 model), decreased the EMw (from 87 to 28 ms) by increasing the duration of the QT interval, without notably affecting the QLVPend. Although the EMw greatly decreased, the QLVPend interval remained longer than the QT interval. No incidences of arrhythmia or of TdP were observed.
In these IKs-blocked dogs, β-adrenoceptor activation was used to trigger adrenergic-dependent TdP. In normal conditions, β-adrenergic receptor activation induces a shortening of the repolarization by increasing cAMP, resulting in an activation of the IKs channel (Lerman et al., 2001). However, in IKs-blocked dogs, isoprenaline did not decrease the repolarization time (QT or QTcV), but, as expected, decreased the QLVPend, giving rise to a temporal gap between the end of the mechanical systole and the end of the electrical systole, that is, a reversal of the normal state where the mechanical systole is significantly longer than the electrical systole. During this negative ‘window’ (–109 ± 6 ms), ‘after-contractions’ were noted, and adrenergic-dependent TdPs subsequently developed.
To study the effects of treatments for LQT1 on the incidence of TdP and the EMw, we defibrillated the dogs after the first challenge and then treated with saline, mexiletine, atenolol or verapamil. Saline had no notable effect on the durations of the QT or QLVPend, the EMw remained large and negative, and isoprenaline challenge induced TdP in all dogs. Mexiletine, a poorly effective treatment in LQT1 patients (Priori et al., 2000), decreased both QT and QLVPend to a similar extent, but did not notably affect the EMw (–126 ± 27 ms), and did not prevent the induction of TdP by isoprenaline challenge.
β-Blockers do not decrease the QT interval in LQT1 patients (Moss et al., 2000), and indeed, atenolol did not relevantly affect the duration of the QT interval in our dogs. However, pre-treatment with atenolol prevented the induction of TdP in all dogs. By increasing QLVPend, the EMw increased (from –109 to –23 ms). In another group of dogs, the L-type calcium blocker, verapamil, also blocked the induction of TdP in all dogs, without notably affecting QLVPend. In contrast to atenolol, verapamil shortened the QT interval, as described in the literature (Aiba et al., 2005), and in this way increased the EMw from –109 to –16 ms. In summary, both treatments protected the dogs from the induction of TdP: atenolol by increasing the length of the mechanical systole and verapamil by shortening the electrical systole, both increasing EMw to less negative values.
These findings suggest that it is not the prolongation of the QTc interval per se, but rather the development of this large negative EMw that provides the conditions necessary for the induction of TdP, at least in the adrenergic-dependent LQT1 model.
Limitations of the study
The EMw described here, and its relationship to the induction of TdP, is of necessity, a short term predictor of TdP. Although it has been emphasized that the direct relationship between the degree of QT prolongation and the occurrence of TdP is weak, and many drugs which increase the QT interval have not been associated with TdP, such as moxifloxacin, almost all drugs which induce TdP increase the QT interval. Therefore, any known drug, or new compound in development, which increases the QT interval must be suspected of being potentially pro-arrhythmic, until proven otherwise. On the other hand, compounds which induce QT prolongation are not by definition pro-arrhythmic. Probucol prolongs QT and QTc intervals, but simultaneously prolongs the electrical systole, resulting in no major effects on the QT/QS2 relationship (Romics et al., 1988). The authors suggested that this may explain why treatment with probucol shows a low overall risk of TdP in humans (Reinoehl et al., 1996). It may also be that compounds with no effect on QT, but with a major shortening effect on the mechanical systole, could be suspected of being pro-arrhythmic.
In this paper, we describe only the adrenergic-dependent induction of TdP, but as we know from our own experience (Towart et al., 2009) and described by others (Viskin et al., 1996; Noda et al., 2004; Tan et al., 2006), in LQT2 (IKr blockade) patients, the onset of TdP is commonly (incidence 2/3) pause-dependent, and induced by a ‘short-long-short’ sequence pattern. Although, in our experience, short-term QT instability and transmural dispersion seem to have a bigger role in pause-dependent TdP, compared with adrenergic-dependent TdP, and a large negative EMw is also observed in pause-dependent induced TdP (LQT2-like) in the FEAB dog (Towart et al., 2009).
Furthermore, this work has been carried out with dogs anaesthetized with fentanyl and etomidate, using a variety of conditions (pacing, body temperature changes, ‘adrenergic dependent’ TdP induction), and the EMw might behave differently in other settings (e.g. other TdP models such as the chronic AV block dog). Nevertheless, when evaluating studies with more commonly used anaesthetic regimes (pentobarbital or α-chloralose, see Van der Linde, 2009), we noted an ‘EMw-dependent’ induction of TdP in these experiments. In conscious dogs, we also noted an increased QT/QS2– indicating a large negative EMw – when adrenergic-dependent or pause-dependent TdP was induced (see Towart et al., 2009). The clinical relevance of these findings is at present uncertain, although there are some parallels with observations made in the clinic (Vincent et al., 1991), and the similarities we observe between our aLQT1 and aLQT2 dog models and the human LQT1 and LQT2 syndromes suggest that a negative EMw may also appear in humans prior to induction of TdP.
We defined the EMw as the time interval between the end of the complete relaxation and the end of the complete depolarization of the heart. In human studies, the QT/QS2 is used, where the QS2 is the time interval from the start of the ventricular contraction until closure of the aortic valve (second heart sound). At that time the heart is not fully relaxed, and mechanical systole is not complete. We therefore suggest that the EMw is a more appropriate [and now feasible, using Doppler echocardiography (Silva et al., 2002)] way to measure electrical-mechanical disturbances.
Conclusions
β-Adrenoceptor stimulation in combination with a diminished ‘repolarisation-reserve’ was applied to induce LQT1- ‘adrenergic dependent’ TdP in an anaesthetized dog model. Our results confirm that prolongation of QT (QTc) per se is not pro-arrhythmic, in these healthy dogs, without cardiac co-morbidities. However, whenever TdP was induced, we observed a time window between the end of a relatively shortened mechanical systole and the end of a relatively prolonged QT. We defined this EMw as the difference between QLVPend and QT. Mexiletine (a sodium channel blocker), which blocked the QT prolongation but also decreased the QLVPend, had no effect on the EMw, and did not block the induction of ‘adrenergic-dependent’ TdP. In contrast, agents such as atenolol (β-blocker) and verapamil (L-type calcium channel blocker) fully blocked the induction of ‘adrenergic-dependent’ TdP, and greatly reduced the size of the negative window, although in different ways. We suggest that the appearance of a negative window is a prerequisite to achieve a condition in which an R on T will induce TdP (present in all 200 inductions of TdP achieved in our laboratory to date using this model).
We have demonstrated that, in contrast to the duration of the QT interval, the EMw is independent of changes in HR and body temperature and that this parameter can serve as an estimate of the risk of ventricular arrhythmia (TdP) in our dog model of LQT1. Moreover, we suggest the use of this parameter as a potential risk-marker in pre-clinical cardiovascular safety studies and as a biomarker to improve clinical management of LQTS patients, especially in patients with borderline QT prolongation.
Acknowledgments
We want to thank Charles Calder for his excellent work on the anaesthetized dog studies and for creating Figure 3. This work was supported financially by the Institute for Encouragement of Innovation by Science and Technology in Flanders (agentschap voor Innovatie door Wetenschap en Technologie; IWT).
Glossary
Abbreviations
- aLQT
acquired (drug-induced) long QT
- EMw
electro-mechanical window
- FEAB
fentanyl/etomidate-anaesthetised beagle
- IKr
rapidly activating potassium membrane current
- IKs
slowly activating potassium membrane current
- ISR
increased sinus rate
- LQT
long QT
- SLS
short-long-short
- TdP
Torsade de Pointes
Conflicts of interest
The authors are all employees of Johnson & Johnson.
References
- Aiba T, Shimizu W, Inagaki M, Noda T, Miyoshi S, Ding W-G. Cellular and ionic mechanism for drug-induced long QT syndrome and effectiveness of Verapamil. J Am College Cardiology. 2005;45:300–307. doi: 10.1016/j.jacc.2004.09.069. [DOI] [PubMed] [Google Scholar]
- Airaksinen J, Ikäheimo M, Kaila J, Linnaluoto M, Takkunen J. Systolic time intervals amd the QT-QS2 interval in young female diabetics. Ann Clin Res. 1984;16:188–191. [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Algra A, Tijssen J, Roelandt J, Pool J, Lubsen J. QTc prolongation measured by standard 12 lead electrocardiogram is an independent risk factor for sudden death. Circulation. 1991;83:1888–1894. doi: 10.1161/01.cir.83.6.1888. [DOI] [PubMed] [Google Scholar]
- Boudoulas H, Geleris P, Lewis RP, Leier CV. Effect of increased adrenergic activity on the relationship between electrical and mechanical systole. Circulation. 1981a;64:28–33. doi: 10.1161/01.cir.64.1.28. [DOI] [PubMed] [Google Scholar]
- Boudoulas H, Geleris P, Lewis RP, Rittgers SF. Linear relationship between electrical systole, mechanical systole and heart rate. Chest. 1981b;80:613–617. doi: 10.1378/chest.80.5.613. [DOI] [PubMed] [Google Scholar]
- Boudoulas H, Sohn YH, O'Neill W, Brown R, Weissler M. The QT>QS2 syndrome: a new mortality risk indicator in coronary artery disease. Am J Cardiology. 1982;50:1229–1235. doi: 10.1016/0002-9149(82)90454-4. [DOI] [PubMed] [Google Scholar]
- Chambers JB, Ward DE. The QT and QS2 intervals in patients with mitral leaflet prolapse. Am Heart J. 1987;114:355–361. doi: 10.1016/0002-8703(87)90503-5. [DOI] [PubMed] [Google Scholar]
- Conrad KA. Effects of atropine on diastolic time. Circulation. 1981;63:371–377. doi: 10.1161/01.cir.63.2.371. [DOI] [PubMed] [Google Scholar]
- Crumb W, Cavero I. QT interval prolongation by non-cardiovascular drugs: issues and solutions for novel drug development. Pharmaceutical Sci & Technology Today. 1999;2:270–280. doi: 10.1016/s1461-5347(99)00172-8. [DOI] [PubMed] [Google Scholar]
- De Caprio L, Ferro G, Cuomo S, Volpe M, Artialo D, De luca N, et al. QT/QS2 ratio as an index of autonomic tone changes. Am J Cardiology. 1984;53:818–822. doi: 10.1016/0002-9149(84)90411-9. [DOI] [PubMed] [Google Scholar]
- De Ferrari GM, Schwartz PJ. Long QT syndrome, a purely electrical disease? Not anymore. Eur Heart J. 2009;30:253–255. doi: 10.1093/eurheartj/ehn587. [DOI] [PubMed] [Google Scholar]
- Fabritz L. Drug-induced Torsade de Pointes – a form of mechano-electric feedback? Cardiovascular Res. 2007;76:202–203. doi: 10.1016/j.cardiores.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Ferrier GR, Howlett SE. Cardiac excitation-contraction coupling: role of membrane potential in regulation of contraction. Am J Physiol Heart Circ Physiol. 2001;280:1928–1944. doi: 10.1152/ajpheart.2001.280.5.H1928. [DOI] [PubMed] [Google Scholar]
- Fridericia L. Die Systolendauer in Electrocardiogramm bei normalen Menschen und bei Herzkranken. Acta Medica Scandinavia. 1920;53:469–486. [Google Scholar]
- Gallacher DJ, Van de Water A, van der Linde H, Hermans A, Lu HR, Towart R, et al. In vivo mechanisms precipitating Torsades de Pointes in a canine model of drug-induced long-QT1 syndrome. Cardiovascular Res. 2007;76:247–256. doi: 10.1016/j.cardiores.2007.06.019. [DOI] [PubMed] [Google Scholar]
- Haugaa KH, Edvardsen T, Leren TP, Gran JM, Smiseth OA, Amlie JP. Left ventricular mechanical dispersion by tissue Doppler imaging: a novel approach for identifying high-risk individuals with long-QT syndrome. Eur Heart J. 2009;30:330–337. doi: 10.1093/eurheartj/ehn466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jost N, Virag L, Bitay M, Takacs J, Lengyel C, Biliczki P. Restricting excessive cardiac action potential and QT prolongation: a vital role for IKs in human ventricular muscle. Circulation. 2005;112:1392–1399. doi: 10.1161/CIRCULATIONAHA.105.550111. [DOI] [PubMed] [Google Scholar]
- Khan MH. Oral class III antiarrhythmics: what is new? Curr Opin Cardiol. 2004;19:47–51. doi: 10.1097/00001573-200401000-00010. [DOI] [PubMed] [Google Scholar]
- Lerman B, Engelstein E, Burkhoff D. Mechanoelectrical feedback. Role of β-adrenergic receptor activation in mediating load-dependent shortening of ventricular action potential and refractoriness. Circulation. 2001;104:486–490. doi: 10.1161/hc2901.091397. [DOI] [PubMed] [Google Scholar]
- Magnano A, Holleran S, Ramakrishnan R, Reiffel J, Bloomfield D. Autonomic nervous system influences on QT interval in normal subjects. J Am College Cardiology. 2002;39:1820–1826. doi: 10.1016/s0735-1097(02)01852-1. [DOI] [PubMed] [Google Scholar]
- Molnar J, Zhang F, Weiss J, Ehlert F, Rosenthal J. Diurnal pattern of QTc interval: how long is prolonged? Possible relation to circadian triggers of cardiovascular events. J Am College Cardiology. 1996;27:76–83. doi: 10.1016/0735-1097(95)00426-2. [DOI] [PubMed] [Google Scholar]
- Moss AJ, Zareba W, Hall WJ, Schwartz PJ, Crampton RS, Benhorin J. Effectiveness and limitations of β-blocker therapy in congenital long-QT syndrome. Circulation. 2000;101:616–623. doi: 10.1161/01.cir.101.6.616. [DOI] [PubMed] [Google Scholar]
- Noda T, Shimizu W, Satomi K, Suyama K, Kurita T, Aihara N, et al. Classification and mechanism of Torsade de Pointes initiation in patients with congenital long QT syndrome. Eur Heart J. 2004;25:2149–2154. doi: 10.1016/j.ehj.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Pasquié J-L, Richard S. Prolongation in QT interval is not predictive of Ca2+-dependent arrhythmias: implications for dug safety. Expert Drug Saf. 2009;8:57–72. doi: 10.1517/14740330802655454. [DOI] [PubMed] [Google Scholar]
- Priori SG, Ronchetti E, Memmi M. Gene specific therapy for arrhythmogenic disorders. Italian Heart J. 2000;1(Suppl. 3):52–54. [PubMed] [Google Scholar]
- Reinoehl J, Frankovich D, Machado C, Kawasaki R, Baga J, Pires LA, et al. Probucol-associated tachyarrhytmic events and QT prolongation: importance of gender. Am Heart J. 1996;131:1184–1191. doi: 10.1016/s0002-8703(96)90095-2. [DOI] [PubMed] [Google Scholar]
- Romics L, Littmann L, Laszlo Z, Fenyvesi T. The effects of probucol on QT/QS2 relation and systolic time intervals. Int J Cardiology. 1988;19:303–308. doi: 10.1016/0167-5273(88)90234-3. [DOI] [PubMed] [Google Scholar]
- Shimizu W, Antzelevitch C. Cellular basis for the ECG features of the LQT1 form of the long-QT syndrome: effects of β-adrenergic agonists and antagonists and sodium channel blockers on transmural dispersion of repolarisation and Torsade de Pointes. Circulation. 1998;98:2314–2322. doi: 10.1161/01.cir.98.21.2314. [DOI] [PubMed] [Google Scholar]
- Silva CES, Ferreira LDC, Peixoto LB, Monaco CG, Gil MMA, Ortiz J. Study of the myocardial contraction and relaxation velocities through Doppler tissue imaging echocardiography. A new alternative in the assessment of the segmental ventricular function. Arquivos Brasileiros Cardiologia. 2002;78:206–211. doi: 10.1590/s0066-782x2002000200009. [DOI] [PubMed] [Google Scholar]
- Tan HL, Bardai A, Shimizu W, Moss AJ, Schilze-Bahr E, Noda T, et al. Genotype-specific onset of arrhythmias in congenital long-QT syndrome. Possible therapy implications. Circulation. 2006;114:2096–2103. doi: 10.1161/CIRCULATIONAHA.106.642694. [DOI] [PubMed] [Google Scholar]
- Towart R, Linders JTM, Hermans AN, Rohrbacher J, van der Linde HJ, Ercken M, et al. Blockade of the IKs potassium channel: an overlooked cardiovascular liability in drug safety screening? J Pharmacol Toxicol Methods. 2009;60:1–10. doi: 10.1016/j.vascn.2009.04.197. [DOI] [PubMed] [Google Scholar]
- Van de Water A, Verheyen J, Xhonneux R, Reneman R. An improved method to correct the QT-interval of the electrocardiogram for changes in heart rate. J Pharmacol Methods. 1989;22:207–217. doi: 10.1016/0160-5402(89)90015-6. [DOI] [PubMed] [Google Scholar]
- Van der Linde H. Induction of β-adrenergic dependent TdP (LQT1-like) in fentanyl, α-chloralose and sodium pentobarbital anaesthetised beagle dogs. J Pharmacol Toxicol Methods. 2009;60:248. [Google Scholar]
- Van der Linde H, Van Deuren B, Teisman A, Towart R, Gallacher DJ. The effect of changes in core body temperature on the QT interval in beagle dogs: a previously ignored phenomenon, with a method for correction. Br J Pharmacol. 2008;154:1474–1481. doi: 10.1038/bjp.2008.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Deuren B, Van Ammel K, Somers Y, Cools F, Straetemans R, van der Linde HJ, et al. The fentanyl/etomidate-anaesthetised beagle (FEAB) dog: a versatile in vivo model in Cardiovascular Safety Research. J Pharmacol Toxicol Methods. 2009;60:11–23. doi: 10.1016/j.vascn.2009.04.195. [DOI] [PubMed] [Google Scholar]
- Vincent GM, Deepak J, Timothy KW. Effects of exercise on heart rate, QT, QTc and QT/QS2 in the Romano-Ward inherited long QT syndrome. Am J Cardiology. 1991;68:498–503. doi: 10.1016/0002-9149(91)90785-j. [DOI] [PubMed] [Google Scholar]
- Viskin S, Alla SR, Barron HV, Heller K, Saxon L, Kitzis I, et al. Mode of onset of Torsade de Pointes in congenital Long QT Syndrome. J Am Coll Cardiol. 1996;28:1262–1268. doi: 10.1016/s0735-1097(96)00311-7. [DOI] [PubMed] [Google Scholar]