

Abstract
BACKGROUND AND PURPOSE
Torsades de pointes (TdP) liability is a stochastic event, which indicates that unidentified factors have an important role in facilitating the initiation of TdP by increasing the probability of TdP occurrence. We sought to identify factors that facilitate drug-induced TdP.
EXPERIMENTAL APPROACH
We studied dofetilide-induced TdP in pentobarbital-anaesthetized, phenylephrine-sensitized rabbits, seeking biomarkers that discriminated between the animals that experienced TdP (‘TdP+’ animals) and those that did not (‘TdP−’ animals). As novel variables, the beat-to-beat variability and instability of ECG intervals were measured at preset times, irrespective of whether or not hearts were in stable sinus rhythm (‘absolute’ variability and instability). Autonomic activity was also determined.
KEY RESULTS
Dofetilide delayed repolarization and induced arrhythmias prior to TdP. The variability of the coupling interval and shape of arrhythmic beats before TdP were significantly greater in the ‘TdP+’ group than in the ‘TdP−’ group. Accordingly, the ‘absolute’ variability and instability of the ECG intervals were significantly elevated in the ‘TdP+’ group. Phenylephrine increased significantly the up-baroreflex sensitivity in the ‘TdP+’ group before dofetilide administration.
CONCLUSIONS AND IMPLICATIONS
‘Preceding’ arrhythmias have characteristics that permit prediction of TdP occurrence: the more chaotic the ventricular rhythm, the greater the probability of TdP initiation. This suggests that complexity of the arrhythmic beats may play an important mechanistic role in TdP genesis. The electrical instability quantified by the novel ‘absolute’ variability and instability parameters correlates with the probability of TdP occurrence. Baroreflex may contribute to TdP genesis in vivo.
Keywords: α1-adrenoceptor stimulation, anaesthetized rabbit, baroreflex, biomarkers, coupling interval, dofetilide, phenylephrine, preceding arrhythmias, absolute beat-to-beat variability and instability, torsades de pointes
Introduction
The pro-arrhythmic liability of any drug under development must be assessed to minimize the risk of life-threatening drug-induced arrhythmias, such as torsades de pointes (TdP) during pharmacotherapy (Farkas and Nattel, 2010).
The most commonly used animal model for the in vivo screening for TdP liability is the α1-adrenoceptor-stimulated anaesthetized rabbit model developed by Carlsson et al. (1990). In this model, the ability of a test agent to evoke TdP is evaluated during co-administration of a ‘priming’ substance, the selective α1-adrenoceptor agonist methoxamine (Carlsson et al., 1990) or phenylephrine (Farkas et al., 1998; 2002;). The role of the priming α1-adrenoceptor stimulation is not completely understood. Interestingly, co-administration of the selective inhibitor of the rapid component of the delayed rectifier K+ current (IKr) dofetilide and the α1-adrenoceptor agonist methoxamine only infrequently evoked TdP in isolated rabbit hearts (Farkas et al., 2006). This has three important caveats. First, in vivo factors may have a crucial role in assisting the genesis of TdP and one of the possible contributors to genesis of TdP in vivo is the autonomic nervous system. A recent study revealed that bilateral vagotomy prevented drug-induced TdP in anaesthetized rabbits (Farkas et al., 2008). Second, it predicates the use of surrogate biomarkers for TdP. Indeed, surrogate biomarkers such as QT prolongation are pre-eminent in TdP liability testing, to the extent that most methodological approaches combine multiple biomarkers in an integrated risk assessment (Pugsley et al., 2008). Third, in this and all TdP liability tests, it is not possible to predict precisely whether or not TdP will occur in any individual animal or isolated heart. This shows that TdP liability is a stochastic event, and indicates that hitherto unidentified factors have an important role in facilitating the initiation of TdP (by increasing the probability of TdP occurrence).
The aim of the present study was to identify factors that facilitate the initiation of drug-induced TdP. We selected dofetilide-induced TdP in anaesthetized, α1-adrenoceptor-stimulated rabbits, as this is perhaps one of the most extensively studied conditions for TdP (i.e. model plus drug). The approach was to identify the biomarkers and the endogenous determinants of TdP that best discriminated between the animals that experienced TdP (‘TdP+’ animals) and those that did not (‘TdP−’ animals). For completeness we included relevant variables including basic haemodynamics, blood gases, repolarization-related parameters, arrhythmias prior to TdP and activity of the autonomic nervous system in the analysis.
Methods
Animals
Animals were handled in accordance with the European Community guidelines for the use of experimental animals, and the protocol was reviewed and approved by the Ethical Committee for the Protection of Animals in Research at the University of Szeged, Hungary. The pro-arrhythmic activity of dofetilide was examined by using the in vivo method of Carlsson et al. (1990) with minor modifications (Vincze et al., 2008). The experiments were performed on female New Zealand White rabbits weighing 2.7 ± 0.04 kg (n = 30).
Anaesthesia
A catheter was introduced into the marginal vein of the left ear for blood sampling and induction of anaesthesia. One millilitre of blood was withdrawn via the catheter to determine serum ion (K+, Na+ and Ca2+) concentrations in the conscious animal. After blood sampling, the animals were anaesthetized with pentobarbital (i.v.). In accordance with our previous study (Vincze et al., 2008), the depth of anaesthesia was carefully assessed throughout the experiment. Surgery started when the pedal withdrawal reflex had disappeared and there was no response to pinching of the ears. The dose of anaesthetic was adjusted to the need of the animal. When there was any sign of pain or consciousness, extra bolus of 6–9 mg pentobarbital was administered. The dose of pentobarbital needed for sufficiently deep anaesthesia was 53 ± 2 mg·kg−1 (at a rate of 30 mg·mL−1·min−1). The rabbits were retrospectively divided into two groups according to the presence or the absence of TdP, that is, the animals that experienced TdP formed the ‘TdP+’ group, and the animals that did not experience this arrhythmia formed the ‘TdP−’ group. The administered doses of pentobarbital did not differ significantly between the ‘TdP+’ and the ‘TdP−’ groups (55 ± 2 mg·kg−1 and 49 ± 2 mg·kg−1 respectively). When anaesthesia was not deep enough after induction and there was any sign of pain or consciousness, an extra bolus of pentobarbital was administered (to two rabbits): 7 and 4.3 mg·kg−1 respectively.
Surgical preparation
A catheter was introduced into the right carotid artery to measure blood pressure and to withdraw arterial blood for blood gas analysis. The catheter was filled with heparin/saline (0.9% w/v NaCl containing 15 I.U.·mL−1 heparin) and connected to a pressure transducer of a National Instruments data acquisition hardware (PC card, National Instruments, Austin, TX, USA) using SPEL Advanced Haemosys software (version 2.76, Experimetria Ltd. and Logirex Software Laboratory, Budapest, Hungary). Blood pressure was recorded at a sampling rate of 1000 Hz. Catheters were introduced into the right jugular vein and the marginal vein of the right ear for the infusion of drugs. After tracheal cannulation, artificial ventilation was started (Harvard rodent ventilator, model 683, Harvard Apparatus, South Natick, MA, USA) with room air at a rate of 40 strokes·min−1, a stroke volume of ∼6 mL·(kg body weight)−1 and a positive end-expiratory pressure of 1–2 cm H2O. Blood gases were monitored with a Stat Profile® Critical Care Xpress pH/blood gas analyser (Nova Biomedical, Waltham, MA, USA). If necessary, the stroke volume of the ventilation pump and the positive end-expiratory pressure were adjusted to maintain the blood gases within the normal range. Subcutaneous needle electrodes were inserted in all four limbs, and leads I, II and III of the ECG were recorded simultaneously at a sampling rate of 1000 Hz with the National Instruments system. The preparation was followed by a minimum 10 min stabilizing period.
Experimental protocol
After a 10 min baseline period, α1-adrenoceptor agonist phenylephrine infusion was started intravenously at increasing rates (i.e. 3, 6, 9, 12 and 15 µg·kg−1·min−1 for 3, 3, 3, 3 and 5 min respectively). From the 27th minute, dofetilide (0.02 mg·kg−1·min−1 i.v., for 30 min) was administered simultaneously with the background phenylephrine infusion (at a rate of 15 µg·kg−1·min−1) until the end of the experiments (Figure 1). The selective IKr inhibitor dofetilide was chosen as its TdP liability is well established in humans (Pedersen et al., 2007) and in experimental models (Farkas et al., 2006; Vincze et al., 2008). The infusion rate of dofetilide was chosen as this rate of the dofetilide infusion induced TdP approximately in half of rabbits in a pilot study.
Figure 1.
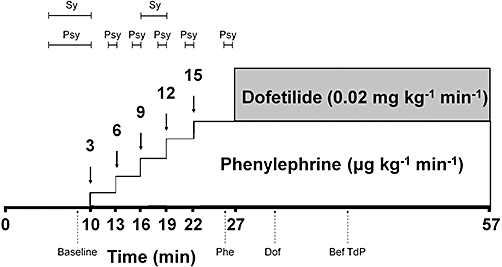
Drug administration protocol. Phenylephrine is administered at increasing rates (3, 6, 9, 12 and 15 µg·kg−1·min−1). Dofetilide is infused at a rate of 0.02 mg·kg−1·min−1. Segments indicate the uninterrupted periods of the measurement of sympathetic (Sy) and parasympathetic (Psy) activity. ECG variability and instability parameter measurements were performed at four time points indicated with upward, dashed arrows. Baseline, value determined from 40 consecutive ventricular beats before drug administration; Phe, value determined from 40 consecutive ventricular beats in the last minute of the 15 µg·kg−1·min−1 phenylephrine infusion rate before the start of dofetilide infusion; Dof, the value determined from 40 consecutive RR intervals shortly after the beginning of the dofetilide infusion in the 2nd–4th minutes of dofetilide infusion (‘Sinus’ variability and instability) or in the 4th minute of dofetilide infusion (‘Absolute’ variability and instability); Bef TdP, the value measured from 40 consecutive ventricular beats immediately before TdP occurred in the ‘TdP+’ group of animals or at an equivalent time point in the ‘TdP−’ group of animals.
In a recent study we found that dofetilide infusion at a rate of 0.04 mg·kg−1·min−1 induced TdP in 40% of rabbits anaesthetized with Na-pentobarbital (Vincze et al., 2008). In contrast, dofetilide at a rate of 0.02 mg·kg−1·min−1 induced TdP in 67% of the rabbits anaesthetized with Na-pentobarbital in the present study. This difference in TdP incidence between the two studies may be due to not fully identical experimental settings in the two studies (e.g. different experimenter, laboratory, time and source of dofetilide). However, the conclusions of the present study and Vincze et al. (Vincze et al., 2008) are not influenced by this difference in TdP incidence, as within-study experimental settings were standard in these two independent investigations.
ECG analysis
After completing experiments the ECG was replayed and the arrhythmia incidences, the time to onset, and the duration of ventricular arrhythmias were obtained by a skilled expert. After completing arrhythmia analysis, the experiments were encoded and the ECG intervals (RR, PQ, QRS, QT and Tpeak–Tend) were measured by the same expert in a blinded and prospective manner without the knowledge that the actual animal did or did not experience TdP later in the course of the experiment.
The blood pressure and the ECG intervals were measured at predetermined time points. The RR, PQ, QRS, QT and Tpeak–Tend intervals were measured by the manual positioning of on-screen markers. The QT interval measurement was performed as we described previously (Farkas et al., 2004). Briefly, the QT interval was defined as the time between the first deviation from the isoelectric line during the PQ interval until the end of the TU wave. The T (or U) wave frequently overlapped the P wave of the following sinus-origin beat, due to the relatively high heart rate of the rabbit or to substantial QT prolongation. In these cases, the extrapolation method was used (Farkas et al., 2004), that is, the end of the TU wave was extrapolated from the curve of the TU wave to the isoelectric line under the P wave. All the values for the QT interval were corrected for the heart rate with the equation QTcL = QT − 0.704 (RR-250) developed for pentobarbital-anaesthetized rabbits (Batey and Coker, 2002). Duration of the JT interval was subsequently calculated by subtraction of the QRS interval from the corresponding QT interval. The ventricular electric diastolic interval was defined as the time between the end of the TU wave until the beginning of the following QRS complex. When the end of the TU wave was chopped off by the subsequent QRS complex, due to substantial QT prolongation or to occurrence of an R-on-T phenomenon, then ventricular electric diastolic interval was arbitrarily taken as 0.000001 ms (not 0 ms). The Tpeak–Tend interval was measured in the standard limb lead II of the ECG in the last minute before the start of the phenylephrine infusion (baseline), in the last minute before the start of the dofetilide infusion and in the 4th minute of the dofetilide infusion (further measurements were prevented by the frequent occurrence of dofetilide-induced arrhythmias). When the end of the Tpeak–Tend interval overlapped the following P wave, the extrapolation method (Farkas et al., 2004) was applied. Four consecutive Tpeak–Tend intervals were measured and averaged at each time point.
The T-wave lability was quantified as a root mean square of the differences in the voltage between corresponding signal values of subsequent beats. The magnitude of aperiodic T-wave lability was quantified by measuring a T-wave lability index with a computer program developed by us following the descriptions of Nemec et al. (2003).
Arrhythmia diagnosis
Ventricular premature beat, bigeminy, salvo and ventricular fibrillation were defined according to the Lambeth Conventions (Walker et al., 1988). When continuous ventricular fibrillation lasted longer than 120 s, the experiment was terminated and the ventricular fibrillation was defined as lethal. TdP was defined as a polymorphic ventricular tachycardia consisting of four or more ventricular complexes where clear twisting of the QRS complexes around the isoelectric axis could be seen in at least one ECG lead (Figure 2). Runs of four or more ventricular premature beats without the TdP-like twisting QRS morphology were differentiated from TdP and were defined as ventricular tachycardia (Figure 2). Blocks in the conduction system were also monitored. Conduction disturbances included atrioventricular blocks and intraventricular conduction defects (right or left bundle branch blocks).
Figure 2.
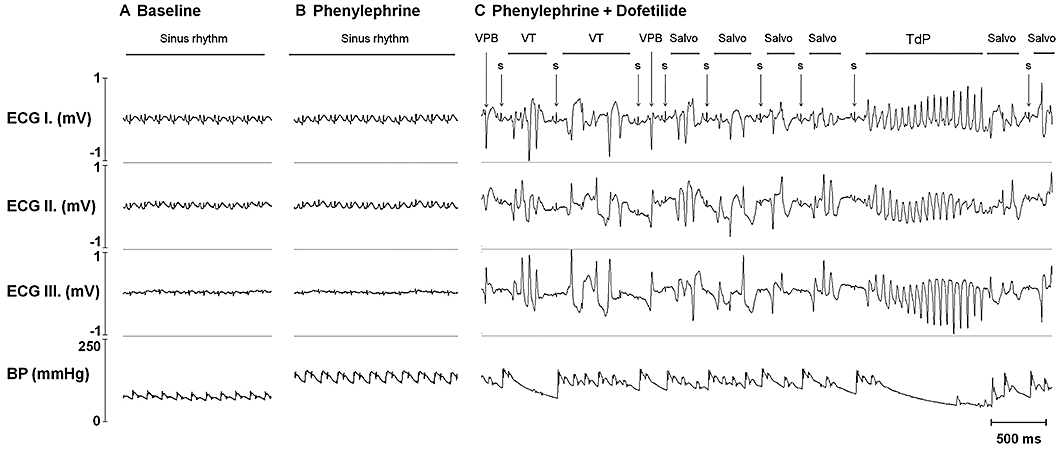
An example of dofetilide-induced torsades de pointes ventricular tachycardia (TdP). ECG and arterial blood pressure signals recorded before drug administration (A), in the last minute of the 15 µg·kg−1·min−1 phenylephrine infusion rate before the start of dofetilide infusion (B) and during dofetilide infusion immediately before the first run of TdP occurred (C). ECG I–III, electrocardiogram leads I–III; BP, arterial blood pressure signal recorded in the right carotid artery; VPB, ventricular premature beat; VT, ventricular tachycardia different from TdP; S, sinus beat. Note the chaotic ventricular rhythm and the corresponding, haemodynamically non-compromising, irregular fluctuation in the blood pressure before TdP.
Measurement of the beat-to-beat variability and instability of the ECG intervals: ‘sinus’ and ‘absolute’ variability and instability
The ‘sinus’ beat-to-beat variability and instability of the RR and QT intervals were determined from the manual measurement data on 40 consecutive RR intervals and the corresponding QT intervals in sinus rhythm (‘sinus’ variability and instability) in the last minute of the drug-free state, in the last minute of the phenylephrine infusion before the dofetilide administration and in the 2nd–4th minutes of the dofetilide infusion, when arrhythmias were still infrequent.
The ‘absolute’ beat-to-beat variability and instability of the RR, QRS, JT, QT and electric diastolic intervals were determined irrespective of the presence or absence of sinus rhythm from manual measurement data on 40 consecutive RR intervals and the corresponding QRS and QT intervals at predetermined time points, that is, in the last minute of the drug-free state, in the last minute of the phenylephrine infusion before the dofetilide administration and in the 4th minute of the dofetilide infusion. The ‘absolute’ beat-to-beat variability and instability of the RR, QRS, JT, QT and electric diastolic interval were also determined at a 4th time point immediately before TdP occurred in the ‘TdP+’ group of animals or at an equivalent time point in the rabbits that did not experience TdP (700 s after the start of dofetilide infusion in the ‘TdP−’ group of animals). That is, 40 consecutive ventricular cycles (RR intervals and contemporaneous QRS and QT intervals) were measured regardless of rhythm at the predetermined time point. In case of arrhythmic ventricular beats, the QT interval was defined as the time between the start of the QRS complex of the arrhythmic ventricular beat until the end of the T wave (or U wave, when it was present). The T (or U) wave frequently overlapped the P wave of the following sinus-origin beat, due to the relatively high heart rate of the rabbit or to substantial QT prolongation. In these cases, the extrapolation method was used (Farkas et al., 2004), that is, the end of the TU wave was extrapolated from the curve of the TU wave to the isoelectric line under the P wave. The extrapolation method was also used in order to measure the length of the QT interval when the end of the TU wave was chopped off by the subsequent QRS complex, due to substantial QT prolongation or to occurrence of an R-on-T phenomenon. In these cases, the end of the TU wave was extrapolated from the curve of the TU wave to the isoelectric line through the subsequent QRS complex.
All of the beat-to-beat variability and instability parameters of the RR and QT intervals were determined in a blinded and prospective manner at the first three time points (see above). Measurement of the ‘absolute’ beat-to-beat variability and instability of the RR and QT intervals at the 4th time point (immediately before TdP occurred or at the mean TdP onset time) was performed subsequently in an unblinded manner.
We developed a computer program in a .NET environment to obtain the following parameters of the beat-to-beat variability and instability of the RR, QRS, JT, QT and electric diastolic intervals: the ‘root mean square’ for a sequence of RR, QRS, JT, QT and electric diastolic interval durations, the ‘root mean square’ and the ‘standard deviation’ of the successive differences of the RR, QRS, JT, QT and electric diastolic intervals, the ‘Instability’ of the RR, QRS, JT, QT and electric diastolic intervals, the ‘short-term variability’ (STV) and the ‘long-term variability’ (LTV) were calculated as described earlier (Hondeghem et al., 2001; Thomsen et al., 2004; Vincze et al., 2008; Farkas et al., 2009). We also calculated the ‘total instability’, the ‘short-term instability’ and the ‘long-term instability’ (LTI) of the consecutive RR, QRS, JT, QT and electric diastolic intervals as described previously (van der Linde et al., 2005).
Characterization of the arrhythmic beats during dofetilide administration
The multiplicity of the arrhythmic beats was quantified by measuring the coupling interval, the R voltage and the duration of the QRS interval from manual measurement data on the first 40 arrhythmic beats after the start of dofetilide infusion and the last 40 arrhythmic beats immediately before TdP occurrence (or at equivalent time point in the ‘TdP−’ group of animals). The R wave of the QRS complex was defined as the wave with the greatest deflection from the isoelectric line within the QRS complex; R wave was regarded as a negative wave, when the greatest deflection of the QRS complex was negative. Coupling interval was defined as the distance between the peak of the R wave of the arrhythmic beat and the peak of the R wave of the preceding QRS complex. R voltage was defined as the voltage of the peak of the R wave from the isoelectric line. Duration of the QRS interval of the arrhythmic beat was defined as the interval from the first deflection of the QRS complex until the J point. Within-animal mean and within-animal standard deviation of the coupling intervals, the R voltages and the QRS durations measured on 40 arrhythmic beats were calculated at every time point in every animal.
Assessing vagal nerve activity and sympathetic activity in sinus rhythm
In order to assess vagal nerve activity, the beat-to-beat variability of the RR interval and the baroreflex sensitivity (BRS) of the spontaneous baroreflex sequences were analysed with a computer software (WinCPRS; Absolute Aliens Oy, Turku, Finland) as described earlier (Vincze et al., 2008). The analysis was performed in the corresponding ECG section and the arterial blood pressure signal of the last 5 min of the drug-free period before the phenylephrine infusion and the last 1 min of each rate of phenylephrine infusion before the dofetilide administration (see Experimental protocol and Figure 1). Only the arrhythmia-free ECG sections with uninterrupted sinus rhythm were analysed (Figure 3), that is, if any arrhythmia occurred within the analysed 5-min- or 1-min-long period at the time point of the measurement, then the whole period was excluded from the analysis. The software calculated the mean and the standard deviation of the RR intervals together with root mean square of the successive differences (see above) and the mean ‘up’ and ‘down’ BRS within a given experimental period.
Figure 3.
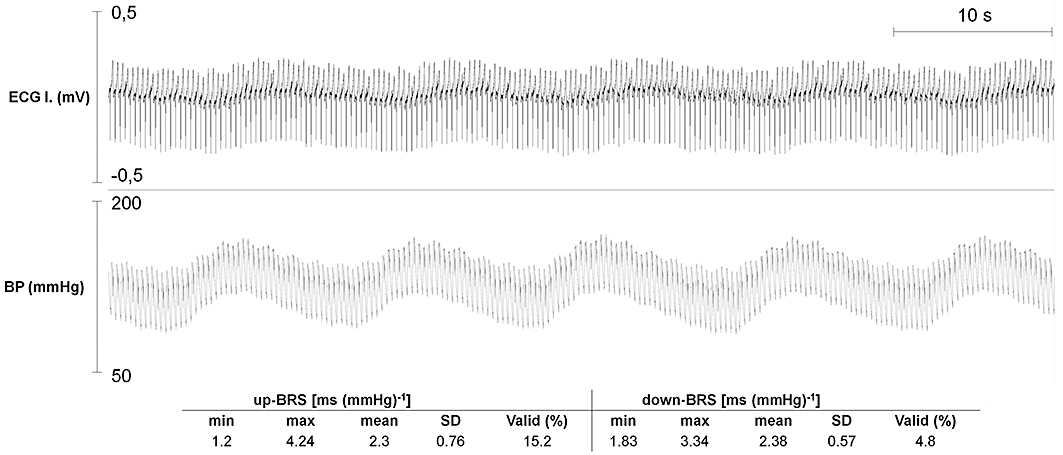
A representative example of the 115 arrhythmia-free, 1-min-long periods used for computerized analysis of the heart rate variability and the baroreflex sensitivity (BRS) during phenylephrine infusion. The period was recorded at the last minute of the highest rate of phenylephrine infusion (15 µg·kg−1·min−1) immediately before the start of dofetilide infusion in an animal that experienced TdP later, during dofetilide infusion. Table 5 contains the typical up- and down-BRS values calculated from the spontaneous baroreflex sequences identified within the period. Min, minimum; max, maximum; SD, standard deviation; Valid: the percentage incidence of RR intervals displaying spontaneous baroreflex sequences used to calculate BRS data.
The spectral power of the systolic arterial pressure was calculated in the frequency range 0.15–0.5 Hz (mid-frequency range) to assess the sympathetic activity as we described recently (Vincze et al., 2008). Spectral analysis was performed in the last 5 min of the drug-free period and the 3 min of the 9 µg·kg−1·min−1 phenylephrine infusion rate. Only the arrhythmia-free, uninterrupted periods were analysed, that is, if any arrhythmia occurred within the analysed 5-min- or 3-min-long period at the time point of the measurement, then the whole period was excluded from the analysis.
Statistical evaluation
Continuous data were expressed as mean ± standard error of the mean (SEM). All data from independent samples, with the exception of arrhythmia incidences, were compared with Kruskal–Wallis tests. Continuous data from the same sample were compared with Wilcoxon tests. Arrhythmia incidences were compared by using the Fisher's exact probability test. Differences were considered statistically significant when P < 0.05.
Materials
Dofetilide was a generous gift from Gedeon Richter PLC. (Budapest, Hungary). The sources of all other drugs and the preparation of drug solutions were described in detail earlier (Vincze et al., 2008). Drug and receptor nomenclature follows Alexander et al. (2009).
Results
Arrhythmia incidences, frequency of arrhythmic beats and onset time of arrhythmias
Torsades de pointes occurred in 20 out of 30 animals (Table 1). The rabbits were retrospectively divided into two groups according to the presence or the absence of TdP, that is, the animals that experienced TdP formed the ‘TdP+’ group (n = 20 animals), and the animals that did not experience this arrhythmia formed the ‘TdP−’ group (n = 10 animals) (Table 1).
Table 1.
Per cent incidence of arrhythmias before and during dofetilide infusion in anaesthetized rabbits
| Incidence of arrhythmias (%) | ||||||||
|---|---|---|---|---|---|---|---|---|
| n | VPB | BG | Salvo | Block | VT | TdP | VF | |
| Before dofetilide | ||||||||
| TdP− | 10 | 30 | 20 | 20 | 0 | 10 | 0 | 0 |
| TdP+ | 20 | 65 | 45 | 35 | 10 | 15 | 0 | 0 |
| During dofetilide | ||||||||
| TdP− | 10 | 90 | 80 | 90 | 60 | 40 | 0 | 0 |
| TdP+ | 20 | 100 | 95 | 100 | 90 | 100* | 100* | 15 |
Values are percentage incidences of arrhythmias.
P < 0.05 as compared with the ‘TdP−’ group, Kruskal–Wallis test.
Before dofetilide, the drug-free period and period when only phenylephrine infusion was administered; During dofetilide, the period when phenylephrine and dofetilide infusions were administered simultaneously. TdP−, group of animals not experiencing torsades de pointes during the experiment; TdP+, group of animals experiencing torsades de pointes during the experiment; VPB, ventricular premature beat; BG, bigeminy; Block, conduction block of any kind; VT, ventricular tachycardia different from torsades de pointes; TdP, torsades de pointes; VF, ventricular fibrillation; n, group size.
Arrhythmias (mostly ventricular premature beats) occurred in some animals during phenylephrine administration before dofetilide infusion (Table 1). However, most of the arrhythmia incidences were low during phenylephrine infusion, which allowed us to detect the pro-arrhythmic effects of dofetilide infusion. There was no significant difference in the group incidence of arrhythmias between the ‘TdP+’ and ‘TdP−’ groups of animals before dofetilide administration (Table 1); TdP and ventricular fibrillation did not occur before dofetilide infusion. Dofetilide strikingly increased the group incidence of arrhythmias in both groups. Interestingly, not only the incidence of TdP, but also the incidence of non-TdP type ventricular tachycardia was significantly higher in the ‘TdP+’ group than in the ‘TdP−’ group during dofetilide infusion. TdP degenerated into sustained ventricular fibrillation leading to death in three animals (15%) in the ‘TdP+’ group (Table 1).
The per cent frequency of arrhythmic beats was calculated within the period of the 40 ventricular cycles used for measurement of the ‘absolute’ beat-to-beat variability and instability parameters (number of arrhythmic beats per 40 beats times 100). There was no statistical difference in the frequency of arrhythmic beats before dofetilide administration between the ‘TdP+’ and ‘TdP−’ groups of animals (Figure 4A). Dofetilide infusion strikingly increased the frequency of arrhythmic beats during the course of its infusion in both the ‘TdP+’ and ‘TdP−’ groups of animals. After 3 min of dofetilide infusion (at the 3rd time point), the frequency of arrhythmic beats were significantly higher in the ‘TdP+’ group than that in the ‘TdP−’ group (Figure 4A). Later, as the number of ventricular arrhythmias increased further, the difference in the frequency of arrhythmic beats became insignificant between the ‘TdP+’ and ‘TdP−’ groups at the 4th time point, immediately before TdP occurred in the ‘TdP+’ group of animals (Figure 4A).
Figure 4.
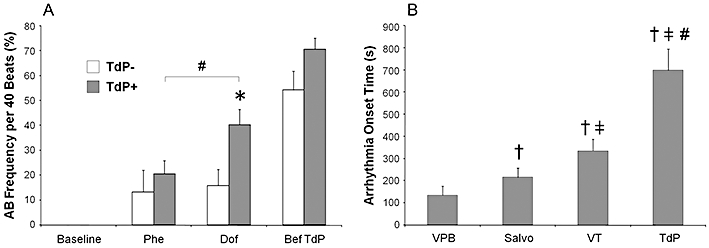
A: Per cent frequency of arrhythmic beats (AB frequency) in the 40 consecutive ventricular beats at the time point of the measurement (number of arrhythmic beats per 40 beats times 100). TdP−, group of animals (n = 10) not experiencing torsades de pointes during the experiment; TdP+, group of animals (n = 20) experiencing torsades de pointes during the experiment. All values shown as means ± SEM. *P < 0.05 versus the ‘TdP−’ group, Kruskal–Wallis test. #P < 0.05 Dof versus Phe, Wilcoxon test. The results of within-group comparisons are shown only between the Phe and Dof time points. For further details, see Figure 1. B: Arrhythmia onset times after the start of dofetilide infusion in the ‘TdP+’ group of animals. Every onset time was determined in all 20 ‘TdP+’ animals; all values shown as means ± SEM. †P < 0.05 versus VPB; ‡P < 0.05 versus Salvo; #P < 0.05 versus VT, Wilcoxon test. For further details, see Figure 2.
Dofetilide infusion caused a cascade in arrhythmia development in every ‘TdP+’ animal; not only the number of arrhythmias, but also the complexity of arrhythmias increased during the course of dofetilide infusion. That is, the arrhythmia onset followed a progressive order: the earliest arrhythmia to occur was ventricular premature beat, then salvo came, later non-TdP type ventricular tachycardia occurred, finally TdP developed (Figure 4B). In fact, the final and most complex arrhythmia was ventricular fibrillation to occur during dofetilide infusion but it occurred only in 3 out of 20 ‘TdP+’ animals, whereas all the other less complex arrhythmias occurred in every ‘TdP+’ animal. There was no statistical difference in the onset times of ventricular premature beat, salvo and non-TdP type ventricular tachycardia between the ‘TdP+’ and the ‘TdP−’ groups (data not shown). The cumulative dose of dofetilide that triggered TdP was 0.23 ± 0.03 mg·kg−1 and TdP occurred at 699 ± 97 s after the start of dofetilide infusion.
Blood pressure
The baseline systolic and diastolic arterial blood pressure values did not differ between the ‘TdP+’ and the ‘TdP−’ groups (Table 2). The stepwise elevation of the rate of infusion of phenylephrine increased the blood pressure. Arrhythmias occurred frequently in all animals approximately 4 min after the start of dofetilide administration, which prevented measurement of the blood pressure in sinus rhythm thereafter. There was no significant difference in the blood pressure values between the ‘TdP+’ and the ‘TdP−’ groups at any time point.
Table 2.
Heart rate, QT and Tpeak–Tend intervals, T-wave lability index and arterial blood pressure in anaesthetized rabbits
| Phenylephrine | Phe + Dof | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| n | 0 min | 10 min | 13 min | 16 min | 19 min | 22 min | 27 min | 29 min | 31 min | |
| TdP− | ||||||||||
| HR | 10 | 275 ± 10 | 275 ± 10 | 270 ± 10 | 255 ± 12 | 253 ± 13 | 242 ± 15 | 236 ± 12 | 229 ± 6 | 222 ± 23 |
| QT | 10 | 160 ± 4 | 167 ± 3 | 163 ± 4 | 172 ± 5 | 172 ± 6 | 184 ± 6 | 188 ± 7 | 196 ± 5 | 213 ± 14 |
| TpTe | 10 | 52 ± 5 | 58 ± 5 | 68 ± 7 | ||||||
| TWLI | 10 | 0.08 ± 0.02 | 0.08 ± 0.01 | n.d. | n.d. | |||||
| DAP | 10 | 85 ± 5 | 94 ± 4 | 99 ± 5 | 105 ± 4 | 110 ± 5 | 114 ± 4 | 115 ± 3 | 126 ± 7 | |
| SAP | 10 | 110 ± 4 | 117 ± 3 | 122 ± 5 | 129 ± 5 | 137 ± 5 | 139 ± 5 | 143 ± 5 | 151 ± 6 | |
| TdP+ | ||||||||||
| HR | 20 | 285 ± 7 | 280 ± 7 | 276 ± 6 | 258 ± 7 | 243 ± 7 | 233 ± 7 | 231 ± 7 | 237 ± 12 | 205 ± 23 |
| QT | 20 | 168 ± 4 | 166 ± 4 | 166 ± 4 | 172 ± 4 | 177 ± 5 | 184 ± 6 | 189 ± 6 | 187 ± 6 | 199 ± 15 |
| TpTe | 20 | 51 ± 3 | 60 ± 5 | 60 ± 5 | ||||||
| TWLI | 18 | 0.08 ± 0.01 | 0.12 ± 0.02 | n.d. | n.d. | |||||
| DAP | 20 | 90 ± 3 | 93 ± 2 | 102 ± 3 | 106 ± 3 | 109 ± 3 | 112 ± 4 | 115 ± 2 | 111 ± 6 | |
| SAP | 20 | 115 ± 3 | 117 ± 3 | 126 ± 3 | 131 ± 4 | 135 ± 3 | 139 ± 4 | 146 ± 4 | 140 ± 6 | |
All values are means ± SEM. There was no significant difference in the respective values of any variable between the ‘TdP+’ and ‘TdP−’ groups of animals at any time point. For further details, see Table 1.
Phenylephrine, time points when phenylephrine is infused at increasing rates; Phe + Dof, time points when phenylephrine and dofetilide are infused simultaneously (see Figure 1 for protocol); n, number of animals in which the parameter was determined at 0 or 10 min (n declined due to arrhythmia occurrence at later time points); HR, sinus heart rate (min−1); QT, the electrocardiographic QT interval (ms); TpTe, the electrocardiographic Tpeak–Tend interval (ms); TWLI, T-wave lability index; DAP, diastolic arterial pressure (mmHg); SAP, systolic arterial pressure (mmHg); n.d. not determined.
Heart rate and ECG intervals in sinus rhythm
Phenylephrine reduced sinus heart rate (Tables 2 and 3) and prolonged the QT interval equally in both groups before the dofetilide infusion (Tables 2 and 4). Arrhythmias occurred frequently in all animals approximately 4 min after the start of the dofetilide administration, which prevented measurement of the sinus heart rate and the ‘sinus’ ECG intervals thereafter.
Table 3.
The ‘sinus’ and ‘absolute’ beat-to-beat variability and instability of the RR interval in anaesthetized rabbits
| n | MeanRR | SD | RMS | RMSSD | SDSD | TI | STI | ||
|---|---|---|---|---|---|---|---|---|---|
| Sinus | TdP− | ||||||||
| Baseline | 10 | 221 ± 8 | 1.6 ± 0.1 | 221 ± 8 | 1.8 ± 0.1 | 1.9 ± 0.1 | 1.9 ± 0.2 | 1.0 ± 0.1 | |
| Phe | 8 | 259 ± 14‡ | 2.3 ± 0.5 | 259 ± 14‡ | 2.2 ± 0.2 | 2.3 ± 0.2 | 2.7 ± 0.6 | 1.0 ± 0.1 | |
| Dof | 8 | 264 ± 15‡ | 2.3 ± 0.4 | 264 ± 15‡ | 2.4 ± 0.4 | 2.4 ± 0.4 | 2.8 ± 0.6 | 1.2 ± 0.2 | |
| TdP+ | |||||||||
| Baseline | 20 | 215 ± 5 | 1.8 ± 0.2 | 215 ± 2 | 1.8 ± 0.1 | 1.8 ± 0.1 | 2.1 ± 0.2 | 0.9 ± 0.1 | |
| Phe | 13 | 265 ± 11‡ | 2.8 ± 0.5 | 265 ± 11‡ | 3.0 ± 0.7 | 3.1 ± 0.7 | 3.4 ± 0.7 | 1.7 ± 0.5 | |
| Dof | 12 | 270 ± 11‡ | 3.1 ± 0.5‡ | 270 ± 11‡ | 3.6 ± 0.7‡ | 3.6 ± 0.7‡ | 4.0 ± 0.8‡ | 1.9 ± 0.5‡ | |
| Absolute | TdP− | ||||||||
| Phe | 10 | 257 ± 11 | 13.4 ± 7.2 | 258 ± 11 | 19.9 ± 2.6 | 20.2 ± 12 | 11.3 ± 5.8 | 7.2 ± 5.4 | |
| Dof | 10 | 259 ± 12 | 18.1 ± 7.9 | 260 ± 12 | 31.8 ± 14.2 | 32.2 ± 14 | 21.5 ± 10.8 | 17.4 ± 8.6 | |
| Bef TdP | 10 | 273 ± 11 | 46.9 ± 9.5#† | 279 ± 12#† | 81.3 ± 17.4# | 82.3 ± 17.7#† | 60.3 ± 13.5# | 50.1 ± 12.8#† | |
| TdP+ | |||||||||
| Phe | 20 | 262 ± 7 | 22.6 ± 4.9 | 263 ± 7 | 39.5 ± 9.2 | 40.1 ± 9.4 | 24.5 ± 6.1 | 22.6 ± 6.2 | |
| Dof | 20 | 265 ± 7 | 38.5 ± 5.6# | 269 ± 7 | 67.9 ± 10.2# | 68.8 ± 10.3# | 44.3 ± 7.8*# | 39.8 ± 7.5*# | |
| Bef TdP | 20 | 291 ± 17#† | 71.5 ± 5.9*#† | 300 ± 17#† | 106.1 ± 9.4#† | 107.4 ± 9.5#† | 76.9 ± 8.4#† | 53.0 ± 6.9#† | |
P < 0.05 versus ‘TdP−’ group, Kruskal–Wallis test.
P < 0.05 versus baseline.
P < 0.05 versus Phe.
P < 0.05 versus Dof, Wilcoxon test.
Sinus, the beat-to-beat variability and instability of the RR interval when measured in sinus rhythm; Absolute, the beat-to-beat variability and instability of the RR interval when arrhythmias were involved in the measurement; n, number of animals in which the RR variability and instability were determined (sinus variability and instability were determined only when there was sinus rhythm at the time point of the measurement); MeanRR, the mean of the measured 40 consecutive RR intervals (ms); SD, the standard deviation of the measured 40 consecutive RR intervals (ms); RMS, the root mean square of the RR intervals (ms); RMSSD, the root mean square of the successive differences of the RR intervals (ms); SDSD, standard deviation of successive differences of the RR intervals (ms); TI, total instability of the RR intervals (ms); STI, short-term instability of the RR intervals (ms).
Table 4.
The ‘sinus’ and ‘absolute’ beat-to-beat variability and instability of the QT interval in anaesthetized rabbits
| n | MeanQT | SD | RMS | RMSSD | SDSD | TI | STI | ||
|---|---|---|---|---|---|---|---|---|---|
| Sinus | TdP− | ||||||||
| Baseline | 10 | 167 ± 6 | 5.6 ± 0.4 | 168 ± 6 | 6.6 ± 0.5 | 6.5 ± 0.5 | 6.6 ± 0.7 | 3.3 ± 0.3 | |
| Phe | 8 | 185 ± 12‡ | 5.8 ± 0.4 | 185 ± 12‡ | 8.1 ± 0.8 | 7.8 ± 0.6 | 7.2 ± 0.7 | 3.8 ± 0.4 | |
| Dof | 8 | 207 ± 10‡ | 7.9 ± 0.8 | 208 ± 10‡ | 10.4 ± 2.4‡ | 9.6 ± 1.2‡ | 8.9 ± 1.0 | 3.9 ± 0.6 | |
| TdP+ | |||||||||
| Baseline | 20 | 169 ± 4 | 5.3 ± 0.4 | 169 ± 4 | 6.7 ± 0.5 | 6.3 ± 0.4 | 6.2 ± 0.6 | 2.8 ± 0.2 | |
| Phe | 13 | 192 ± 7‡ | 7.1 ± 0.7‡ | 192 ± 7‡ | 9.2 ± 0.9‡ | 8.4 ± 0.5‡ | 8.2 ± 0.7 | 4.2 ± 0.4‡ | |
| Dof | 12 | 193 ± 7‡ | 7.2 ± 0.8‡ | 193 ± 7‡ | 8.6 ± 1.1‡ | 8.6 ± 1.1‡ | 8.5 ± 1.1 | 3.8 ± 0.3‡ | |
| Absolute | TdP− | ||||||||
| Phe | 10 | 190 ± 9 | 8.7 ± 1.6 | 190 ± 9 | 12.1 ± 2.6 | 12.3 ± 2.6 | 10.0 ± 1.9 | 5.5 ± 1.2 | |
| Dof | 10 | 190 ± 6 | 14.1 ± 3.4 | 191 ± 6 | 21.7 ± 7.0 | 22.0 ± 7.1 | 16.3 ± 5.6 | 12.1 ± 5.3 | |
| Bef TdP | 10 | 232 ± 7#† | 19.0 ± 2.8# | 233 ± 7#† | 28.9 ± 4.6# | 29.2 ± 4.7# | 24.0 ± 4.1# | 15.3 ± 3.1# | |
| TdP+ | |||||||||
| Phe | 20 | 195 ± 5 | 10.9 ± 1.3 | 195 ± 5 | 16.3 ± 2.6 | 16.5 ± 2.6 | 12.7 ± 1.9 | 8.5 ± 1.7 | |
| Dof | 20 | 205 ± 5* | 21.7 ± 3.4# | 206 ± 5*# | 34.4 ± 6.2# | 34.9 ± 6.3# | 23.3 ± 4.1# | 17.7 ± 4.2# | |
| Bef TdP | 20 | 258 ± 11#† | 34.3 ± 2.6*#† | 260 ± 11*#† | 45.1 ± 3.5*# | 45.7 ± 3.5*# | 37.4 ± 2.8*#† | 21.8 ± 1.5*# | |
Dofetilide prolonged the QT (Table 2) and the rate-corrected QT intervals (QTc) in the ‘TdP+’ and the ‘TdP−’ groups up until the time point at which these intervals could be measured (see above). There was no significant difference in the QT and the QTc intervals between the ‘TdP+’ and the ‘TdP−’ groups, when QT interval was measured in sinus rhythm. There was no qualitative difference in the effects of the drugs on the QT and the QTc intervals at any time point in both the ‘TdP+’ and the ‘TdP−’ groups, thus QTc data are not shown.
There was no significant difference in the Tpeak–Tend intervals between the ‘TdP+’ and the ‘TdP−’ groups at any of the three time points of the measurement (Table 2). The mean baseline ‘sinus’ PQ interval was 50 ± 1 ms in ‘TdP+’ and the ‘TdP−’ groups; the mean baseline ‘sinus’ QRS interval was 47 ± 1 ms and 49 ± 3 ms in the ‘TdP+’ and the ‘TdP−’ groups respectively. The ‘sinus’ PQ and QRS intervals remained stable up until the time point at which these could no longer be measured (4 min of dofetilide infusion); there were no statistical differences in the ‘sinus’ PQ or QRS intervals between the ‘TdP+’ and the ‘TdP−’ groups at any time point of the measurement (data not shown).
T-wave lability index could be measured only until the end of phenylephrine infusion, as dofetilide evoked many arrhythmias, which did not allow assessment of 100 subsequent T waves during normal sinus rate. Phenylephrine slightly increased the T-wave lability index in the ‘TdP+’ group, but the change was not significant (Table 2).
Beat-to-beat variability and instability of the RR and the QT intervals in sinus rhythm
There was a small, but significant increase in the ‘sinus’ beat-to-beat variability and instability of the RR and QT intervals in the ‘TdP+’ group during the course of the experiment (Tables 3 and 4); however, the increase in the ‘sinus’ beat-to-beat variability and instability of the QT interval was caused mostly by the phenylephrine infusion prior to dofetilide administration. Nevertheless, neither ‘sinus’ RR nor ‘sinus’ QT variability and instability parameters differed significantly between the ‘TdP+’ and ‘TdP−’ groups of animals at baseline, during the phenylephrine infusion and at the beginning of the dofetilide infusion (Figures 5 and 6, Tables 3 and 4).
Figure 5.
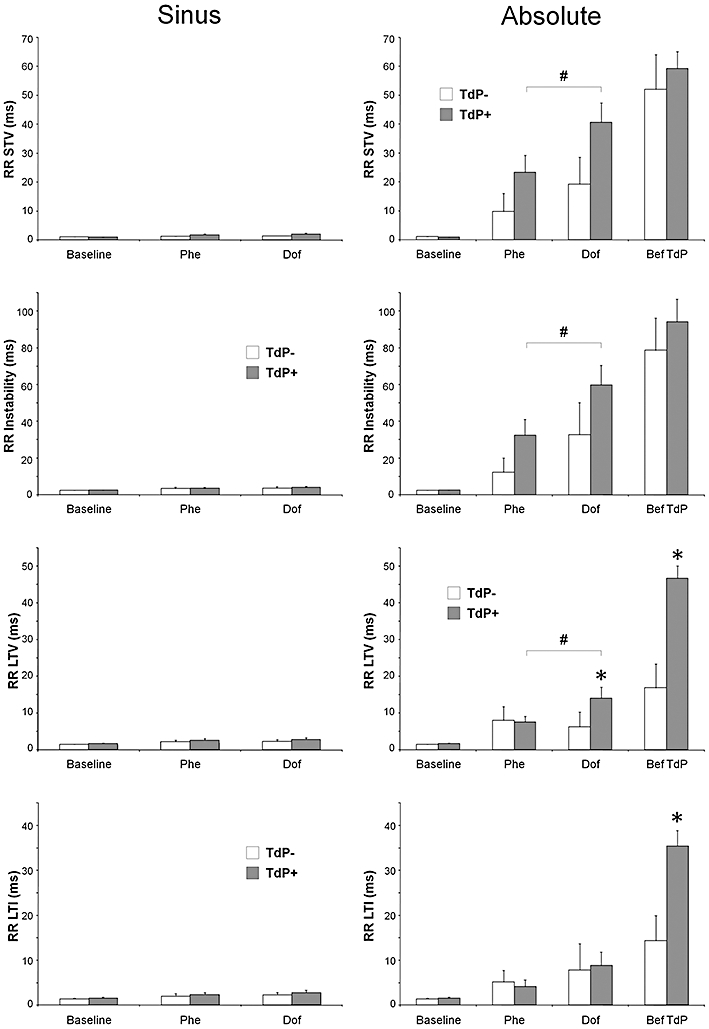
The ‘sinus’ and ‘absolute’ beat-to-beat variability and instability of the RR interval in anaesthetized rabbits. Sinus (figures on the left side): the beat-to-beat variability and instability of the RR interval when measured in sinus rhythm. Absolute (figures on the right side): the beat-to-beat variability and instability of the RR interval when arrhythmias were involved in the measurement. RR STV, short-term variability of the RR intervals; RR instability, instability of the RR intervals; RR LTV, long-term variability of the RR intervals (ms); RR LTI, long-term instability of the RR intervals (ms). All values shown as means ± SEM. *P < 0.05 versus the ‘TdP−’ group, Kruskal–Wallis test. #P < 0.05 Dof versus Phe, Wilcoxon test. The results of within-group comparisons are shown only between the Phe and Dof time points. For further details, see Figures 1 and 4.
Figure 6.
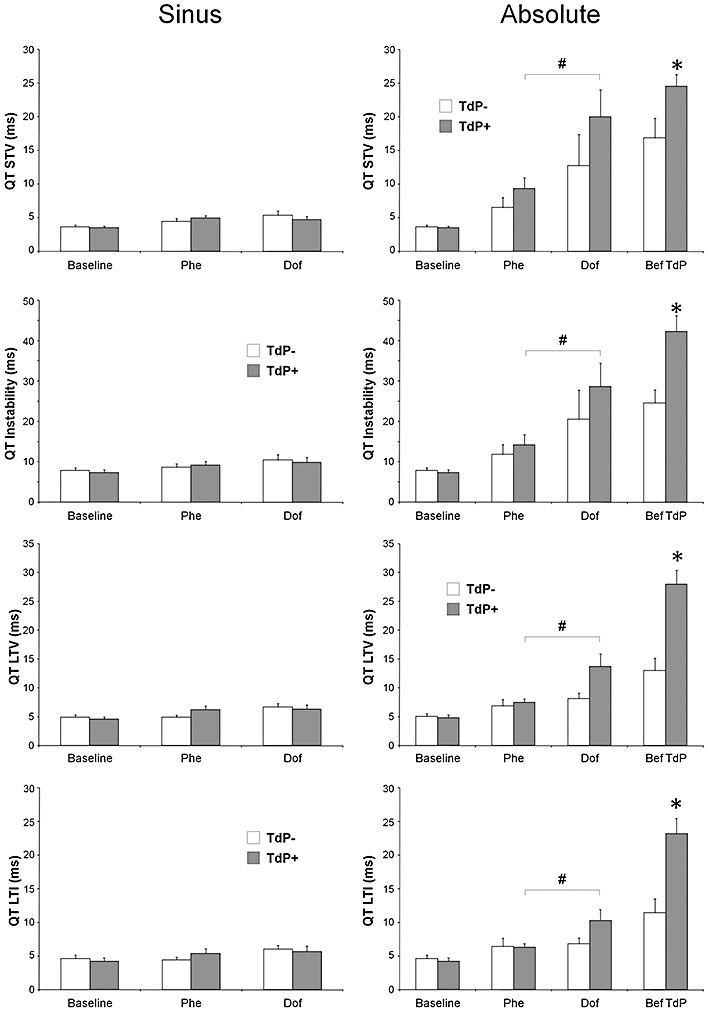
The ‘sinus’ and ‘absolute’ beat-to-beat variability and instability of the QT interval in anaesthetized rabbits. All values shown as means ± SEM. *P < 0.05 versus the ‘TdP−’ group, Kruskal–Wallis test. #P < 0.05 Dof versus Phe, Wilcoxon test. The results of within-group comparisons are shown only between the Phe and Dof time points. For further details, see Figures 1,4 and 5.
The ‘absolute’ beat-to-beat variability and instability of the ECG intervals
When the ECG intervals in 40 consecutive cardiac cycles were analysed irrespective of the presence or absence of sinus rhythm during the measurement, a remarkable effect of the drugs was revealed. Phenylephrine infusion increased the ‘absolute’ beat-to-beat variability and instability of all ECG intervals as compared with baseline in both the ‘TdP+’ and the ‘TdP−’ groups, but these parameters did not differentiate between the ‘TdP+’ and ‘TdP−’ groups of animals before dofetilide administration (Figures 5–7, Tables 3 and 4).
Figure 7.
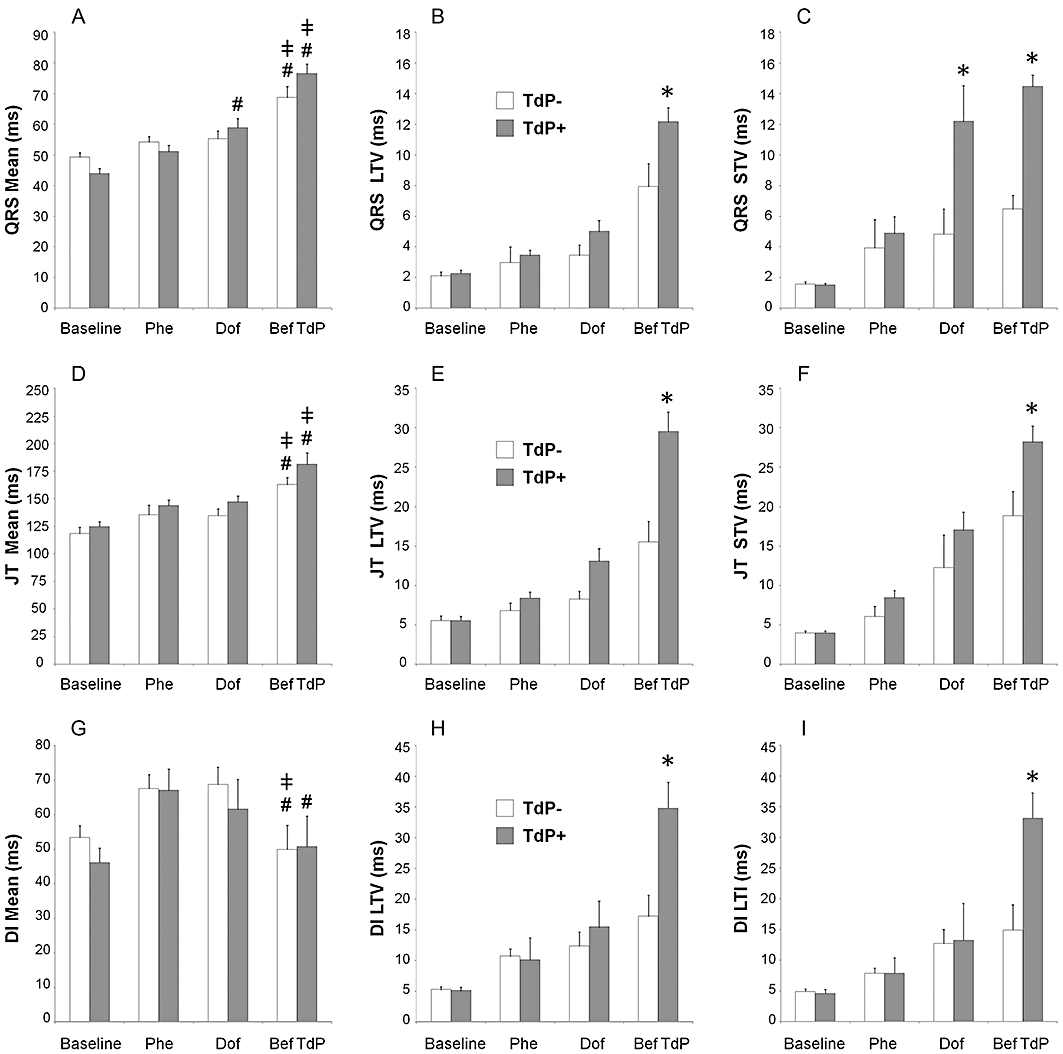
The ‘absolute’ beat-to-beat variability of the QRS interval (A–C) and the JT interval (D–F), and the ‘absolute’ beat-to-beat variability and instability of the ventricular electric diastolic interval (DI, G–I) in anaesthetized rabbits. Mean, the mean of the measured 40 consecutive QRS, JT or DIs. All values shown as means ± SEM. *P < 0.05 versus the ‘TdP−’ group, Kruskal–Wallis test. #P < 0.05 Dof versus Phe, Wilcoxon test. ‡P < 0.05 Bef TdP versus Dof, Wilcoxon test. Within group comparisons were performed only with the QRS mean, the JT mean and the DI mean values (A, D and G); the results of within-group comparisons are shown only among the Phe, Dof and Bef TdP time points. For further details, see Figures 1,4 and 5. Note that the ‘absolute’ instability, STI, LTI, TI, SD, SDSD, RMSSD parameters of the QRS and the JT intervals were significantly increased in the ‘TdP+’ group immediately before TdP occurrence as compared with the respective values in the ‘TdP−’ group (Kurskal–Wallis test, data not shown). Apart from ‘absolute’ LTV and LTI, no other beat-to-beat variability and instability parameters of the DI differentiated between the ‘TdP+’ and ‘TdP−’ groups of animals (data not shown).
Dofetilide progressively increased the ‘absolute’ beat-to-beat variability and instability of the RR and QT intervals in the ‘TdP+’ and ‘TdP−’ groups, but the dofetilide-induced increase in these parameters reached statistical significance only in the ‘TdP+’ group after 3 min of dofetilide infusion (3rd time point vs. 2nd time point, within-group analysis, Figures 4 and 5, Tables 3 and 4).
Dofetilide infusion progressively increased the ‘absolute’ beat-to-beat variability and instability parameters of the RR, QRS, JT, QT and electric diastolic intervals in both the ‘TdP+’ and the ‘TdP−’ groups. Importantly, most of these parameters differentiated between the ‘TdP+’ and ‘TdP−’ groups of animals; that is, the ‘absolute’ beat-to-beat variability and instability parameters of the RR, QRS, JT, QT, and electric diastolic intervals were significantly greater in the ‘TdP+’ group of animals than in the ‘TdP−’ group of animals immediately before TdP occurrence (Figures 5–7, Tables 3 and 4). LTV and LTI were the most sensitive variables out of the numerous variability and instability parameters we calculated; ‘absolute’ LTV and ‘absolute’ LTI showed significant difference in every ECG interval (the RR, QRS, QT, JT and electric diastolic interval) between the ‘TdP+’ and the ‘TdP−’ groups of animals immediately before TdP occurrence (Figures 5–7, Tables 3 and 4).
Repolarization prolongation measured by the ‘absolute’ mean JT interval
Dofetilide infusion significantly prolonged the mean JT interval measured in 40 consecutive cardiac cycles irrespective of the presence or absence of the sinus rhythm (the ‘absolute’ mean JT) in both the ‘TdP+’ and ‘TdP−’ groups of animals, but there was no statistical difference in the JT intervals between the ‘TdP+’ and ‘TdP−’ groups of animals at any time point of the measurement (Figure 7D).
Characterization of the arrhythmic beats during dofetilide administration
The arrhythmic beats immediately before TdP in the ‘TdP+’ group of animals were significantly more variable in terms of coupling interval and shape than those measured at equivalent time point in the ‘TdP−’ group of animals; the within-animal standard deviations of the coupling interval, R voltage and QRS duration measured from the last 40 arrhythmic beats before TdP were significantly greater in the ‘TdP+’ group of animals than in the ‘TdP−’ group of animals (Figure 8). No such difference was seen between the ‘TdP+’ and ‘TdP−’ groups of animals when the first 40 arrhythmic beats after the start of dofetilide infusion were analysed. The within-animal mean coupling interval, R voltage and the QRS duration of the arrhythmic beats did not differ between the ‘TdP+’ and ‘TdP−’ groups of animals at any time point of the measurement.
Figure 8.
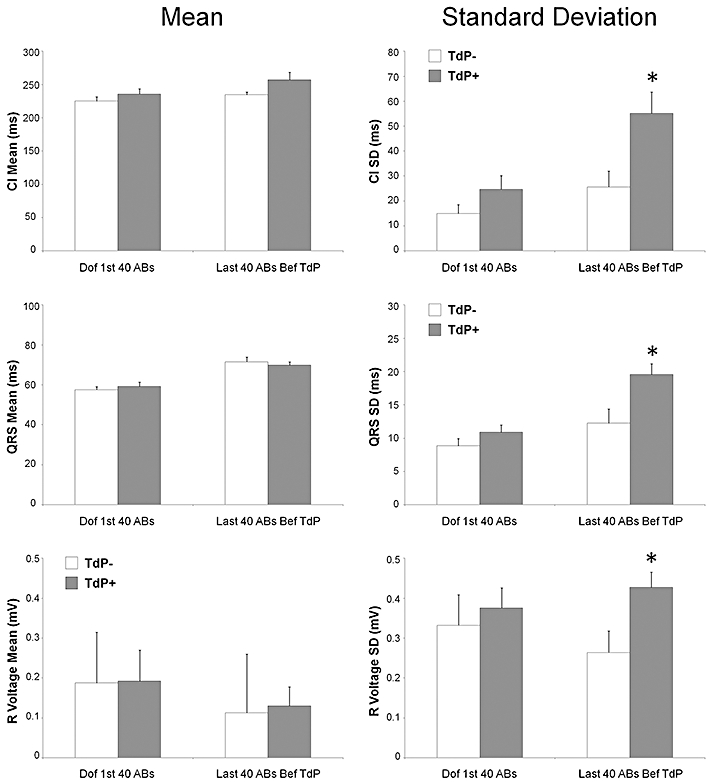
Characterization of the first 40 arrhythmic beats (ABs) after the start of dofetilide infusion (Dof 1st 40 ABs) and the last 40 ABs immediately before torsades de pointes (TdP) occurred in the ‘TdP+’ group of animals or at an equivalent time point in the ‘TdP−’ group of animals (last 40 ABs Bef TdP). Within-animal mean (mean) and within-animal standard deviation (SD) of the coupling intervals (CI), QRS intervals and R voltages calculated from 40 ABs measured at the time point. *P < 0.05 versus the ‘TdP−’ group, Kruskal–Wallis test.
Assessing vagal nerve activity: ‘heart rate variability’ and BRS in sinus rhythm
The computerized ‘heart rate variability’ analysis produced quantitatively and qualitatively similar data to that gained with the manual measurement of 40 consecutive RR intervals (see above and Table 3), that is, ‘sinus’ RR variability parameters did not differ statistically between the ‘TdP+’ and the ‘TdP−’ groups at baseline and during the phenylephrine infusion (data not shown). However, in the ‘TdP+’ group of animals, there was a significant increase in heart rate variability during dofetilide infusion as compared with the baseline (Table 3, ‘sinus’ beat-to-beat variability parameters of the RR interval), which indicates that vagal nerve activity increased during drug infusion before TdP occurred. No such increase was detected in the ‘TdP−’ group of animals.
The mean up-baroreflex sensitivity (up-BRS) was slightly, but non-significantly higher at all predetermined time points in the ‘TdP+’ group as compared with the respective values in the ‘TdP−’ negative group. However, when only a single between-group analysis was performed using the pooled BRS data on all five 1-min time periods during the phenylephrine infusion, a significantly higher up-BRS was found in the ‘TdP+’ group than in the ‘TdP−’ group (Table 5). There was no statistical difference in the down-BRS between the ‘TdP+’ and the ‘TdP−’ groups at any time point of the measurement (data not shown).
Table 5.
Baroreceptor sensitivity before dofetilide administration in anaesthetized rabbits
| Baseline | Phe3 | Phe6 | Phe9 | Phe12 | Phe15 | Phe total | ||
|---|---|---|---|---|---|---|---|---|
| TdP− | Up-BRS | 1.66 ± 0.31 | 1.76 ± 0.23 | 2.07 ± 0.34 | 1.81 ± 0.38 | 2.29 ± 1.01 | 1.85 ± 0.63 | 1.95 ± 0.24 |
| n = 9 | n = 10 | n = 9 | n = 9 | n = 8 | n = 8 | K = 44 | ||
| TdP+ | Up-BRS | 1.83 ± 0.32 | 1.86 ± 0.24 | 2.24 ± 0.32 | 2.42 ± 0.29 | 2.56 ± 0.92 | 2.59 ± 0.46 | 2.27 ± 0.14* |
| n = 16 | n = 19 | n = 17 | n = 14 | n = 14 | n = 7 | K = 71 |
P < 0.05 versus ‘TdP−’ group, Kruskal–Wallis test.
Up-BRS, the mean baroreceptor sensitivity [ms·(mmHg)−1] measured in spontaneous sequences of constantly widening RR interval as a result of rises in the systolic arterial blood pressure; Baseline, baseline baroreflex sensitivity (BRS) determined by spontaneous up-BRS sequences found in the 5-min period before the start of phenylephrine infusion; Phe3, Phe6, Phe9, Phe12 and Phe15, BRS determined by spontaneous up-BRS sequences found in the last 1-min period of the phenylephrine infusion at a rate of 3, 6, 9, 12 and 15 µg·kg−1·min−1 respectively; Phe total, the pooled BRS data of Phe3, Phe6, Phe9, Phe12 and Phe15. n, number of animals in which spontaneous up-BRS sequences were determined at the time point of the measurement; K, the cumulative number of 1-min periods in which spontaneous up-BRS sequences were measured during stepwise elevation of the phenylephrine infusion rate.
Assessing sympathetic activity in sinus rhythm
The mid-frequency spectral power of the systolic arterial pressure, which is a good marker of the sympathetic activity in sinus rhythm, did not differ significantly between the ‘TdP+’ and ‘TdP−’ groups at baseline [10.1 ± 4.8 vs. 7.4 ± 2.7 (mmHg)2 respectively] and during phenylephrine infusion [6.4 ± 3.0 vs. 3.7 ± 1.6 (mmHg)2 respectively].
Blood gases and serum ion levels
The blood gases and the serum levels of K+, Na+ and Ca2+ were in the physiological range at baseline, during the phenylephrine infusion and at the beginning of the dofetilide infusion; there were no statistical differences in the serum levels of K+, Na+ or Ca2+, or the serum pH+ or blood gas values between the ‘TdP+’ and the ‘TdP−’ groups at baseline, during the phenylephrine infusion and at the beginning of the dofetilide infusion (data not shown).
Discussion
Dofetilide prolonged the repolarization and induced a large number of arrhythmias prior to TdP in our study. Data showed that ‘preceding’ arrhythmias may play a significant role in the genesis of TdP in anaesthetized, α1-adrenoceptor-stimulated rabbits. The arrhythmic beats immediately before TdP in the ‘TdP+’ group of animals were significantly more variable in terms of coupling interval and shape than those measured in the ‘TdP−’ group of animals at equivalent time point. Accordingly, the ‘absolute’ beat-to-beat variability and instability parameters of the ECG intervals were significantly elevated in the ‘TdP+’ group of animals immediately before TdP onset (Tables 3 and 4 and Figures 5–7). Our data indicate that in the setting of prolonged repolarization the more chaotic the ventricular rhythm, the greater the ‘absolute’ beat-to-beat variability and instability of the ECG intervals and the probability of TdP development.
Those animals that subsequently experienced TdP had more responsive baroreflex during phenylephrine infusion before dofetilide administration and their heart rate variability significantly increased during dofetilide infusion, which suggests vagal nerve involvement in the genesis of TdP. Dofetilide-induced TdP was not determined by the baseline haemodynamics, repolarization properties, autonomic status, blood gases or serum ion concentrations.
Preceding arrhythmias
In our study, dofetilide induced large number of arrhythmias prior to TdP and not only the number but also the complexity of these ‘preceding’ arrhythmias increased before TdP. However, the cascade of ‘preceding’ arrhythmias during the administration of the test drug is not unique to our experimental settings. Ventricular premature beats developing minutes before TdP (Carlsson et al., 1992; 1993;) and the cascade of drug-induced arrhythmias prior to TdP onset were reported in the anaesthetized rabbit model of TdP (Buchanan et al., 1993; Lu et al., 2000; Farkas et al., 2002; Philp et al., 2007) as well as in clinical studies (Locati et al., 1995). Thus, the preceding arrhythmias in our study probably did not critically depend on the arrhythmogenic substrate provided by the combination of phenylephrine infusion and pentobarbital anaesthesia. Non-TdP type ventricular tachycardia preceded TdP occurrence and the incidence of non-TdP type ventricular tachycardia was significantly higher in the ‘TdP+’ group than in the ‘TdP−’ group during dofetilide infusion, which suggest that non-TdP type ventricular tachycardia can serve as a biomarker of TdP in the applied model.
The present study is the first that measured the frequency and the multiplicity of the arrhythmias prior to TdP and quantified the beat-to-beat electrical instability caused by the ‘preceding’ arrhythmias. The data indicate that in the setting of delayed repolarization the more chaotic the ventricular rhythm (arrhythmic beats and ‘absolute’ beat-to-beat variability and instability), the greater the probability of TdP development. That is, ‘preceding’ arrhythmias have characteristics that permit prediction of TdP occurrence. This has previously been overlooked.
Beat-to-beat variability of the QT interval in sinus rhythm
Dynamic ECG beat-to-beat variability parameters, such as instability or STV, have been proposed recently as better surrogates than QT interval for TdP (Shah and Hondeghem, 2005; Thomsen et al., 2007). ‘Sinus’ QT STV did not predict TdP in our study with α1-adrenoceptor-stimulated, anaesthetized rabbits, which is in a good agreement with the results of earlier studies utilizing the same model (Michael et al., 2007; Farkas et al., 2008; Vincze et al., 2008). However, Carlsson et al. (2009) reported that dofetilide at a rate of 0.883 µg·kg−1·min−1 (vs. 20 µg·kg−1·min−1 in our study) increased QT STV in sinus rhythm prior to the occurrence of ventricular premature beats and TdP in methoxamine-sensitized, α-chloralose-anaesthetized rabbits. This suggests that the predictive value of the ‘sinus’ beat-to-beat variability of the QT interval in the rabbit model may depend on the experimental conditions, for example, anaesthesia, α1-adrenergic stimulant and rate of infusion of the test drug.
‘Absolute’ beat-to-beat variability and instability ECG interval parameters and electrical instability
The novel ‘absolute’ variability and instability parameters are determined by measuring the beat-to-beat variability and instability of the ECG intervals at precise times before the window for TdP manifestation, even during the occurrence of ventricular arrhythmias. Thus, ‘absolute’ variability and instability parameters are able to quantify the beat-to-beat electrical instability even during arrhythmias. In our study, the ‘absolute’ beat-to-beat variability and instability parameters differentiated between the ‘TdP+’ and ‘TdP−’ groups of animals and predicted TdP occurrence (Tables 3 and 4 and Figures 5–7). The data showed that the predictive sensitivity of these variables was greater than the sensitivity of equivalent variables measured earlier, during stable sinus rhythm. Beat-to-beat electrical instability quantified by the novel ‘absolute’ variability and instability parameters may represent a precise biomarker for TdP risk. This novel approach may facilitate early rejection of hazardous candidates in preclinical drug discovery. Moreover, the data array indicates that the stochastic determinants of TdP are manifested in the beat-to-beat variability and instability of the ECG intervals immediately prior to TdP manifestation, indicating a mechanistic link.
‘Absolute’ beat-to-beat variability and instability of the ECG intervals elaborates the mechanism of TdP in rabbits
Dofetilide increased the ‘absolute’ beat-to-beat variability and instability of all measured ECG parameters indicating the development of chaotic ventricular repolarization, conduction and multiple ectopic activity. The electrical instability measured by the ‘absolute’ beat-to-beat variability and instability of the ECG intervals correlated with the probability of TdP development in our model. Our data suggest that the ‘absolute’ beat-to-beat variability and instability parameters of the ECG intervals were greatly affected by the multiplicity of the arrhythmic beats in terms of number, coupling interval and shape. Interestingly, immediately before TdP, the number (frequency) of arrhythmic beats did not differ between the ‘TdP+’ and the ‘TdP−’ groups of animals. Nevertheless, at same time point immediately before TdP, the arrhythmic beats were significantly more variable in terms of coupling interval and shape in the ‘TdP+’ group of animals than in the ‘TdP−’ group of animals. It is widely accepted that the site of origin determines the electrocardiographic shape of the ectopic beat. Thus, our data suggest that immediately before TdP, the number of sites of origin of the arrhythmic beats in the ‘TdP+’ group of animals was greater than that in the ‘TdP−’ group of animals at equivalent time point. Vos et al. (2000) suggested that multiple ventricular ectopic beats contributed to TdP development by increasing spatial dispersion of repolarization in chronic atrioventricular-blocked dogs in vivo. However, that study did not examine the relationship between TdP development and the multiplicity of the ectopic beats in terms of coupling interval and the shape. Our data indicate that in the setting of prolonged repolarization the more sites (myocardial cells) producing ectopic beats in the heart in an unsynchronized way with different coupling intervals, the greater the ‘absolute’ beat-to-beat variability and instability of the ECG intervals and the probability of TdP development.
According to the most widely accepted theory, the mechanism of TdP initiation (trigger) involves an early afterdepolarization-induced ectopic beat, while the mechanism of the arrhythmia maintenance (substrate) involves re-entry circuits produced by an increase in spatial dispersion of the repolarization of the ventricular wall (Antzelevitch, 2007). Earlier investigations showed that single and multiple premature beats may increase spatial inhomogeneity of repolarization (Laurita et al., 1998; Vos et al., 2000), thus the dofetilide-induced cascade of the ‘preceding’ arrhythmias might enhance the spatial dispersion and produced an ideal substrate for re-entrant arrhythmias, for example, TdP. Furthermore, immediately before TdP, the coupling intervals of the arrhythmic beats were more variable in the ‘TdP+’ group than those in the ‘TdP−’ group of animals. This implies that a greater variability in coupling intervals provides a greater probability that one of the ectopic beats can work as a trigger by falling in the vulnerable period of the existing re-entry circuits. The electrophysiological substrate produced by the ‘preceding’ arrhythmias may be the link between drug-induced early afterdepolarizations and TdP in the anaesthetized, α1-adrenergically stimulated rabbit model of TdP.
Vagal nerve activity contributes to drug-induced TdP in rabbits
In the present study, heart rate variability significantly increased from baseline in the ‘TdP+’ group of animals before TdP during simultaneous administration of phenylephrine and dofetilide, which indicates vagal nerve activation prior to TdP occurrence. Furthermore, phenylephrine infusion increased significantly the up-BRS in all of the animals before dofetilide infusion, but the increase was significantly higher in the ‘TdP+’ group, which indicates that the vagal nerve (and the baroreflex) was more responsive to the rise in blood pressure in the animals experiencing TdP than that in the animals not developing TdP. Nevertheless, the reason for the observed increase in vagal nerve activity in the ‘TdP+’ group of animals and the role of the vagal nerve activity in TdP development need further investigations to clarify.
Phenylephrine induces bradycardia via activation of the baroreflex resulting in an increase in vagal nerve activity and a reduction in cardiac sympathetic activity in pentobarbital-anaesthetized rabbits (Farkas et al., 2008). Importantly, as the present study shows, although bradycardia makes the heart susceptible to drug-induced TdP, phenylephrine-induced bradycardia was not the key factor for TdP induction, as heart rate did not differ between the ‘TdP+’ and the ‘TdP−’ groups.
Recently, we reported that vagotomy prevented clofilium-induced TdP in α1-adrenoceptor-stimulated, pentobarbital-anaesthetized rabbits (Farkas et al., 2008). Likewise, inhalation of formaldehyde vapour initiated TdP in conscious, dofetilide-treated rabbits with a mechanism involving reflex co-activation of the parasympathetic and the sympathetic outflows to the heart (Nalivaiko et al., 2004). These earlier studies and our present heart rate variability and BRS results imply that vagal nerve activity may contribute to the development of drug-induced TdP.
Sympathetic activity
In our study, the parasympathetic and sympathetic activities of the animals were estimated only in long, uninterrupted periods of sinus rhythm, as the methods required. According to our results, the sympathetic activity during sinus rhythm did not predict TdP occurrence. However, it is widely accepted that an increased sympathetic activity may contribute to the genesis of TdP (Verrier and Antzelevitch, 2004). The progressive increase in the frequency of arrhythmic beats and the ‘absolute’ beat-to-beat variability and instability of the RR interval during dofetilide infusion shows that the number of arrhythmic beats and the irregularity of the ventricular rhythm progressively increased in the ‘TdP+’ group prior to TdP occurrence. In humans, ventricular premature beats were consistently followed by a burst of sympathetic nerve activation (Welch et al., 1989). Furthermore, it has been shown recently that greater degrees of the irregularity of the ventricular rhythm caused greater sympathoexcitation (Segerson et al., 2007). As premature ventricular beats occurred frequently and the irregularity of the ventricular rhythm progressively increased in the animals before TdP, we cannot exclude the possibility that sympathetic activity contributed to dofetilide-induced TdP in our study.
Conclusion
‘Preceding’ arrhythmias have characteristics that permit prediction of TdP occurrence: the more chaotic the ventricular rhythm, the greater the probability of TdP initiation. This suggests that complexity of the arrhythmic beats may play an important mechanistic role in TdP genesis. The beat-to-beat electrical instability quantified by the novel ‘absolute’ variability and instability parameters correlates with the probability of TdP occurrence in our model. Furthermore, baroreflex and vagal nerve activity may contribute to dofetilide-induced TdP in the applied in vivo model.
Acknowledgments
Mr Gábor Komáromi is acknowledged for his help with calculation of the beat-to-beat variability and instability parameters. Dr Michael J. Curtis (King's College, London, UK) is thanked for his helpful comments on the manuscript. The generous gift of dofetilide by Gedeon Richter PLC. (Budapest, Hungary) is gratefully acknowledged. This work was supported by the Hungarian Ministry of Health [ETT 203/2003, 353/2006]; the Hungarian National Research Fund [OTKA F-046776, OTKA K-69018, NI-61902]; the National Research and Development Programmes [NKFP 1A/046/2004]; and the Hungarian Academy of Sciences.
Glossary
Abbreviations
- BRS
baroreflex sensitivity
- DI
ventricular electric diastolic interval
- ECG
electrocardiogram
- IKr
the rapid component of the delayed rectifier potassium current
- LTI
long-term instability
- LTV
long-term variability
- QTc
heart rate-corrected QT interval
- RMS
root mean square
- RMSSD
root mean square of the successive differences
- SDSD
standard deviation of the successive differences
- STI
short-term instability
- STV
short-term variability
- TdP
torsades de pointes
- TI
total instability
- Tpeak–Tend
the period between the peak and the end of T wave
Conflict of interest
None.
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (4th edn.) 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C. Ionic, molecular, and cellular bases of QT-interval prolongation and torsade de pointes. Europace. 2007;9(Suppl 4):iv4–iv15. doi: 10.1093/europace/eum166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batey AJ, Coker SJ. Proarrhythmic potential of halofantrine, terfenadine and clofilium in a modified in vivo model of torsade de pointes. Br J Pharmacol. 2002;135:1003–1012. doi: 10.1038/sj.bjp.0704550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan LV, Kabell G, Brunden MN, Gibson JK. Comparative assessment of ibutilide, d-sotalol, clofilium, E-4031, and UK-68,798 in a rabbit model of proarrhythmia. J Cardiovasc Pharmacol. 1993;22:540–549. [PubMed] [Google Scholar]
- Carlsson L, Almgren O, Duker G. QTU-prolongation and torsades de pointes induced by putative class III antiarrhythmic agents in the rabbit: etiology and interventions. J Cardiovasc Pharmacol. 1990;16:276–285. doi: 10.1097/00005344-199008000-00014. [DOI] [PubMed] [Google Scholar]
- Carlsson L, Abrahamsson C, Drews L, Duker G. Antiarrhythmic effects of potassium channel openers in rhythm abnormalities related to delayed repolarization. Circulation. 1992;85:1491–1500. doi: 10.1161/01.cir.85.4.1491. [DOI] [PubMed] [Google Scholar]
- Carlsson L, Drews L, Duker G, Schiller-Linhardt G. Attenuation of proarrhythmias related to delayed repolarization by low-dose lidocaine in the anesthetized rabbit. J Pharmacol Exp Ther. 1993;267:1076–1080. [PubMed] [Google Scholar]
- Carlsson L, Andersson B, Linhardt G, Lofberg L. Assessment of the ion channel-blocking profile of the novel combined ion channel blocker AZD1305 and its proarrhythmic potential versus dofetilide in the methoxamine-sensitized rabbit in vivo. J Cardiovasc Pharmacol. 2009;54:82–89. doi: 10.1097/FJC.0b013e3181ac62c9. [DOI] [PubMed] [Google Scholar]
- Farkas A, Leprán I, Papp JG. Comparison of the antiarrhythmic and the proarrhythmic effect of almokalant in anaesthetised rabbits. Eur J Pharmacol. 1998;346:245–253. doi: 10.1016/s0014-2999(98)00067-3. [DOI] [PubMed] [Google Scholar]
- Farkas A, Leprán I, Papp JG. Proarrhythmic effects of intravenous quinidine, amiodarone, d-sotalol, and almokalant in the anesthetized rabbit model of torsade de pointes. J Cardiovasc Pharmacol. 2002;39:287–297. doi: 10.1097/00005344-200202000-00016. [DOI] [PubMed] [Google Scholar]
- Farkas A, Batey AJ, Coker SJ. How to measure electrocardiographic QT interval in the anaesthetized rabbit. J Pharmacol Toxicol Methods. 2004;50:175–185. doi: 10.1016/j.vascn.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Farkas A, Dempster J, Coker SJ. Importance of vagally mediated bradycardia for the induction of torsade de pointes in an in vivo model. Br J Pharmacol. 2008;154:958–970. doi: 10.1038/bjp.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas AS, Nattel S. Minimizing repolarization-related proarrhythmic risk in drug development and clinical practice. Drugs. 2010;70:573–603. doi: 10.2165/11535230-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Farkas AS, Acsai K, Tóth A, Dézsi L, Orosz S, Forster T, et al. Importance of extracardiac α1-adrenoceptor stimulation in assisting dofetilide to induce torsade de pointes in rabbit hearts. Eur J Pharmacol. 2006;537:118–125. doi: 10.1016/j.ejphar.2006.03.014. [DOI] [PubMed] [Google Scholar]
- Farkas AS, Makra P, Csík N, Orosz S, Shattock MJ, Fülöp F, et al. The role of the Na+/Ca2+ exchanger, INa and ICaL in the genesis of dofetilide-induced torsades de pointes in isolated, AV-blocked rabbit hearts. Br J Pharmacol. 2009;156:920–932. doi: 10.1111/j.1476-5381.2008.00096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondeghem LM, Carlsson L, Duker G. Instability and triangulation of the action potential predict serious proarrhythmia, but action potential duration prolongation is antiarrhythmic. Circulation. 2001;103:2004–2013. doi: 10.1161/01.cir.103.15.2004. [DOI] [PubMed] [Google Scholar]
- Laurita KR, Girouard SD, Akar FG, Rosenbaum DS. Modulated dispersion explains changes in arrhythmia vulnerability during premature stimulation of the heart. Circulation. 1998;98:2774–2780. doi: 10.1161/01.cir.98.24.2774. [DOI] [PubMed] [Google Scholar]
- van der Linde H, Van de Water A, Loots W, Van Deuren B, Lu HR, Van Ammel K, et al. A new method to calculate the beat-to-beat instability of QT duration in drug-induced long QT in anesthetized dogs. J Pharmacol Toxicol Methods. 2005;52:168–177. doi: 10.1016/j.vascn.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Locati EH, Maison-Blanche P, Dejode P, Cauchemez B, Coumel P. Spontaneous sequences of onset of torsade de pointes in patients with acquired prolonged repolarization: quantitative analysis of Holter recordings. J Am Coll Cardiol. 1995;25:1564–1575. doi: 10.1016/0735-1097(95)00100-i. [DOI] [PubMed] [Google Scholar]
- Lu HR, Remeysen P, De Clerck F. Nonselective I(Kr)-blockers do not induce torsades de pointes in the anesthetized rabbit during α1-adrenoceptor stimulation. J Cardiovasc Pharmacol. 2000;36:728–736. doi: 10.1097/00005344-200012000-00007. [DOI] [PubMed] [Google Scholar]
- Michael G, Dempster J, Kane KA, Coker SJ. Potentiation of E-4031-induced torsade de pointes by HMR1556 or ATX-II is not predicted by action potential short-term variability or triangulation. Br J Pharmacol. 2007;152:1215–1227. doi: 10.1038/sj.bjp.0707513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalivaiko E, De Pasquale CG, Blessing WW. Ventricular arrhythmias triggered by alerting stimuli in conscious rabbits pre-treated with dofetilide. Basic Res Cardiol. 2004;99:142–151. doi: 10.1007/s00395-003-0448-1. [DOI] [PubMed] [Google Scholar]
- Nemec J, Hejlik JB, Shen WK, Ackerman MJ. Catecholamine-induced T-wave lability in congenital long QT syndrome: a novel phenomenon associated with syncope and cardiac arrest. Mayo Clin Proc. 2003;78:40–50. doi: 10.4065/78.1.40. [DOI] [PubMed] [Google Scholar]
- Pedersen HS, Elming H, Seibaek M, Burchardt H, Brendorp B, Torp-Pedersen C, et al. Risk factors and predictors of Torsade de pointes ventricular tachycardia in patients with left ventricular systolic dysfunction receiving Dofetilide. Am J Cardiol. 2007;100:876–880. doi: 10.1016/j.amjcard.2007.04.020. [DOI] [PubMed] [Google Scholar]
- Philp KL, Hart G, Coker SJ. A gender-independent proarrhythmic action of 17beta-estradiol in anaesthetized rabbits. Eur J Pharmacol. 2007;575:113–121. doi: 10.1016/j.ejphar.2007.07.035. [DOI] [PubMed] [Google Scholar]
- Pugsley MK, Authier S, Curtis MJ. Principles of safety pharmacology. Br J Pharmacol. 2008;154:1382–1399. doi: 10.1038/bjp.2008.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segerson NM, Sharma N, Smith ML, Wasmund SL, Kowal RC, Abedin M, et al. The effects of rate and irregularity on sympathetic nerve activity in human subjects. Heart Rhythm. 2007;4:20–26. doi: 10.1016/j.hrthm.2006.09.017. [DOI] [PubMed] [Google Scholar]
- Shah RR, Hondeghem LM. Refining detection of drug-induced proarrhythmia: QT interval and TRIaD. Heart Rhythm. 2005;2:758–772. doi: 10.1016/j.hrthm.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Thomsen MB, Verduyn SC, Stengl M, Beekman JD, de Pater G, van Opstal J, et al. Increased short-term variability of repolarization predicts d-sotalol-induced torsades de pointes in dogs. Circulation. 2004;110:2453–2459. doi: 10.1161/01.CIR.0000145162.64183.C8. [DOI] [PubMed] [Google Scholar]
- Thomsen MB, Oros A, Schoenmakers M, van Opstal JM, Maas JN, Beekman JD, et al. Proarrhythmic electrical remodelling is associated with increased beat-to-beat variability of repolarisation. Cardiovasc Res. 2007;73:521–530. doi: 10.1016/j.cardiores.2006.11.025. [DOI] [PubMed] [Google Scholar]
- Verrier RL, Antzelevitch C. Autonomic aspects of arrhythmogenesis: the enduring and the new. Curr Opin Cardiol. 2004;19:2–11. doi: 10.1097/00001573-200401000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincze D, Farkas AS, Rudas L, Makra P, Csík N, Leprán I, et al. Relevance of anaesthesia for dofetilide-induced torsades de pointes in α1-adrenoceptor-stimulated rabbits. Br J Pharmacol. 2008;153:75–89. doi: 10.1038/sj.bjp.0707536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos MA, Gorenek B, Verduyn SC, van der Hulst FF, Leunissen JD, Dohmen L, et al. Observations on the onset of torsade de pointes arrhythmias in the acquired long QT syndrome. Cardiovasc Res. 2000;48:421–429. doi: 10.1016/s0008-6363(00)00192-9. [DOI] [PubMed] [Google Scholar]
- Walker MJA, Curtis MJ, Hearse DJ, Campbell RWF, Janse MJ, Yellon DM, et al. The Lambeth Conventions: guidelines for the study of arrhythmias in ischaemia infarction, and reperfusion. Cardiovasc Res. 1988;22:447–455. doi: 10.1093/cvr/22.7.447. [DOI] [PubMed] [Google Scholar]
- Welch WJ, Smith ML, Rea RF, Bauernfeind RA, Eckberg DL. Enhancement of sympathetic nerve activity by single premature ventricular beats in humans. J Am Coll Cardiol. 1989;13:69–75. doi: 10.1016/0735-1097(89)90551-2. [DOI] [PubMed] [Google Scholar]