

Abstract
BACKGROUND AND PURPOSE
Emodin [1,3,8-trihydroxy-6-methylanthraquinone] has been reported to exhibit vascular anti-inflammatory properties. However, the corresponding mechanisms are not well understood. The present study was designed to explore the molecular target(s) of emodin in modifying lipopolysaccharide (LPS)-associated signal transduction pathways in endothelial cells.
EXPERIMENTAL APPROACH
Cultured primary human umbilical vein endothelial cells (HUVECs; passages 3–5) were pre-incubated with emodin (1–50 µg·mL−1). LPS-induced expression of pro-inflammatory cytokines [interleukin (IL)-1β, IL-6] and chemokines (IL-8; CCL2/MCP-1) were determined by reverse transcription-PCR and elisa. Nuclear factor-κB (NF-κB) activation, inhibitor of κB (IκB)α degradation and Toll-like receptor-4 (TLR-4) were detected by immunocytochemistry and Western blotting. Cholesterol depletion [by methyl β-cyclodextrin (MBCD), a specific cholesterol binding agent] and cholesterol replenishment were further used to investigate the roles of lipid rafts in activation of HUVECs.
KEY RESULTS
Emodin inhibited, concentration-dependently, the expression of LPS-induced pro-inflammatory cytokines (IL-1β, IL-6) and chemokines (IL-8, CCL2) and, in parallel, inhibited NF-κB activation and IκBα degradation in HUVECs. However, emodin did not inhibit the NF-κB activation and IκBα degradation induced by IL-1β. The cholesterol binding agent, MBCD, inhibited LPS-induced NF-κB activation in passaged HUVECs [which also lack the LPS receptor, membrane CD14 (mCD14)], showing that lipid rafts played a key role in LPS signalling in mCD14-negative HUVECs. Moreover, emodin disrupted the formation of lipid rafts in cell membranes by depleting cholesterol.
CONCLUSIONS AND IMPLICATIONS
Lipid rafts were crucial in facilitating inflammatory responses of mCD14-negative HUVECs to LPS. Emodin disrupted lipid rafts through depleting cholesterol and, consequently, inhibited inflammatory responses in endothelial cells.
Keywords: emodin, TLR-4, anti-inflammation, NF-κB, HUVECs
Introduction
Atherosclerosis, a multifactorial cardiovascular disease and the leading cause of death in modern society, involves multiple processes, including endothelial dysfunction, inflammation, vascular proliferation, leucocyte adhesion and transmigration. The vascular endothelium is situated on the internal surface of all blood vessels and serves as a physical barrier between the blood and tissues. These cells also exhibit a number of physiological functions, including the mediation of vascular tone, blood flow, leucocyte trafficking, angiogenesis and coagulation (Wort and Evans, 1999). Activation of endothelial cell plays a particularly important role in inflammation via production of pro-inflammatory cytokines/chemokines, cell adhesion molecules and nitric oxide (Kim et al., 2006). There are few effective therapeutic options available to treat atherosclerosis because of its pathological complexity. The key problem in the cardiovascular studies is the demonstration of atherogenesis and its prevention. Among the many biochemical and pathophysiological hallmarks, inflammation is one of the most important features and generally considered as the critical target for prevention of atherosclerosis (Xia et al., 2007).
Several medicinal herbs have been shown to inhibit specific cellular and humoral immune responses (Chen et al., 2005; Xia et al., 2007; Singh et al., 2008). Emodin [1,3,8-trihydroxy-6-methylanthraquinone], extracted from Frangula bark, has been shown to display a wide range of biological activities including antiviral, antimicrobial, anti-tumour, anti-fibrosis, elimination of reactive oxygen species and anti-inflammatory (Li et al., 2005; Huang et al., 2009). Treatment of RAW 264.7 macrophages with emodin (20 µg·mL−1) inhibited the expression of a panel of inflammatory-associated genes, including tumour necrosis factor-α (TNF-α), inducible nitric oxide synthase (iNOS), interleukin-10 (IL-10), cytosolic inhibitor of κB (IκB)α, IκB kinase (IKK)-α and IKK-γ, to different extents, as well as the nuclear translocation of nuclear factor-κB (NF-κB) (Li et al., 2005) In the case of endothelial cells, very few studies have involved emodin. Although, more than 10 years ago, there was a report that 50 µg·mL−1 emodin inhibited TNF-induced NF-κB activation and adhesion molecule expression in endothelial cells (Kumar et al., 1998), the molecular target(s) of the anti-inflammatory actions of emodin in endothelium remain mostly unknown.
Lipopolysaccharide (LPS), or endotoxin, is the major component of the outer surface of Gram-negative bacteria. LPS is a potent activator of cells in the immune and inflammatory systems, including macrophages, monocytes and endothelial cells, and contributes to the systemic response to bacterial infection known as septic shock (Zhang et al., 1999). LPS-induced activation of monocytes/macrophages is mediated through a cell surface receptor glycoprotein, known as membrane CD14 (mCD14). The binding of LPS to mCD14 is enhanced by LPS-binding protein, a plasma protein. However, passaged human umbilical vein endothelial cells (HUVECs) do not express mCD14 and respond to LPS only in the presence of soluble CD14 (Lloyd and Kubes, 2006). The vascular endothelium is a key host target of LPS and the first tissue barrier. Endothelial cells express the Toll-like receptor-4 (TLR-4), an innate immune, pattern recognition receptor that is activated by LPS (Qureshi et al., 1999; Takeda and Akira, 2005). Endothelial cell activation is dependent on the assembly on the cell surface of a multi-protein recognition complex consisting of soluble CD14, MD-2, TLR-4 and other molecules (Bannerman and Goldblum, 2003; Bowdish et al., 2004). Following activation of this receptor complex, the interaction of IL-1 receptor associate kinase (IRAK) family members with TNF receptor associated factor-6 (TRAF-6) results, subsequently, in the activation of a downstream kinase cascade involving NF-κB-inducing kinase (NIK) and IKK. IKK-mediated phosphorylation of IκB leads to IκB degradation, thus enabling NF-κB nuclear translocation and the induction of new gene expression. Many transcription factors are important in orchestrating the inflammatory response (Morris et al., 2006; Parker et al., 2007). NF-κB is an essential transcription factor for expression of inflammation-related proteins. The extensively studied signal pathways induced by LPS provide means of exploring the molecular target(s) of the immunosuppressive and anti-inflammatory effects of emodin.
The aim of the present investigation was to find out whether emodin could suppress LPS-induced pro-inflammatory responses and NF-κB activation in HUVECs and to identify the molecular target(s) of emodin in the TLR-4 signalling pathway. Moreover, the effect of emodin on pro-inflammatory cytokines and chemokines induced by LPS would also be addressed. Our results showed that emodin inhibited NF-κB dependent transcriptional activation induced by LPS, but not that induced by IL-1β, in mCD14-negative HUVECs. This action was due to the inhibition of LPS-receptor complex formation by emodin, through disrupting the structure of cell membrane lipid rafts.
Methods
Endothelial cell culture and cell treatment
Human umbilical vein endothelial cells along with the culture medium were purchased from ScienCell Research Laboratories (San Diego, CA, USA). The culture medium was composed of basal medium, 5% fetal bovine serum, 1% endothelial cell growth supplement and 1% penicillin/streptomycin solution. Cells were seeded in poly-L-lysine coated 75 cm2 cell culture flask (Corning, NY, USA), and maintained in an incubator at 37°C with 95% humidity and 5% CO2. HUVECs were subcultured after trypsinization (0.025% trypsin, 0.5 mM EDTA, 1 mM sodium pyruvate and 10 mM HEPES) when grew to approximately 90% confluence, and used throughout the passages 3 to 6.
The cultured HUVECs were preincubated with various concentrations of emodin. Briefly, emodin stock solution was prepared by dissolving emodin in dimethyl sulfoxide (DMSO) (Sigma, St. Louis, MO, USA) to a concentration of 10 mg·mL−1, and was diluted with cell culture medium to reach final concentrations 1–50 µg·mL−1 before experiments. Cells seeded in different culture plates were preincubated with 1–50 µg·mL−1 emodin for 30 min. The control sample was treated with 0.5% DMSO in culture medium, which is equal to the maximal DMSO concentration in emodin-preincubated samples. Finally, cells were stimulated with 0.1 µg·mL−1 or 1 µg·mL−1 LPS in the presence of emodin.
Cell viability assay
Cell viability was assessed by WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfopheny)-2H-tetrazolium] cytotoxic assay kit (Beyotime, Beijing, China) according to the manufacturer's instructions. Briefly, HUVECs were seeded with 5 × 103 cells per well in 96 well plates. Confluent HUVECs were treated as mentioned above, and the supernatants in each well were replaced by fresh culture medium (100 µL per well) after cells were exposed to LPS for 5 h, and then WST-8 solution (10 µL) was added to each well and incubated for 1 h. The absorbance values of all the samples were recorded by a microplate reader (Model 680, Bio-Rad, Philadelphia, PA, USA) at 450 nm.
Total RNA isolation and RT-PCR
Total RNA was isolated from cultured HUVECs by using TriZol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions after the cells were stimulated with 0.1 µg·mL−1 LPS for 4 h. One microgram of total RNA obtained from each sample was used for reverse transcription-PCR (RT-PCR) in a 20 µL reaction mixture, and the reverse transcription was performed at 42°C for 90 min. The reverse transcriptase was inactivated at 70°C for 5 min. The cDNA reaction mixture (3 µL) was amplified in a 25 µL standard PCR reaction. PCR amplification was performed on a thermal cycler by 30 cycles, each cycle consisting of 94°C for 1 min, 60°C for 1 min and 72°C for 2 min. The PCR productions were separated by 1% agarose gel electrophoresis. The following sets of primers were used in the PCR amplification. Primer sequences for IL-1β were 5′-GGATATGGAGCAACAAGTGG-3′ and 5′-ATGTACCAGTTGGGGAACTG-3′ for the sense and antisense primers respectively (Bowdish et al., 2004). Primers for IL-6, IL-8, CCL2, TLR-4 and β-actin were, for IL-6, sense, 5′-TCAATGAGGAGACTTG CCTG-3′ and antisense, 5′-GATGAGTTGTCATGTCCTGC-3′ (Chang et al., 2004); for IL-8, sense, 5′-ATG ACTTCCAAGCTGGCCGTGGCT-3′ and antisense, 5′-TCTC AGCCCTCTTCAAA AACTTCTC-3′ (Li et al., 2004); for CCL2, sense, 5′-CCAATT CTCAAACTGAA GCTCGC-3′, and antisense, 5′-CTTAGCTGCAGATTCTTGGGTTGTG-3′ (Lo et al., 1998); for TLR4, sense, 5′-TGGATACGTTTCCTTATAAG-3′, and antisense, 5′-GA AATGGAGGCACCCCTTC-3′; for β-actin, sense, 5′-CTGGGAC GACATGGAG AAA-3′, and antisense, 5′-AAGGAAGGCTGGAAGAGTGC-3′ (Faure et al., 2000).
elisa assay
The cultured HUVECs were seeded in 48 well plates and preincubated with emodin for 30 min. Supernatants were collected after 5 h stimulation, by 0.1 µg·mL−1 LPS at 37°C, and stored at −20°C until assay for cytokines. IL-6 and CCL2 protein levels were determined by elisa assay kit according to the manufacturer's instructions (Senxiong Biotech. Shanghai, China). Absorbance was detected with a microplate reader (Model 680, Bio-Rad) at 490 nm, and the data processing was accomplished with the software of Curve Expert 1.3 (Boster, Wuhan, China).
Immunocytochemistry and lipid rafts staining
The treated HUVECs were washed with phosphate buffered saline (PBS) and fixed in 4% paraformaldehyde for 15 min, following by washing three times in Tris buffered saline with Triton (TBST, 20 mM Tris, 150 mM NaCl, 0.1% Triton X-100) for 5 min, and incubating with blocking buffer (TBST with 5% bovine serum albumin) for 2 h at room temperature. The cells were incubated at 4°C overnight with rabbit anti-human p65 polyclonal antibody (1:200). After washing in TBST with gentle shaking, the cells were stained with Cy3-labelled goat anti-rabbit IgG (1:1000) for 2 h at room temperature, and then the cells were washed three times in TBST for 8 min again. The fluorescent images were obtained using an inverted fluorescent microscope (Nikon TE-2000, Japan).
For labelling membrane lipid rafts with cholera toxin subunit B (CTxB), the treated HUVECs were fixed in 4% paraformaldehyde for 20 min at room temperature, and incubated with Alexa Fluor 488-conjugated CTxB (5 µg·mL−1) for 20 min. After washing with TBST, cell nuclei were stained by 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI). Fluorescent images were captured by the fluorescent microscope mentioned above.
Protein extraction and Western blotting
Total proteins were extracted from cultured HUVECs with lysis buffer (Beyotime, Beijing, China) containing 20 mM Tris, 150 mM NaCl, 1% Triton X-100 and several protease inhibitors. After the cells were lysed, supernatants were collected after centrifugation (12 000×g for 5 min) at 4°C. Nuclear and cytoplasmic proteins of HUVECs were extracted by nuclear and cytoplasmic protein extraction kits according to the manufacturer's instructions (Beyotime, Beijing, China).
Protein concentrations were determined by a bicinchoninic acid protein assay kit (Beyotime, Beijing, China). Equal amounts (15–30 µg) of protein extracted from cultured cells were run on 10% SDS-PAGE, followed by electrotransferring to polyvinylidene fluoride membranes (Millipore, Bedford, MA, USA). The membranes were blocked with 5% skim milk powder for 1 h, and sequentially incubated with rabbit anti-human p65 (1:1000), rabbit anti-human IκB (1:1000), rabbit anti-human TLR-4 (1:600) polyclonal antibodies or mouse anti-human β-actin monoclonal antibody. After washing, the membranes were further probed with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:5000) or horseradish peroxidase-conjugated goat anti-mouse IgG (1:5000). The oxidase components were detected by chemiluminescence (BeyoECL Plus) (Beyotime, Beijing, China).
Cholesterol replenishment experiment
Human umbilical vein endothelial cells were treated with culture medium alone or medium containing emodin (20, 30 and 50 µg·mL−1), DMSO (0.5%) or MBCD (10 mM) at 37°C for 60 min. Subsequently the cells were washed with PBS and incubated with medium alone or medium containing water-soluble cholesterol (84 µg·mL−1) for 30 min. The cells were exposed to LPS (0.1 µg·mL−1) for another 30 min. To detect the effect of cholesterol itself on HUVECs activation, cells were treated with cholesterol alone for 30 min. The nuclear translocation of p65 and expression of IκBα were analysed as mentioned above.
Presentation of data and statistical analysis
Data are presented as mean ± standard deviation (SD). The statistical significance of differences was tested using one-way anova followed by post hoc Bonferroni tests and P < 0.05 was considered significant.
Materials
Lipopolysaccharide (from Escherichia coli 055:B5), emodin (extract from Frangula bark), water-soluble cholesterol and methyl-β-cyclodextrin (MBCD) were obtained from Sigma (St. Louis, MO, USA). Alexa Fluor 488-conjugated cholera toxin subunit B (CTxB) was purchased from Invitrogen Life Technologies (Carlsbad, CA, USA). IL-1β was purchased from R&D Systems (Minneapolis, MN, USA). Rabbit anti-human TLR-4 polyclonal antibody and mouse anti-human β-actin monoclonal antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). NF-κB inhibitor BAY11-7082, DAPI, rabbit anti-human p65 polyclonal antibody, rabbit anti-human IκBα polyclonal antibody and Cy3-labeled goat anti-rabbit IgG were from Beyotime Institute of Biotechnology (Beijing, China). Peroxidase conjugated goat anti-mouse and goat anti-rabbit IgG were obtained from ZSGB-BIO (Beijing, China). The drug/molecular target nomenclature in this study follows Alexander et al. (2009).
Results
Emodin inhibits secretion of inflammatory cytokines, concentration-dependently
To determine whether emodin affected expressions of inflammatory cytokines in LPS-stimulated HUVECs, the levels of mRNA for IL-1β, IL-6, IL-8 and CCL2 were detected by RT-PCR. Our studies showed that the expression of mRNA for IL-1β, IL-6, IL-8 and CCL2 in LPS-stimulated HUVECs were down-regulated by emodin in a dose-dependent manner (Figure 1A). This effect was clearer when the emodin concentrations exceeded 10 µg·mL−1. DMSO (0.5%) had no influence on expression of these inflammatory cytokines (Figure 1A). We also tested for cytotoxicity with emodin in these concentrations and found no obvious cytotoxicity towards HUVECs (data not shown). Expression of IL-6 and CCL2 protein was determined by elisa after exposure of HUVECs to LPS, in the absence or presence of emodin (5–50 µg·mL−1) for 5 h. IL-6 and CCL2 proteins were also dose-dependently inhibited by high concentrations of emodin (10–50 µg·mL−1) (Figure 1B and C), which agreed well with the effects on mRNA expression. DMSO (0.5%) did not affect IL-6 and CCL2 expression. The NF-κB inhibitor BAY11-7082 markedly inhibited IL-6 and CCL2 expression in LPS-stimulated HUVECs (Figure 1B and C).
Figure 1.
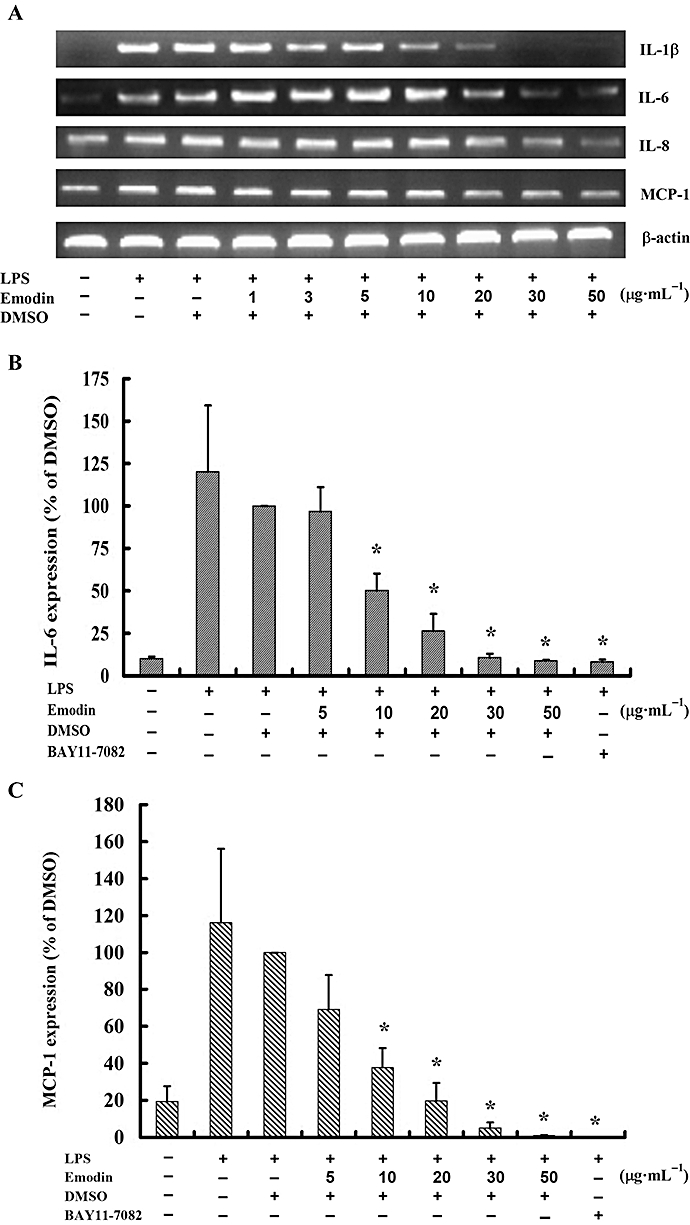
Emodin inhibits lipopolysaccharide (LPS)-induced cytokine production concentration-dependently. (A) Cells were preincubated with emodin for 30 min and then treated with LPS (0.1 µg·mL−1) for 4 h. mRNA expression of cytokines (IL-1β, IL-6, IL-8 and CCL2) was detected by reverse transcription-PCR and agarose gel electrophoresis. (B) Cells were preincubated with emodin or the NF-κB inhibitor, BAY11-7082 (40 µM) for 30 min and then treated with LPS (0.1 µg·mL−1) for 5 h. The protein expression level of IL-6 and CCL2 were detected by elisa. Values represent mean ± SD of three replicate experiments. *P < 0.05 for emodin or NF-κB inhibitor versus dimethyl sulfoxide.
Emodin blocks LPS-induced NF-κB activation and IκBα degradation
Nuclear factor-κB is a multi-subunit transcription factor which rapidly activates the transcription of various cytokines and chemokines (Caamano and Hunter, 2002), playing key roles in the inflammatory response. The NF-κB dimers (p65 and p50) bound to its inhibitor, IκB, was found mainly in the cytoplasm in unactivated cells. Upon activation, IκB is degraded, allowing NF-κB to translocate into the nucleus where it can regulate gene transcription. Here, immunocytochemistry and Western blotting were used to test whether emodin could inhibit LPS-induced NF-κB activation in HUVECs. The immunocytochemical data were consistent with those from Western blotting. The results obtained from the two methods showed that emodin markedly blocked the translocation of p65 from cytoplasm into the nucleus in a dose-dependent manner (Figure 2). However, the expression of total p65 was not affected by a range of concentrations of emodin (Figure 2B). The degradation of IκBα in HUVECs induced by LPS was also detected and we found that IκBα degradation was markedly inhibited by emodin (Figure 2C).
Figure 2.
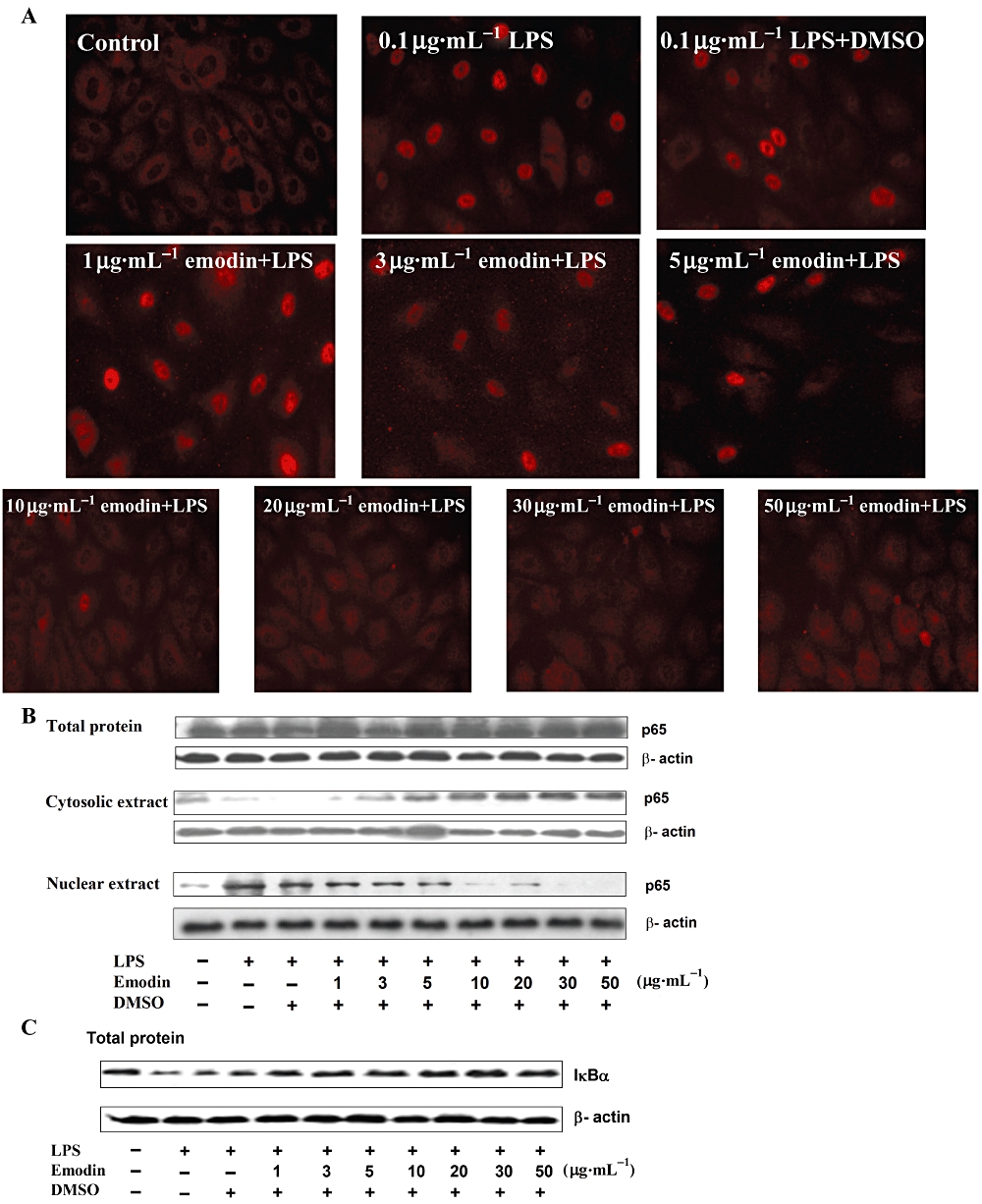
Emodin inhibits lipopolysaccharide (LPS)-induced NF-κB activation and IκBα degradation. (A) Cells were preincubated with emodin for 30 min, and then treated with LPS (0.1 µg·mL−1) for 30 min. Cellular distribution of the NF-κB subunit p65 in human umbilical vein endothelial cells was detected by immunofluorescence staining. (B) and (C) Cells were preincubated with emodin for 30 min and then treated with LPS (0.1 µg·mL−1) for 30 min. Cells were lysed with lysis buffer. The levels of p65 in total cell extract, cytosolic extract and nuclear extract (B), and IκBα degradation (C), were detected by Western blotting.
Emodin did not inhibit IL-1β-induced NF-κB activation and IκBα degradation
To explore the molecular target(s) of emodin action, we firstly hypothesized that targets of emodin action were in the cytoplasm. LPS- and IL-1β-induced signalling pathways share many of the same intracellular steps, from myeloid differentiation factor 88 (MyD88) to NF-κB (see Figure 3A) (Zhang et al., 1999). There are also two MyD88-independent pathways of late-phase NF-κB and the transcription factor IFN response factor 3 (IRF-3) mediated by TLR-4, which modulates the expressions of CXCL10 and IFN-β respectively (Figure 3A) (Youn et al., 2005). However, the MyD88-independent pathways are dependent on mCD14 and passaged endothelial cells do not express mCD14 (Lloyd-Jones et al., 2008). Herein, to disclose the target(s) of emodin, it is reasonable to compare the two signalling pathways induced by LPS and IL-1β after emodin treatment. As shown in Figure 3B, emodin did not inhibit IL-1β-induced NF-κB activation. To further determine the effect of emodin on the translocation of p65 from the cytoplasm to the nucleus, the distribution of p65 in cytoplasm and nucleus was detected by Western blotting (Figure 3C). This result agreed well with that of the immunocytochemical assay, as shown in Figure 3B. Moreover, the degradation of IκBα was not inhibited by emodin in IL-1β-induced HUVECs (Figure 3D).
Figure 3.
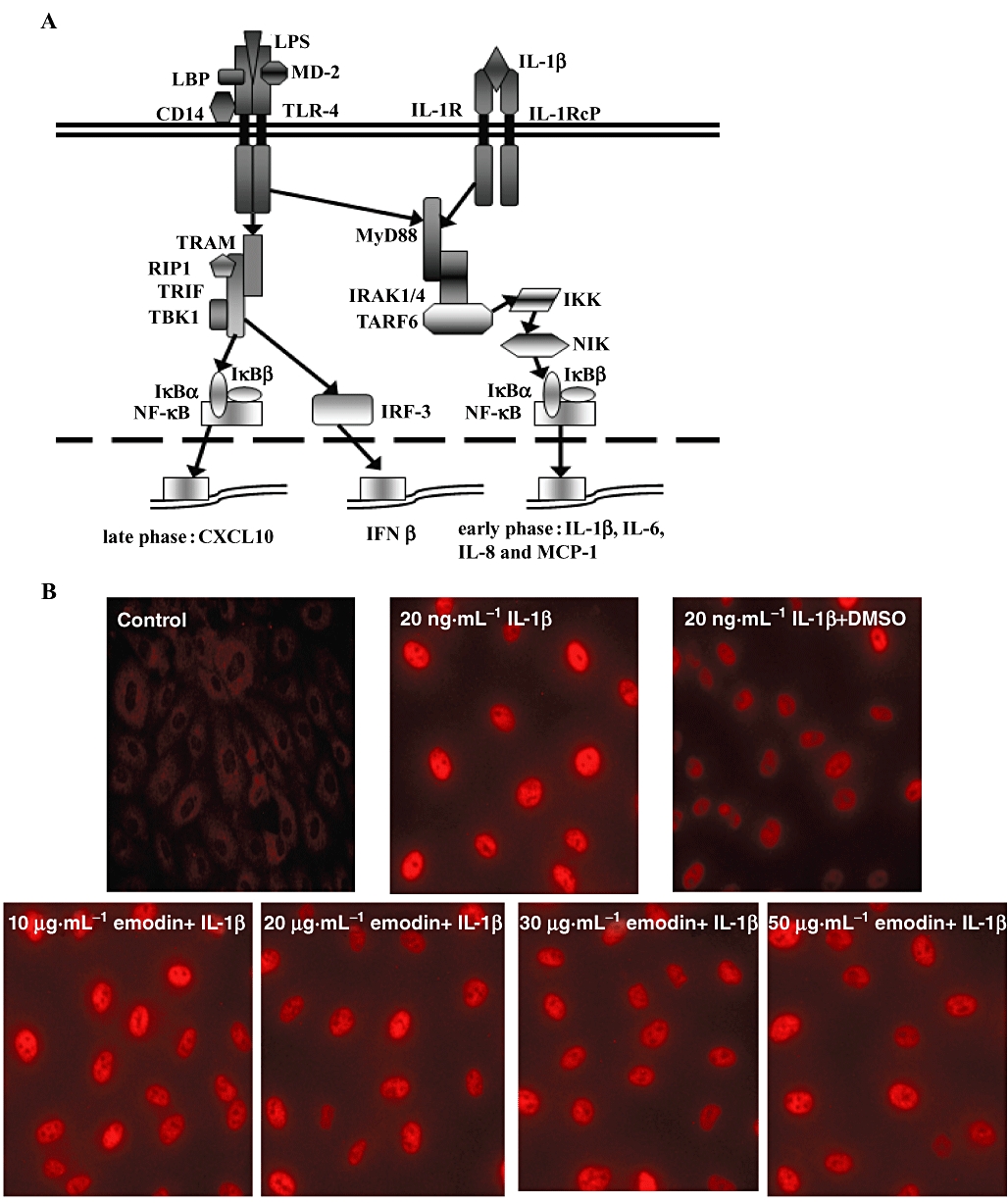
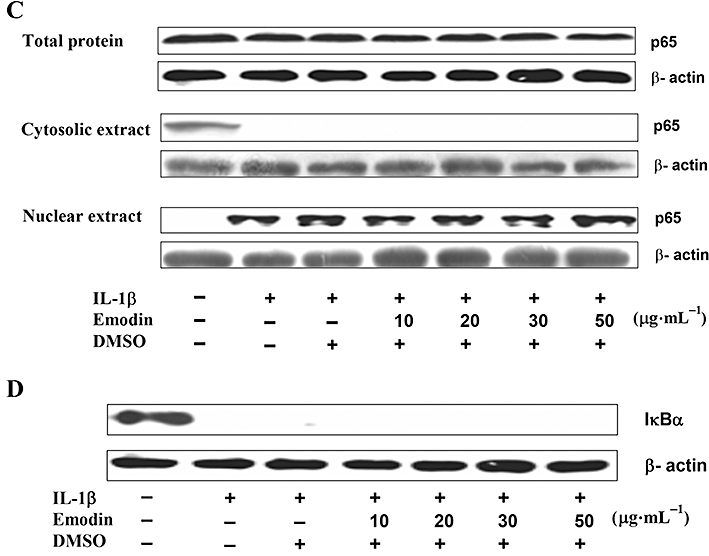
Emodin did not inhibit IL-1β-induced NF-κB activation and IκBα degradation. (A) Scheme of signalling pathways initiated by lipopolysaccharide and IL-1β (adapted from Youn et al., 2005) with some modifications. Cells were preincubated with emodin for 30 min and then treated with IL-1β (20 ng·mL−1) for an additional 30 min. The cellular distribution and expression of NF-κB subunit p65 in human umbilical vein endothelial cells was detected by immunofluorescence staining (B). The levels of p65 in total cell extracts, cytosolic extracts and nuclear extracts (C), and IκBα degradation (D), were detected by Western blotting.
Taking together the effects of emodin on NF-κB activation and IκBα degradation induced by the two stimuli, LPS and IL-1β, it was clear that emodin did not directly act on the NF-κB dimer or other parts of the signalling pathway that are shared by activation of TLR-4 and the IL-1 receptor. This suggested to us that emodin may act on the plasma membrane components of the LPS signalling pathway. In the plasma membrane, LPS-induced signal transduction is initiated through a complex structure comprising CD14, TLR-4, MD-2, lipid rafts and other molecules. Our subsequent experiments focused on different components of this membrane complex.
Emodin did not act through affecting the function of sCD14 or TLR-4 expression
CD14, a cell surface glycoprotein, resident in lipid rafts in monocytes and macrophages (Szabo et al., 2007), plays an important role in clustering of TLR-4 in lipid rafts. Passaged endothelial cells do not express the membrane-bound form of CD14 (mCD14), but they can utilize the soluble form of CD14 (sCD14) which is present in serum, and has a similar function to that of mCD14 (Walton et al., 2003; Lloyd-Jones et al., 2008). High concentrations (1 µg·mL−1) of LPS can induce a pro-inflammatory response in endothelial cells, independent of sCD14, through the TLR-4 pathway (Lloyd-Jones et al., 2008). We found that emodin inhibited NF-κB activation induced by 1 µg·mL−1 LPS, showing that the inhibitory effect of emodin did not involve sCD14 binding to TLR-4 (Figure 4A and B).
Figure 4.
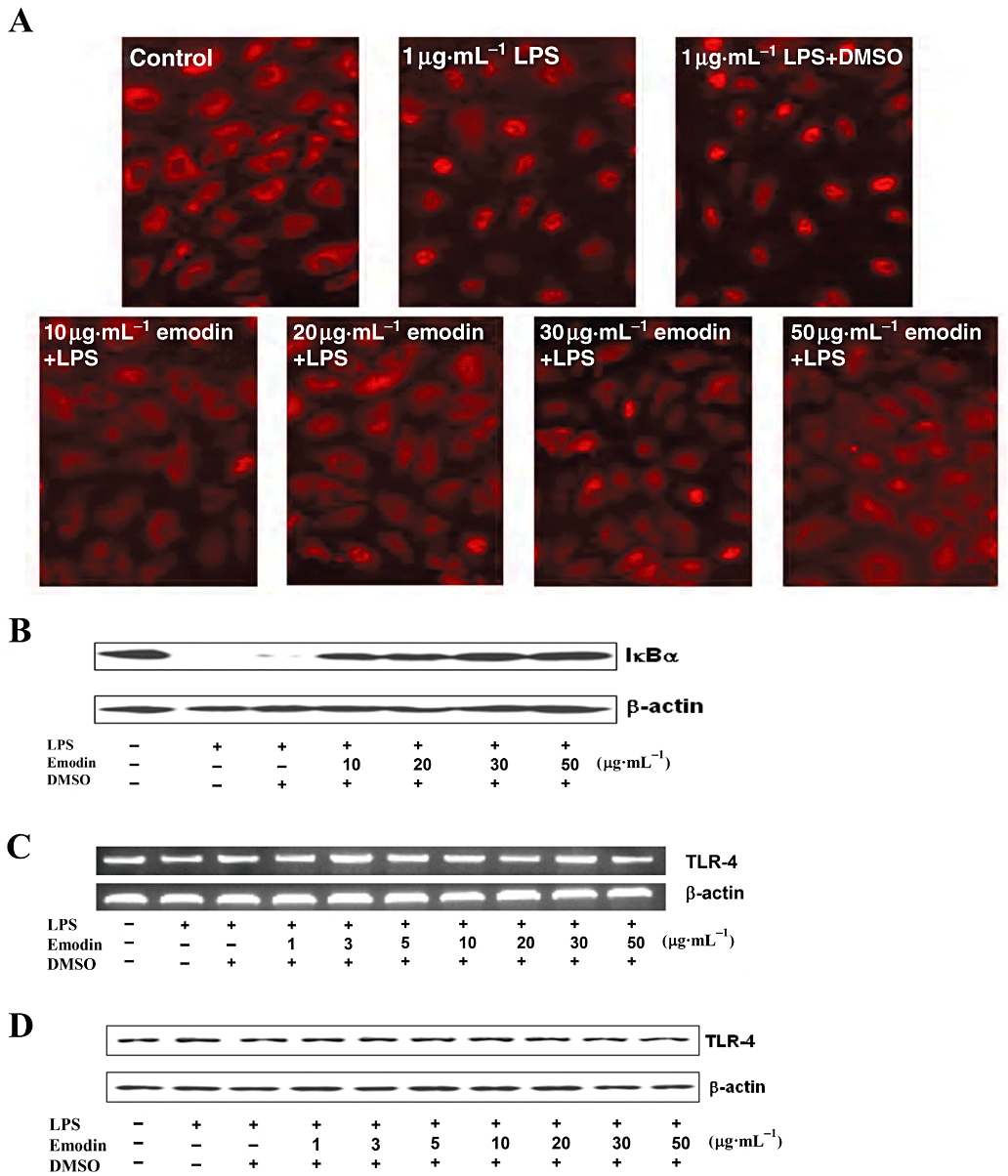
Inhibition of emodin was not through affecting the function of sCD14 and expression of Toll-like receptor-4 (TLR-4). (A) Cells were preincubated with emodin for 30 min and then treated with lipopolysaccharide (LPS) (1 µg·mL−1) for an additional 30 min. Nuclear translocation of p65 and the expression level of IκBα were detected by immunofluorescence staining and Western blotting (B) respectively. (C) Cells were preincubated with emodin for 30 min and then treated with LPS (0.1 µg·mL−1) for 4 h. mRNA expression level of TLR-4 was detected by reverse transcription-PCR. (D) Cells were preincubated with indicated concentrations of emodin for 30 min and then treated with LPS (0.1 µg·mL−1) for 5 h. TLR-4 protein expression was analysed by Western blotting.
Toll-like receptor-4 (LPS receptor) is a membrane protein and its expression will directly affect NF-κB activation in HUVECs exposed to LPS. Thus, emodin could suppress LPS-induced pro-inflammatory responses and NF-κB activation by down-regulating TLR-4 expression. To assess this possibility, HUVECs were stimulated by LPS in the absence and presence of emodin (1–50 µg·mL−1) for 4 or 5 h and TLR-4 expression was detected by RT-PCR and Western blotting. As shown in Figure 4C and D, there were no changes in TLR-4 expression in LPS-stimulated HUVECs, after emodin treatment, at RNA or protein levels.
Emodin disrupts lipid rafts to inhibit activation of HUVECs induced by LPS
Lipid rafts, which localize in the cell membrane and internal membrane, are microdomains that contain high concentrations of cholesterol and glycosphingolipids. It is well known that lipid rafts provide platforms for the formation of receptor complexes, their internalization and triggering of signalling pathways, including the LPS-induced mCD14-dependent signalling pathway, finally leading to cell activation. However, the role of lipid rafts in the activation of passaged endothelial cells induced by LPS is not fully defined. To address and demonstrate the relationship between lipid rafts and NF-κB activation induced by LPS, passaged HUVECs was pretreated with the cholesterol binding agent MBCD, known to disrupt lipid rafts, for 1 h, and then the cells were stimulated with 1 µg·mL−1 LPS in the presence of MBCD for 30 min. As shown in Figure 5A and B, NF-κB activation in HUVECs induced by LPS was significantly inhibited by MBCD, implying that lipid rafts make an important contribution to LPS-induced activation of passaged HUVECs.
Figure 5.
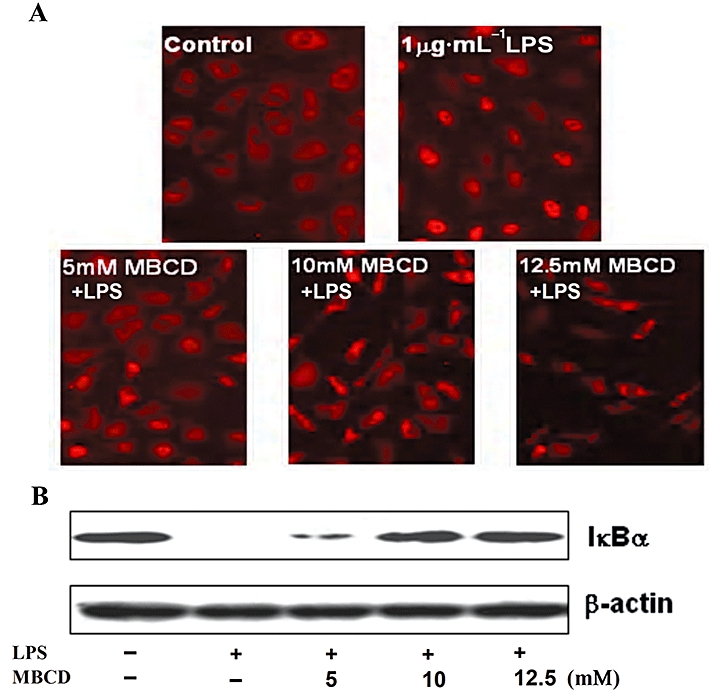
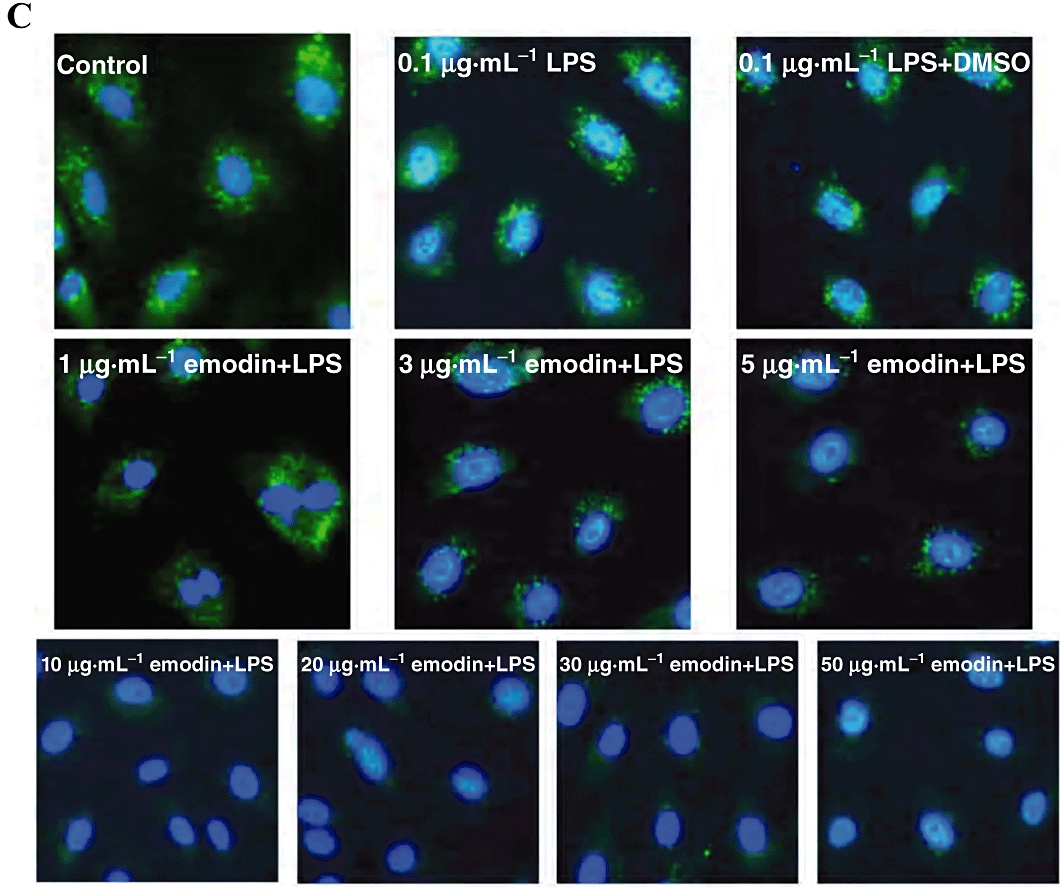
Emodin disrupts lipid rafts to inhibit activation of human umbilical vein endothelial cells (HUVECs) by lipopolysaccharide (LPS). (A) Lipid rafts contribute to HUVECs activation by LPS. Cells were preincubated with methyl-β-cyclodextrin (5, 10 and 12.5 mM) for 1 h and then treated with LPS (1 µg·mL−1) for additional 30 min. Nuclear translocation of p65 was detected by immunofluorescence staining and IκBα was detected by Western blotting (B). (C) Cells were preincubated with emodin for 30 min and then treated with LPS (0.1 µg·mL−1) for 4 h. The lipid rafts (green) and nucleus (blue) were stained by Alexa Fluor 488-conjugated CTxB and DAPI respectively.
GM1-ganglioside is a marker for lipid rafts. Disruption of lipid rafts would cause redistribution of GM1-ganglioside from lipid raft to non-lipid raft domains of cell membranes (Triantafilou et al., 2002). Therefore, the effect of emodin on GM1 distribution would be a measure of the disruption of lipid rafts formation. HUVECs were incubated with emodin and LPS for 4 h and Alexa Fluor 488-conjugated CTxB, which specifically binds GM1, was used to label and visualize lipid rafts in HUVECs. Using this assay, few lipid rafts were detected in the cells treated with 10–50 µg·mL−1 emodin, showing that emodin clearly disrupted the formation of lipid rafts (Figure 5C). One interpretation of these results is that emodin dispersed GM1-ganglioside in lipid rafts to non-lipid raft domains of membranes or internal membranes and, consequently, few lipid rafts were visualized. However, lower concentrations of emodin (1–5 µg·mL−1), LPS and DMSO had no obvious effect on the formation of lipid rafts, using this marker.
Cholesterol replenishment prevents the effect of emodin on NF-κB activation induced by LPS
Cholesterol is another major component of lipid rafts (Pike, 2003) and cholesterol enrichment makes lipid rafts more tightly packed than the surrounding phospholipid-rich, non-raft, phase of the cell membrane (Song et al., 2007). The cholesterol binding agent, MBCD, can disrupt lipid rafts by depleting cholesterol from lipid rafts and our results showed that emodin disrupted lipid rafts (Figure 5). To check if emodin and MBCD inhibited LPS-induced HUVEC activation by the same mechanism, cholesterol replenishment experiments were carried out. Cells were treated with 20, 30 and 50 µg·mL−1 emodin for 60 min, followed by 30 min of cholesterol replenishment. Cells were stimulated by LPS (0.1 µg·mL−1) for 30 min. Immunocytochemical assay of p65 and Western blot detection of IκBα revealed that cholesterol replenishment abolished the inhibitory effect of emodin. However, cholesterol alone had no effect on NF-κB activation (Figure 6). Moreover, cholesterol also restored NF-κB activation in HUVECs pre-incubated with MBCD (Figure 6).
Figure 6.
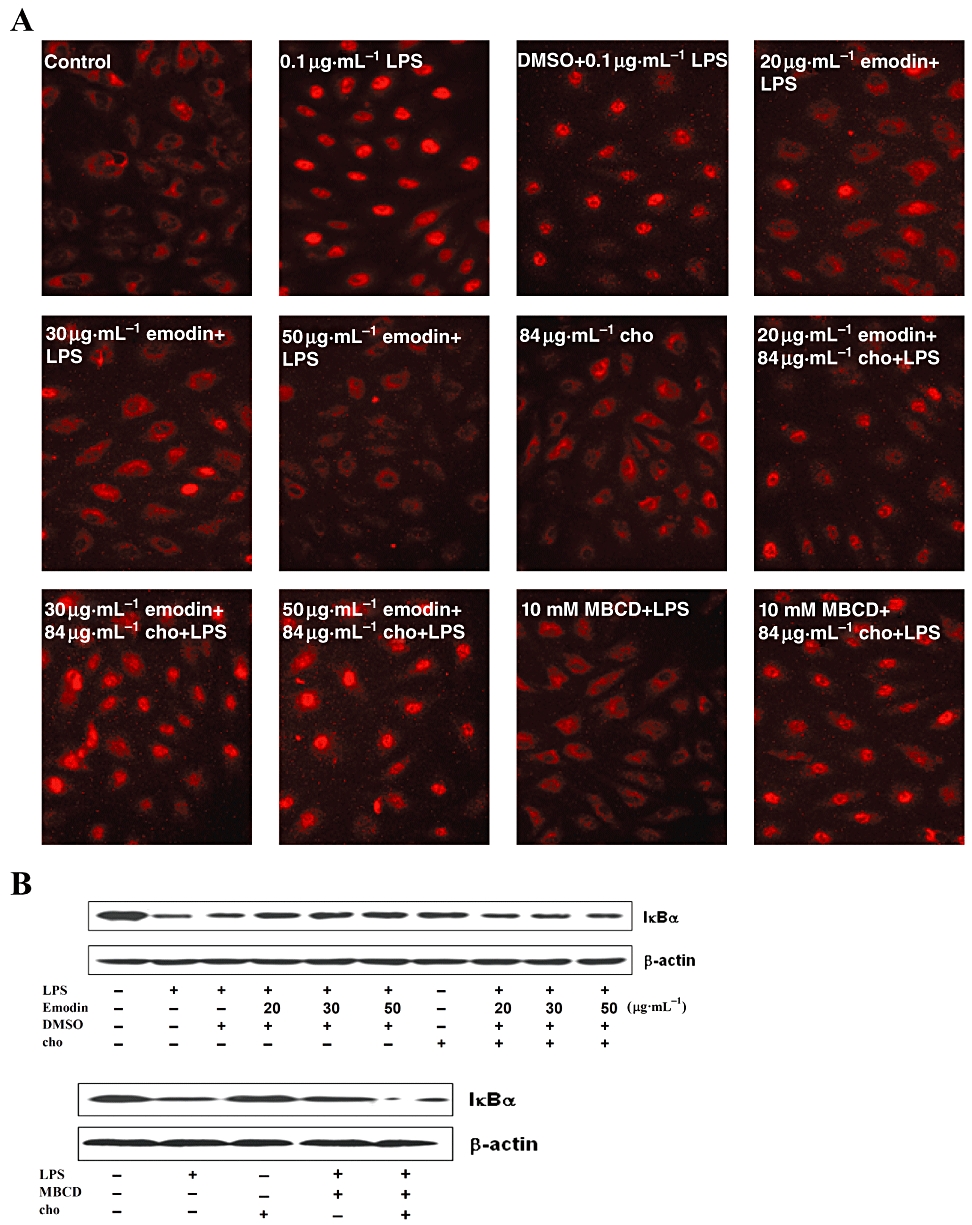
Cholesterol (cho) replenishment prevents the inhibitory effect of emodin on NF-κB activation induced by lipopolysaccharide (LPS). Cells were treated with emodin (20, 30 and 50 µg·mL−1), methyl-β-cyclodextrin (10 mM) or culture medium alone for 60 min, replaced by fresh cell culture medium, and followed by cholesterol replenishment (84 µg·mL−1) or not for 30 min. The cells were then stimulated with 0.1 µg·mL−1 LPS for 30 min. Cells treated with cholesterol alone for 30 min served as another control. Nuclear translocation of p65 and expression of IκBα were detected by immunofluorescence staining and Western blotting respectively.
As NF-κB regulates expression of some cytokines, such as IL-6, we also explored whether IL-6 expression would be restored after cholesterol replenishment. As shown in Figure 7, IL-6 expression was increased after cholesterol replenishment in HUVECs treated with emodin or MBCD. To further confirm that emodin disrupted lipid rafts by depleting cholesterol, we next examined the lipid rafts using Alexa Fluor 488-conjugated CTxB. Cholesterol replenishment completely restored the destruction of lipid rafts induced by emodin and MBCD (Figure 8). Taken together, these results from the cholesterol replenishment experiments indicate that emodin disrupted the lipid rafts by depleting cholesterol, leading to the inhibition of NF-κB activation in LPS-induced HUVECs.
Figure 7.
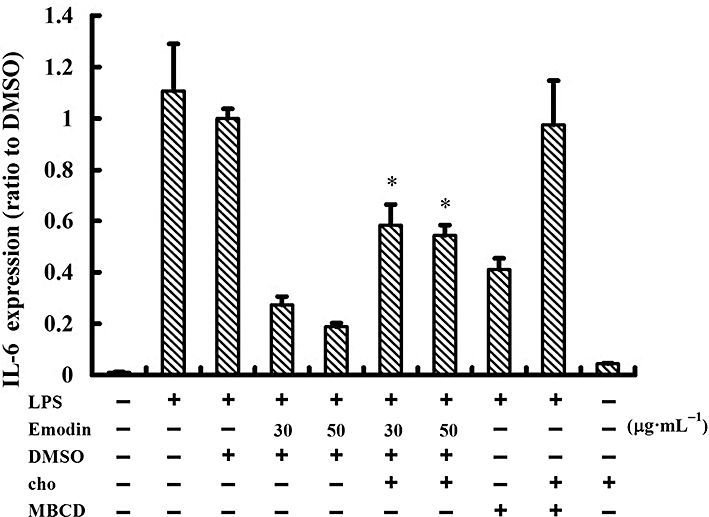
IL-6 expression was increased after cholesterol replenishment in human umbilical vein endothelial cells treated with emodin and methyl-β-cyclodextrin (MBCD). Cells were treated with emodin (30 and 50 µg·mL−1), MBCD (10 mM) or culture medium alone for 60 min, replaced by fresh cell culture medium, and followed by cholesterol (cho) replenishment (80 µg·mL−1) or not for 60 min. The cells were then stimulated with 0.1 µg·mL−1 lipopolysaccharide for 4 h. Cells treated with cholesterol alone for 4 h served as another control. IL-6 expression was detected by elisa.
Figure 8.
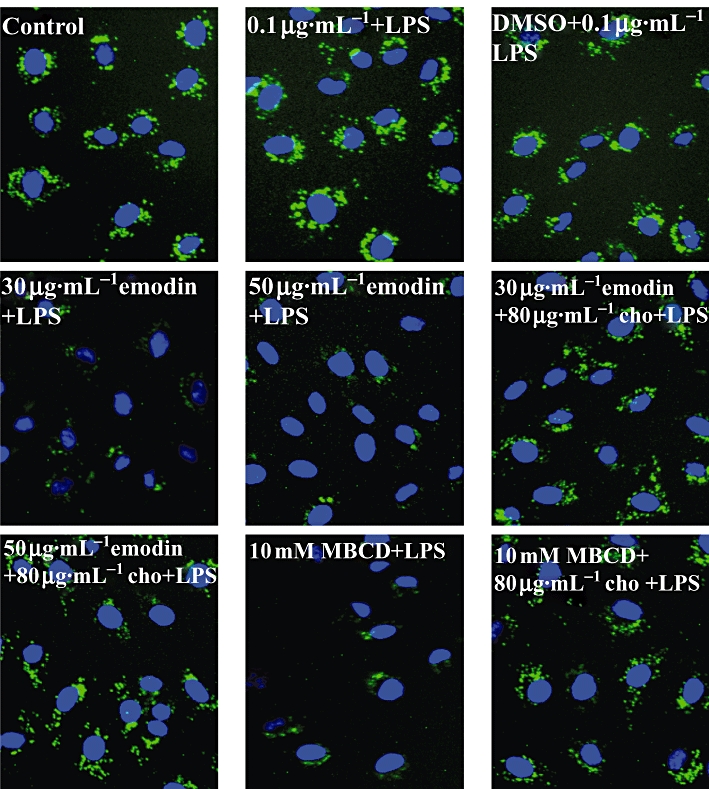
Lipid raft formation was restored after cholesterol replenishment in human umbilical vein endothelial cells treated with emodin and methyl-β-cyclodextrin (MBCD). Cells were treated with emodin (30 and 50 µg·mL−1), MBCD (10 mM) or culture medium alone for 60 min, replaced by fresh cell culture medium, and followed by cholesterol (cho) replenishment (80 µg·mL−1) or not for 60 min. The cells were then stimulated with 0.1 µg·mL−1 lipopolysaccharide for 30 min. The lipid rafts (green) and nucleus (blue) were stained by Alexa Fluor 488-conjugated CTxB and DAPI respectively.
Discussion
The work presented here describes two important findings. Firstly, we provide further evidence for the crucial role of lipid rafts in facilitating the inflammatory responses of mCD14-negative cells (passaged HUVECs) to LPS. Secondly, we showed that emodin disrupted lipid rafts through depleting cholesterol from these structures, leading to the inhibition of inflammatory responses induced by LPS in HUVECs.
Our studies demonstrated that emodin clearly inhibited the expression of IL-1β, IL-6, IL-8 and CCL2 when the HUVECs were stimulated by LPS (Figure 1). Because of the importance of this mode of activating HUVECs, it is essential to discover the mechanism, that is, the molecular target(s) of emodin's action. We first examined the effect of emodin on the intracellular signalling pathway from MyD88 to NF-κB activation and found that emodin inhibited NF-κB activation induced by LPS (Figure 2), but not by IL-1β (Figure 3). It is well known that these two signalling pathways, induced by LPS or IL-1β, share the same intracellular signalling components (from MyD88 to NF-κB) in the MyD88-dependent pathway (Figure 3A). Thus it was unlikely that the site of action of emodin was on the pathway from MyD88 to NF-κB activation. In other words, the molecular target(s) of the anti-inflammatory action of emodin was not within the shared stages of LPS- and IL-1β-induced signalling pathways. Therefore, we postulated that the molecular target(s) of emodin anti-inflammatory action was the LPS receptor complex and its constituent molecules. In the later investigations, we focused on the cell membrane and greater attention was paid to the possible effects of emodin on the formation of the LPS-receptor complex in the cell membrane.
Lipopolysaccharide (1 µg·mL−1) induced a pro-inflammatory endothelial response, independent of sCD14, through the TLR-4 pathway (Lloyd-Jones et al., 2008), comparable to that found in monocytes, at a lower concentration of LPS (10 ng·mL−1) (Lynn et al., 1993; Cohen et al., 1995). Our results showed that emodin inhibited inflammatory responses of HUVECs induced by high LPS concentration (1 µg·mL−1) (Figure 4A and B), allowing us to conclude that inhibition of pro-inflammatory responses by emodin did not involve the function of sCD14. Endothelial cells also express TLR-4, an innate immune pattern recognition receptor that is activated by LPS. We next addressed the question of whether emodin inhibited pro-inflammatory responses through down-regulation of TLR-4 expression. However, the expression of RNA or protein for TLR-4 was not changed by emodin treatment over a range of concentrations (Figure 4C and D).
We then provided evidence that emodin inhibited HUVECs activation induced by LPS through disrupting lipid rafts. MBCD is a cholesterol binding agent, which can deplete cholesterol from the lipid rafts, and in our experiments, we found MBCD inhibited LPS-induced NF-κB activation (Figure 5A and B) and that emodin could also disrupt lipid rafts (Figure 5C). These results indicated that lipid rafts played a key role in the activation of HUVECs by LPS. Furthermore, our cholesterol replenishment results confirmed that cholesterol depletion contributed to the inhibition of HUVEC activation by emodin. After cholesterol replenishment, NF-κB activation and IL-6 expression both increased (Figures 6 and 7) in HUVECs treated with emodin or MBCD. These changes are likely to be due to the replenishment of lipid raft cholesterol (Figure 8), because these results from cell activation and from IL-6 expression agreed well with the restoration of lipid rafts. Overall, our results demonstrated that emodin could disrupt lipid rafts to inhibit endothelial cell activation, by depleting cholesterol from the lipid rafts.
It is well established that lipid rafts constitute dynamic platforms that float in cell membranes and are enriched in signalling molecules. One of these, mCD14, localized in lipid rafts, participates in the recruitment of TLR-4 to lipid rafts, and leads to activation of CD14-positive cells induced by LPS. But for CD14-negative cells (passaged HUVECs), the roles of lipid rafts in LPS-induced signalling pathway have been less clearly defined. Our studies demonstrated that lipid rafts regulated the LPS pathway in passaged HUVECs (Figures 5 and 6). Other investigations have reported that heat-shock protein 70 (HSP 70) and HSP 90, resident in lipid rafts of monocytes and endothelial cells were also involved in LPS-induced signal transduction, as incubation of cells with polyclonal antibodies to HSP 70 and 90, prior to LPS stimulation, abolished IL-6 secretion (Triantafilou et al., 2001a,b; 2002;). One possible explanation for the inhibitory action of emodin is that it prevented the formation of the LPS-receptor complex, including HSP 70, 90 and other molecules, and thus prevented cell activation by LPS. Our current findings may serve to show that the integrity of lipid rafts are critical for inflammatory activation of CD14-negative cells, stimulated by LPS, as well as for the CD14-positive cells.
Lipid rafts are complex microdomains of the cell membrane serving many functions. They play an important role not only in receptor-mediated signal transduction, but also in ion channel-mediated signal transduction. Recently, it has been reported that lipid rafts are also enriched in ion channels. The large-conductance Ca2+-activated K+ channel, MaxiK, appears to play an important role in the NF-κB dependent inflammatory response of macrophages to LPS as blockade of MaxiK down-regulated cytokine expression in LPS stimulated macrophages through inhibiting NF-κB activation, without affecting mitogen-activated protein kinases (Papavlassopoulos et al., 2006). MaxiK is also expressed in HUVECs and is resident in lipid rafts (Maguy et al., 2006; Dong et al., 2007). So, it is possible that emodin could influence ion channel functions through disrupting lipid rafts, leading to inhibition of HUVECs activation by LPS. But further studies are needed to fully assess this possibility.
In summary, the present study has strongly suggested that lipid rafts play a vital role in LPS-induced signalling pathway in CD14-negative, passaged HUVECs. Emodin inhibited the pro-inflammatory activation of HUVECs through directly disrupting lipid rafts by depleting cholesterol, and suppressed LPS induced NF-κB activation and cytokine secretion. This novel anti-inflammatory action of emodin demonstrates that it has promising applications in the therapy of atherosclerosis or other inflammation-related cardiovascular diseases.
Acknowledgments
The financial support provided by the National Natural Science Foundation of China (30700151, 81071257), New Century Excellent Talents Program in Chinese Universities (NCET-09-0263), Sichuan Youth Science and Technology Fundation (2010JQ0004), Visiting Scholar Foundation of Key Lab of Biorheological Science and Technology in Chongqing University (to Yiyao Liu), and Innovative Talent Incubation Program for Youth (Y02018023601062) from UESTC, are greatly appreciated.
Glossary
Abbreviations
- elisa
enzyme-linked immunosorbent assay
- HUVECs
human umbilical vein endothelial cells
- IκB
inhibitor of NF-κB
- LPS
lipopolysaccharide
- MBCD
methyl-β-cyclodextrin
- mCD14
membrane CD14
- MyD88
myeloid differentiation factor 88
- NF-κB
nuclear factor-κB
- RT-PCR
reverse transcription-polymerase chain reaction
- TLR-4
Toll-like receptor-4
Conflicts of interest
None to declare.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (4th edn.) 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannerman DD, Goldblum SE. Mechanisms of bacterial lipopolysaccharide-induced endothelial apoptosis. Am J Physiol Lung Cell Mol Physiol. 2003;284:L899–L914. doi: 10.1152/ajplung.00338.2002. [DOI] [PubMed] [Google Scholar]
- Bowdish DM, Davidson DJ, Speert DP, Hancock RE. The human cationic peptide LL-37 induces activation of the extracellular signal-regulated kinase and p38 kinase pathways in primary human monocytes. J Immunol. 2004;172:3758–3765. doi: 10.4049/jimmunol.172.6.3758. [DOI] [PubMed] [Google Scholar]
- Caamano J, Hunter CA. NF-kappaB family of transcription factors: central regulators of innate and adaptive immune functions. Clin Microbiol Rev. 2002;15:414–429. doi: 10.1128/CMR.15.3.414-429.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YC, Hsu TL, Lin HH, Chio CC, Chiu AW, Chen NJ, et al. Modulation of macrophage differentiation and activation by decoy receptor 3. J Leukoc Biol. 2004;75:486–494. doi: 10.1189/jlb.0903448. [DOI] [PubMed] [Google Scholar]
- Chen W, Esselman WJ, Jump DB, Busik JV. Anti-inflammatory effect of docosahexaenoic acid on cytokine-induced adhesion molecule expression in human retinal vascular endothelial cells. Invest Ophthalmol Vis Sci. 2005;46:4342–4347. doi: 10.1167/iovs.05-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen L, Haziot A, Shen DR, Lin XY, Sia C, Harper R, et al. CD14-independent responses to LPS require a serum factor that is absent from neonates. J Immunol. 1995;155:5337–5342. [PubMed] [Google Scholar]
- Dong DL, Zhang Y, Lin DH, Chen J, Patschan S, Goligorsky MS, et al. Carbon monoxide stimulates the Ca2(+)-activated big conductance k channels in cultured human endothelial cells. Hypertension. 2007;50:643–651. doi: 10.1161/HYPERTENSIONAHA.107.096057. [DOI] [PubMed] [Google Scholar]
- Faure E, Equils O, Sieling PA, Thomas L, Zhang FX, Kirschning CJ, et al. Bacterial lipopolysaccharide activates NF-kappaB through toll-like receptor 4 (TLR-4) in cultured human dermal endothelial cells. Differential expression of TLR-4 and TLR-2 in endothelial cells. J Biol Chem. 2000;275:11058–11063. doi: 10.1074/jbc.275.15.11058. [DOI] [PubMed] [Google Scholar]
- Huang Z, Chen G, Shi P. Effects of emodin on the gene expression profiling of human breast carcinoma cells. Cancer Detect Prev. 2009;32:286–291. doi: 10.1016/j.cdp.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Kim JH, Jeong JH, Jeon ST, Kim H, Ock J, Suk K, et al. Decursin inhibits induction of inflammatory mediators by blocking nuclear factor-kappaB activation in macrophages. Mol Pharmacol. 2006;69:1783–1790. doi: 10.1124/mol.105.021048. [DOI] [PubMed] [Google Scholar]
- Kumar A, Dhawan S, Aggarwal BB. Emodin (3-methyl-1,6,8-trihydroxyanthraquinone) inhibits TNF-induced NF-kappaB activation, IkappaB degradation, and expression of cell surface adhesion proteins in human vascular endothelial cells. Oncogene. 1998;17:913–918. doi: 10.1038/sj.onc.1201998. [DOI] [PubMed] [Google Scholar]
- Li HL, Chen HL, Li H, Zhang KL, Chen XY, Wang XW, et al. Regulatory effects of emodin on NF-kappaB activation and inflammatory cytokine expression in RAW 264.7 macrophages. Int J Mol Med. 2005;16:41–47. [PubMed] [Google Scholar]
- Li J, Moran T, Swanson E, Julian C, Harris J, Bonen DK, et al. Regulation of IL-8 and IL-1beta expression in Crohn's disease associated NOD2/CARD15 mutations. Hum Mol Genet. 2004;13:1715–1725. doi: 10.1093/hmg/ddh182. [DOI] [PubMed] [Google Scholar]
- Lloyd KL, Kubes P. GPI-linked endothelial CD14 contributes to the detection of LPS. Am J Physiol Heart Circ Physiol. 2006;291:H473–H481. doi: 10.1152/ajpheart.01234.2005. [DOI] [PubMed] [Google Scholar]
- Lloyd-Jones KL, Kelly MM, Kubes P. Varying importance of soluble and membrane CD14 in endothelial detection of lipopolysaccharide. J Immunol. 2008;181:1446–1453. doi: 10.4049/jimmunol.181.2.1446. [DOI] [PubMed] [Google Scholar]
- Lo SK, Bovis L, Matura R, Zhu B, He S, Lum H, et al. Leishmania lipophosphoglycan reduces monocyte transendothelial migration: modulation of cell adhesion molecules, intercellular junctional proteins, and chemoattractants. J Immunol. 1998;160:1857–1865. [PubMed] [Google Scholar]
- Lynn WA, Liu Y, Golenbock DT. Neither CD14 nor serum is absolutely necessary for activation of mononuclear phagocytes by bacterial lipopolysaccharide. Infect Immun. 1993;61:4452–4461. doi: 10.1128/iai.61.10.4452-4461.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguy A, Hebert TE, Nattel S. Involvement of lipid rafts and caveolae in cardiac ion channel function. Cardiovasc Res. 2006;69:798–807. doi: 10.1016/j.cardiores.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Morris GE, Parker LC, Ward JR, Jones EC, Whyte MK, Brightling CE, et al. Cooperative molecular and cellular networks regulate Toll-like receptor-dependent inflammatory responses. FASEB J. 2006;20:2153–2155. doi: 10.1096/fj.06-5910fje. [DOI] [PubMed] [Google Scholar]
- Papavlassopoulos M, Stamme C, Thon L, Adam D, Hillemann D, Seydel U, et al. MaxiK blockade selectively inhibits the lipopolysaccharide-induced I kappa B-alpha /NF-kappa B signaling pathway in macrophages. J Immunol. 2006;177:4086–4093. doi: 10.4049/jimmunol.177.6.4086. [DOI] [PubMed] [Google Scholar]
- Parker LC, Prince LR, Sabroe I. Translational mini-review series on Toll-like receptors: networks regulated by Toll-like receptors mediate innate and adaptive immunity. Clin Exp Immunol. 2007;147:199–207. doi: 10.1111/j.1365-2249.2006.03203.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res. 2003;44:655–667. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]
- Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, Gros P, et al. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4) J Exp Med. 1999;189:615–625. doi: 10.1084/jem.189.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Kumar S, Singh P, Raj HG, Prasad AK, Parmar VS, et al. Piper longum Linn. Extract inhibits TNF-alpha-induced expression of cell adhesion molecules by inhibiting NF-kappaB activation and microsomal lipid peroxidation. Phytomedicine. 2008;15:284–291. doi: 10.1016/j.phymed.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Song JH, Tse MC, Bellail A, Phuphanich S, Khuri F, Kneteman NM, et al. Lipid rafts and nonrafts mediate tumor necrosis factor related apoptosis-inducing ligand induced apoptotic and nonapoptotic signals in non small cell lung carcinoma cells. Cancer Res. 2007;67:6946–6955. doi: 10.1158/0008-5472.CAN-06-3896. [DOI] [PubMed] [Google Scholar]
- Szabo G, Dolganiuc A, Dai Q, Pruett SB. TLR4, ethanol, and lipid rafts: a new mechanism of ethanol action with implications for other receptor-mediated effects. J Immunol. 2007;178:1243–1249. doi: 10.4049/jimmunol.178.3.1243. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol. 2005;17:1–14. doi: 10.1093/intimm/dxh186. [DOI] [PubMed] [Google Scholar]
- Triantafilou K, Triantafilou M, Dedrick RL. A CD14-independent LPS receptor cluster. Nat Immunol. 2001a;2:338–345. doi: 10.1038/86342. [DOI] [PubMed] [Google Scholar]
- Triantafilou K, Triantafilou M, Ladha S, Mackie A, Dedrick RL, Fernandez N, et al. Fluorescence recovery after photobleaching reveals that LPS rapidly transfers from CD14 to hsp70 and hsp90 on the cell membrane. J Cell Sci. 2001b;114:2535–2545. doi: 10.1242/jcs.114.13.2535. [DOI] [PubMed] [Google Scholar]
- Triantafilou M, Miyake K, Golenbock DT, Triantafilou K. Mediators of innate immune recognition of bacteria concentrate in lipid rafts and facilitate lipopolysaccharide-induced cell activation. J Cell Sci. 2002;115:2603–2611. doi: 10.1242/jcs.115.12.2603. [DOI] [PubMed] [Google Scholar]
- Walton KA, Cole AL, Yeh M, Subbanagounder G, Krutzik SR, Modlin RL, et al. Specific phospholipid oxidation products inhibit ligand activation of toll-like receptors 4 and 2. Arterioscler Thromb Vasc Biol. 2003;23:1197–1203. doi: 10.1161/01.ATV.0000079340.80744.B8. [DOI] [PubMed] [Google Scholar]
- Wort SJ, Evans TW. The role of the endothelium in modulating vascular control in sepsis and related conditions. Br Med Bull. 1999;55:30–48. doi: 10.1258/0007142991902286. [DOI] [PubMed] [Google Scholar]
- Xia M, Ling W, Zhu H, Wang Q, Ma J, Hou M, et al. Anthocyanin prevents CD40-activated proinflammatory signaling in endothelial cells by regulating cholesterol distribution. Arterioscler Thromb Vasc Biol. 2007;27:519–524. doi: 10.1161/01.ATV.0000254672.04573.2d. [DOI] [PubMed] [Google Scholar]
- Youn HS, Lee JY, Fitzgerald KA, Young HA, Akira S, Hwang DH. Specific inhibition of MyD88-independent signaling pathways of TLR3 and TLR4 by resveratrol: molecular targets are TBK1 and RIP1 in TRIF complex. J Immunol. 2005;175:3339–3346. doi: 10.4049/jimmunol.175.5.3339. [DOI] [PubMed] [Google Scholar]
- Zhang FX, Kirschning CJ, Mancinelli R, Xu XP, Jin Y, Faure E, et al. Bacterial lipopolysaccharide activates nuclear factor-kappaB through interleukin-1 signaling mediators in cultured human dermal endothelial cells and mononuclear phagocytes. J Biol Chem. 1999;274:7611–7614. doi: 10.1074/jbc.274.12.7611. [DOI] [PubMed] [Google Scholar]