

Abstract
BACKGROUND AND PURPOSE
By controlling intracellular cyclic nucleotide levels, phosphodiesterases (PDE) serve important functions within various signalling pathways. The PDE2 and PDE5 families are allosterically activated by their substrate cGMP via regulatory so-called GAF domains. Here, we set out to identify synthetic ligands for the GAF domains of PDE2 and PDE5.
EXPERIMENTAL APPROACH
Using fluorophore-tagged, isolated GAF domains of PDE2 and PDE5, promising cGMP analogues were selected. Subsequently, the effects of these analogues on the enzymatic activity of PDE2 and PDE5 were analysed.
KEY RESULTS
The PDE2 ligands identified, 5,6-DM-cBIMP and 5,6-DCl-cBIMP, caused pronounced, up to 40-fold increases of the cAMP- and cGMP-hydrolysing activities of PDE2. The ligand for the GAF domains of PDE5, 8-Br-cGMP, elicited a 20-fold GAF-dependent activation and moreover revealed a time-dependent increase in PDE5 activity that occurred independently of a GAF ligand. Although GAF-dependent PDE5 activation was fast at high ligand concentrations, it was slow at physiologically relevant cGMP concentrations; PDE5 reached its final catalytic rates at 1 µM cGMP after approximately 10 min.
CONCLUSIONS AND IMPLICATIONS
We conclude that the delayed activation of PDE5 is required to shape biphasic, spike-like cGMP signals. Phosphorylation of PDE5 further enhances activity and conserves PDE5 activation, thereby enabling PDE5 to act as a molecular memory balancing cGMP responses to nitric oxide or natriuretic peptide signals.
Keywords: phosphodiesterases, cyclic guanosine monophosphate, cyclic nucleotide analogues, GAF domains, phosphodiesterase 2, phosphodiesterase 5
Introduction
The second messenger molecules cAMP and cGMP regulate a variety of physiological processes, particularly in the cardiovascular and nervous systems. The intracellular levels of cAMP and cGMP in a tissue depend on the activity of the cyclic nucleotide-forming adenylyl and guanylyl cyclases but are also profoundly determined by the action of the cyclic nucleotide-degrading phosphodiesterases present.
In mammals, 11 families of PDEs exist that either degrade cGMP, cAMP or both and are characterized by different regulatory properties (Bender and Beavo, 2006). With the exception of the photoreceptor PDE6, all the phosphodiesterases are homodimers with C-terminal conserved catalytic domains and N-terminal regions containing different regulatory modules. Five of the 11 PDE families (PDE2, 5, 10, 11 and the photoreceptor PDE6) contain a tandem of GAF domains in their N-terminal region designated GAF-A and GAF-B respectively. GAF domains are small molecule-binding motifs identified by sequence homology in more than 7400 proteins mostly from lower organisms (Aravind and Ponting, 1997). In most cases, their ligands are unknown. The acronym GAF originates from the proteins in which these domains were first identified (cGMP-specific and cGMP-stimulated phosphodiesterases, Anabaenaadenylyl cyclases, Escherichia coliFhlA). In mammals, PDEs are the only proteins containing GAF domains. PDE2 and PDE5 are stimulated by cGMP binding to the GAF domains (Beavo et al., 1971; Russell et al., 1973; Moss et al., 1977; Corbin et al., 2003; Mullershausen et al., 2003; Rybalkin et al., 2003). The GAF domains of PDEs 10 and 11 have been reported to mediate stimulation of the catalytic domain of Anabaena adenylyl cyclases by cAMP and cGMP, respectively, in chimeric proteins (Gross-Langenhoff et al., 2006).
Apparently, the cyclic nucleotide only binds to one of the tandem GAF domains, for example, to GAF-B in PDE2A and in GAF-A in PDE5A (Martinez et al., 2002; Rybalkin et al., 2003; Zoraghi et al., 2005). Whereas stimulation of the dual substrate PDE2 by cGMP was reported as long ago as the 1970s (Beavo et al., 1971; Russell et al., 1973; Moss et al., 1977; for review see Manganiello et al., 1990), cGMP activation of the cGMP-specific PDE5 was described only a few years ago (Corbin et al., 2003; Mullershausen et al., 2003; Rybalkin et al., 2003). Unexpectedly, the kinetics of GAF domain-mediated activation differ considerably: activation of PDE2 was fast while PDE5 activation was reported to occur after a lag phase of several minutes (Rybalkin et al., 2003). In the cGMP-bound state, PDE5 is phosphorylated at serine-102 by the cGMP- and cAMP-dependent protein kinases in vivo and vitro, and this stabilizes cGMP binding (Wyatt et al., 1998; Corbin et al., 2000; Francis et al., 2002; Rybalkin et al., 2002). As PDE5 degrades cGMP and is activated by cGMP, the GAF-mediated activation is difficult to monitor precisely. Therefore, specific ligands for the GAF domains are highly desirable and would be valuable tools for a better understanding of GAF-mediated PDE regulation.
Here, we report on a screen for GAF domain ligands using fluorophore-tagged GAF domains derived from PDE2 and PDE5. Whereas agonists specific for PDE2 or PDE5 GAF domains were identified, of the nucleotide analogues tested no GAF domain antagonists were discovered. By use of the ligand specific for PDE5, a GAF domain-dependent activation was differentiated from an additional time-dependent increase in activity together causing unusual enzyme kinetics.
Methods
Expression of PDE2 and 5 and of fluorescence resonance energy transfer constructs of their GAF domains
The 3′,5′-cyclic nucleotide phosphodiesterases (E.C.3.1.4.17; Alexander et al., 2009) human PDE5A (NM_001083.2) and murine PDE2A1 (NR_026574) were amplified from human or mouse cDNA libraries and subcloned into pcDNA3 (Invitrogen). DNA encoding the catalytic domain of PDE5 (S531..N880) was PCR-amplified and subcloned into pcDNA3.
PCR-amplified DNA encoding the GAF domains of human PDE5 (V93-A515) was subcloned into pcDNA3 containing the coding regions of CFP and YFP as described previously (Russwurm et al., 2007). PCR-amplified DNA encoding the GAF domains of murine PDE2A1 (T199F200..S540H541) was subcloned into the same vector using an EcoRI site located between the BsmBI sites. In the resulting protein, the C-terminal end of CFP (. . . T226A227A228) is consecutively followed by a linker (GDGIH), the tandem GAF domains (see above), a second linker (RIRL) and YFP (MVS . . .).
HEK 293 cells were grown in 75 cm2 flasks as described previously (Russwurm et al., 2009) and transfected with 8 µg of the respective plasmid and 24 µL FuGene6 (Roche) according to the instructions of the manufacturer. Cells were harvested 48–72 h post transfection, lysed in 500 µL of 50 mM NaCl, 50 mM triethanolamine/HCl, pH 7.4 containing 2 mM D,L-dithiothreitol, 1 mM EDTA and protease inhibitor cocktail (mammalian, Sigma-Aldrich) and a cytosolic fraction was obtained by centrifugation (100 000×g, 40 min, 4°C). Protein concentrations were determined using the Bradford Protein Assay (Bio-Rad).
Analysis of isolated GAF domains by fluorescence resonance energy transfer measurements
Samples, 5 µL, of the cytosolic fractions containing the isolated GAF domains as fluorescence resonance energy transfer (FRET) constructs, obtained as described above, were analysed in a total volume of 100 µL of buffer (25 mM triethanolamine/HCl, pH 7.4 containing 2 mM D,L-dithiothreitol and 10 mM MgCl2) using a Cary Eclipse spectrofluorometer with a microplate accessory (Varian) and white half area 96 well microplates (Greiner). Nucleotides were added at the concentrations indicated and emissions were recorded for 30 min [excitation 436 nm, emissions 475 nm (CFP) and 525 nm (YFP), 5 nm excitation and emission slits]. To identify antagonists, a half-maximally effective concentration of cGMP (0.1 µM) was added subsequently and emission changes were again recorded for 30 min. Background values were obtained from a water-containing well and subtracted.
PDE activity measurements
The cGMP- and cAMP-hydrolysing activities were determined essentially as described by Friebe et al. (1998). In short, PDE-containing cytosolic fractions were incubated for 5 min at 37°C in a total volume of 100 µL of 50 mM triethanolamine/HCl, pH 7.4 containing 0.5 g·L−1 bovine serum albumin, 3 mM D,L-dithiothreitol, 3 mM MgCl2, 32P-cGMP (∼3 kBq) at the concentration indicated, and 1 U calf intestinal alkaline phosphatase (Sigma-Aldrich). Reactions were stopped by addition of 900 µL 30% (v/v) charcoal in 50 mM KH2PO4, pH 2.3 and after centrifugation, formed 32P in the supernatant was determined as Čerenkov radiation in a liquid scintillation counter.
For concentration–response curves, between 4 ng and 0.15 µg of cytosolic protein of PDE2- or PDE5-expressing HEK cells were used; the actual amount used was adjusted to yield a product formation between 2× the value determined in the absence of enzyme and 20% of total substrate used.
To determine the time courses of the activity, PDE-containing cytosolic fractions were pre-warmed for 5 min in a total volume of 500 µL of 50 mM triethanolamine/HCl, pH 7.4, containing 0.5 g·L−1 bovine serum albumin and 3 mM D,L-dithiothreitol and subsequently mixed with 500 µL of substrate [32P-cGMP (∼3 kBq) at the indicated concentration in 50 mM triethanolamine/HCl, pH 7.4, containing 3 mM MgCl2 and 10 U calf intestinal alkaline phosphatase]. At the time points indicated, aliquots were withdrawn, stopped and any product formed was determined as described above. The amounts of enzyme, adjusted as described above, were between 0.2 and 1.2 µg of cytosolic protein. All data are presented as means ± SEM of at least three independent experiments performed in duplicate. To obtain average enzymatic activities (Figures 4 and S2), the GMP formed was determined at the time points indicated by the vertical lines (10, 20, 30, 45, 60, 90, 120 s …). Average PDE activity during the intervals is depicted by the bars and was calculated by dividing the amount of GMP formed during the interval by the length of the interval; for example, to obtain the average PDE activity between 30 and 45 s, GMP already present at the beginning of the interval (30 s) was subtracted from GMP at the end of the interval (45 s) to obtain the additional GMP formed in the interval and subsequently divided by the length of the interval (15 s = 45 s − 30 s).
Figure 4.
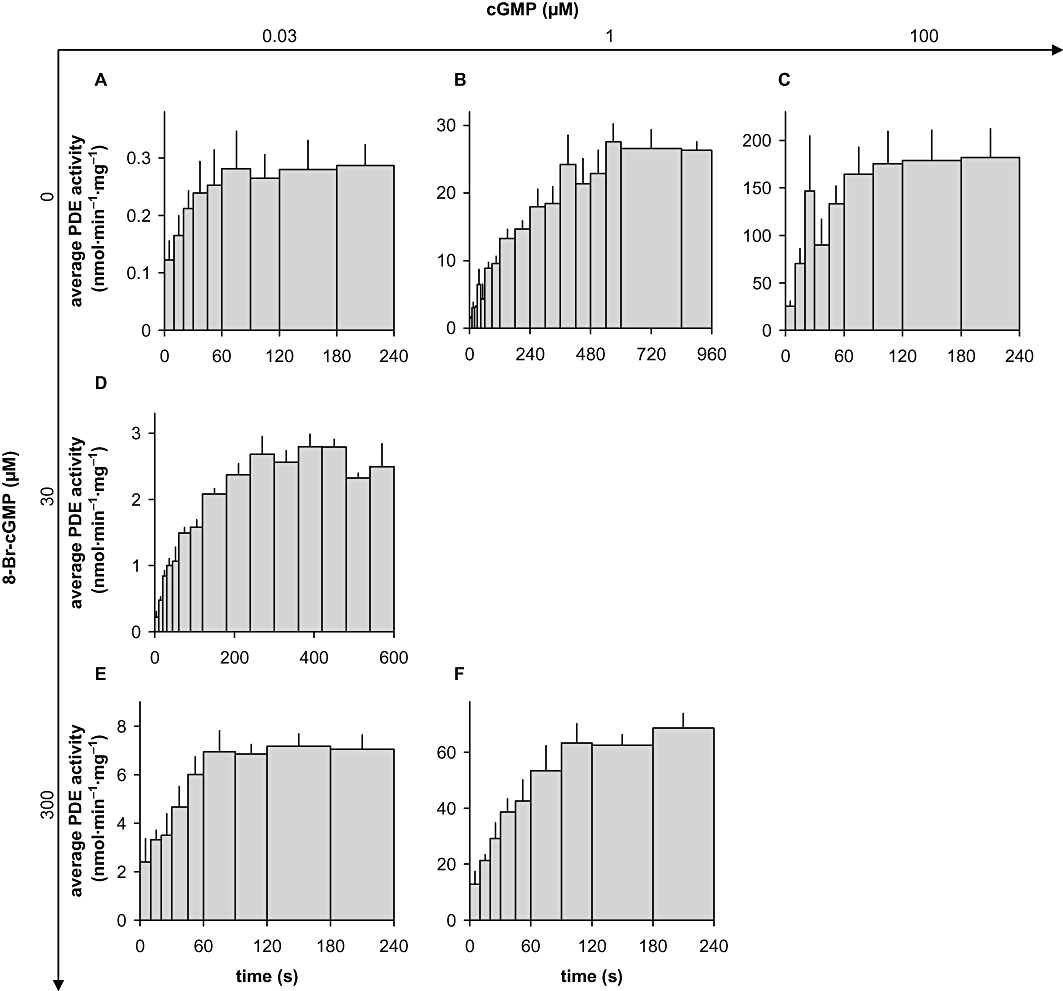
Kinetics of PDE5 activation. Time courses of PDE activation were obtained by measuring GMP accumulation in cytosolic fractions of PDE5 overexpressing HEK cells at 0.03 µM (A, D, E), 1 µM (B, F) or 100 µM (C) cGMP as substrate either in the absence (A, B, C) or in the presence of 30 µM (D) or 300 µM (E, F) 8-Br-cGMP. The reactions were carried out as described in detail in Methods. Average PDE activity in the indicated time intervals was calculated as described in Methods. The width of a bar depicts the length of the respective interval, the height represents the average enzymatic velocity during the interval and the area of the bar is therefore equivalent to GMP formed during the interval. Data shown are mean ± SD of at least three independent experiments performed in duplicate.
For statistical analysis of product accumulation time courses, the integral
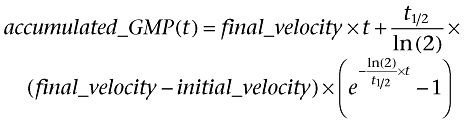 |
of an exponential ‘decay’ (which is actually an increase) of enzymatic velocity from initial_velocity to final_velocity
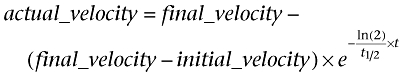 |
was fitted using non-linear regression analysis to the measured GMP accumulation curves to obtain values for half-life and 95% confidence intervals (Frieden, 1979).
Phosphorylation of PDE5
In vitro phosphorylation of 25 µL of PDE5-containing cytosolic fractions (approx. 60 µg of protein) was performed for 10 min at 37°C in a total volume of 100 µL containing 50 mM triethanolamine/HCl, pH 7.4, 0.5 g·L−1 bovine serum albumin, 3 mM D,L-dithiothreitol, 3 mM MgCl2, 500 µM ATP, 300 µM 8-Br-cGMP and catalytic subunit of cAMP-dependent protein kinase (total activity 1.6 nmol·min−1, Jena Bioscience). For control incubations, ATP was omitted and the kinase was replaced by 2 U of calf intestinal alkaline phosphatase to degrade residual ATP. After the phosphorylation experiment, aliquots were saved for Western blots to check successful phosphorylation (Mullershausen et al., 2003) and either cGMP-degrading activity was determined directly or deactivation of the enzyme was monitored after dilution of the sample to reduce the 8-Br-cGMP concentration.
Direct determination of cGMP-degrading activity was performed in 10 s incubations using aliquots corresponding to 0.04–6 µg of cytosolic protein (depending on the substrate concentration, adjusted as described above) without alkaline phosphatase. Subsequently, reactions were stopped by incubating the sample for 5 min at 95°C, and [32P]-GMP formed was degraded to 32P and guanosine by incubating the sample with 2 U alkaline phosphatase for 20 min at 37°C.
Deactivation was monitored after a 1000-fold dilution to reduce the 8-Br-cGMP concentration to 0.3 µM, and PDE activity was determined at the indicated time points in 10 s incubations using a substrate concentration of 0.03 µM [32P]-cGMP, subsequent heating to 95°C and [32P]-GMP degradation as described above. All data presented are means ± SEM of at least three independent experiments performed in duplicate.
Results
Analysis of PDE2 and PDE5 activation induced by GAF domain ligands is hampered by the concomitant degradation of cGMP at the catalytic domains. Therefore, we set out to identify GAF ligands that do not serve as substrate or at least display a higher selectivity for the GAF versus the catalytic domains. To identify these ligands, we used FRET constructs composed of tandem GAF domains of PDE2 and PDE5 sandwiched between two fluorescent proteins, CFP and YFP. The FRET construct derived from the GAF domains of PDE2 was generated according to the crystal structure of the PDE2 GAF domains available (Martinez et al., 2002) and displayed cGMP-dependent FRET changes. The first attempts to generate PDE5 constructs based on the respective regions of PDE5 were unsuccessful, that is, the constructs did not display cGMP-dependent FRET changes. Therefore, the constructs were sequentially elongated, that is, amino acids preceding GAF-A were added at ∼10 amino acid intervals. Interestingly, 51 amino acids had to be added to yield cGMP-dependent FRET changes (Russwurm et al., 2007). Using these constructs, we screened for cGMP analogues eliciting conformational changes similar to cGMP, thereby qualifying as GAF agonists and potential PDE activators. In addition, we searched for analogues that block cGMP activation thereby acting as GAF antagonists and inhibitors of PDE activation.
Screening for GAF ligands of PDE2 and PDE5 with FRET constructs
Figure 1 depicts the cAMP- and cGMP-binding properties of the GAF-derived PDE2 and PDE5 constructs. The PDE5 GAF domains bound cGMP with an EC50 of approximately 0.07 µM and a high specificity, as up to 100 µM cAMP did not elicit any signals (see Figure 1A). The PDE2 GAF domains showed similar high affinity towards cGMP (EC50[cGMP]∼0.04 µM) but a much lower cGMP specificity (EC50[cAMP]∼ 2 µM). The PDE5 and PDE2 FRET constructs were then used to test which of the cyclic nucleotide analogues (100 µM) shown in Table 1 were able to elicit FRET changes indicating agonistic properties (full chemical names and structures of the analogues are listed in Table S1 and Figure S1). Subsequently, a half-maximally effective cGMP concentration was added to reveal possible antagonistic activity (0.1 µM). However, none of the substances tested (100 µM) prevented the FRET changes induced by the half-maximally active cGMP concentration indicating that none of the analogues possessed antagonistic properties.
Figure 1.
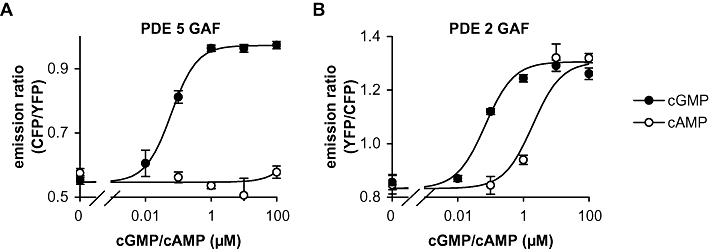
Cyclic nucleotide binding properties of the GAF domains as fluorescence resonance energy transfer (FRET) constructs. Changes in the emission ratio of PDE5 (A) and PDE2 (B) GAF domain FRET constructs induced by cGMP and cAMP were recorded as described in Methods. Data shown are mean ± SD of three independent experiments performed in duplicate.
Table 1.
GAF binding properties of cGMP analogues
| Analogue | EC50(µM) PDE2-GAF | EC50(µM) PDE5-GAF |
|---|---|---|
| cGMP | 0.042 (0.023–0.075) | 0.065 (0.048–0.090) |
| cAMP | 2.4 (1.0–5.4) | – |
| 2-NH2-cPuMP | 0.71 (0.27–1.87) | 3.5 (2.2–5.7) |
| 8-AET-cGMP | – | – |
| 8-APT-cGMP | – | – |
| 1-NH2-cGMP | 4.9 (2.1–11.1) | >100 |
| 2′-AHC-cGMP | – | – |
| Sp-2′-AHC-cGMPS | – | – |
| 8-Br-cGMP | – | 0.56 (0.42–0.74) |
| Rp-8-Br-cGMPS | – | >100 |
| Sp-8-Br-cGMPS | – | 2.7 (1.4–5.4) |
| 8-pCPT-cGMP | >100 | >100 |
| Rp-8-pCPT-cGMPS | – | – |
| Sp-8-pCPT-cGMPS | – | – |
| Rp-8-pCPT-PET-cGMPS | – | – |
| Sp-8-pCPT-PET-cGMPS | – | – |
| 5,6-DM-cBIMP | 0.10 (0.035–0.28) | – |
| DB-cGMP | 3.8 (1.2–11) | – |
| 5,6-DCl-cBIMP | 0.28 (0.12–0.65) | >100 |
| Sp-5,6-DCl-cBIMPS | 19 (7–50) | – |
| 2′-dcGMP | 3.5 (1.3–9.4) | 1.9 (1.2–3.0) |
| Rp-cGMPS | 6.2 (1.8–21) | 3.3 (2.2–5.2) |
| Sp-cGMPS | >100 | 26 (18–35) |
| cIMP | 0.72 (0.33–1.6) | 0.32 (0.21–0.48) |
| MANT-cGMP | 8.2 (2.6–26) | 4.5 (1.2–17) |
| 2′-O-MS-cGMP | 2.5 (0.9–6.4) | 0.62 (0.41–0.93) |
| 2′-O-MS-TME-cGMP | 0.22 (0.08–0.64) | 0.20 (0.11–0.37) |
| 2′-O-ME-cGMP | 28 (15–53) | 47 (23–95) |
| PET-cGMP | 0.35 (0.13–0.89) | 3.2 (1.1–9.2) |
| 8-Br-PET-cGMP | – | >100 |
| Rp-8-Br-PET-cGMPS | – | – |
| Sp-8-Br-PET-cGMPS | – | – |
| cPuMP | 8.0 (2.7–23.7) | – |
| cXMP | >100 | 70 (38–129) |
The half-maximally effective concentrations (EC50) of different cGMP analogues that elicit conformational changes of the GAF domains of PDE2 and PDE5 are presented. Conformational changes were assessed by in vitro recording of fluorescence resonance energy transfer (FRET) between two fluorescent proteins fused to the GAF domains. EC50 values given were calculated from at least three independent determinations performed in duplicate; 95% confidence intervals of the EC50 values are given in parentheses. –, no FRET change detectable at a concentration of 100 µM of the analogue. Full chemical names and structures of the analogues are listed in Table S1 and Figure S1.
Among the agonists investigated, six exhibited a reasonable affinity (<1 µM) for PDE2 and four for PDE5 (see Table 1). Of these, two were specific for PDE2 GAF domains (5,6-DM-cBIMP and 5,6-DCl-cBIMP) and only one displayed specificity for PDE5 GAF domains (8-Br-cGMP).
Activation of PDE2 by the GAF ligands 5,6-DM-cBIMP and 5,6-DCl-cBIMP
Next, the agonistic properties of the GAF ligands identified were studied with the respective holoenzymes. Both PDE2 and PDE5 were recombinantly expressed in HEK cells. Instead of purified enzymes, freshly prepared cytosolic fractions were used to study GAF-dependent PDE activation, because cGMP stimulation of PDE5 has only been demonstrated in freshly prepared crude preparations and is lost during storage or purification (Rybalkin et al., 2003). First, 5,6-DM-cBIMP and 5,6-DCl-cBIMP were chosen as the most promising agonists for PDE2 as they displayed the highest specificity for PDE2 compared with PDE5. PDE2 hydrolyses both nucleotides, cAMP and cGMP, with comparable Km and Vmax (Manganiello et al., 1990; Bender and Beavo, 2006). The concentration-dependent activation of PDE2 by 5,6-DM-cBIMP and 5,6-DCl-cBIMP at different cAMP concentrations is shown in Figure 2 and for comparison, activation of cAMP-hydrolysing activity of PDE2 by cGMP is also shown (Figure 2C). At substrate concentrations below 10 µM cAMP, all agonists caused an approximately 40-fold activation of PDE2 with a comparable EC50 of 1 µM. cGMP-induced activation displayed a biphasic behaviour with a decline at higher concentrations reflecting competition of cGMP with the substrate cAMP at the catalytic domain. This competition did not occur with 5,6-DCl-cBIMP and 5,6-DM-cBIMP indicating a much lower affinity of these compounds for the catalytic domain. The latter finding allowed us to study GAF domain-induced activation of cGMP-hydrolysing activity of PDE2 (see Figure 2D,E). Both agonists induced an approximately 30-fold activation of cGMP hydrolysis at low cGMP concentrations (0.01 µM), which declined at higher substrate concentrations (10-fold at 0.1 µM cGMP; 1.6-fold at 1 µM cGMP) at which the substrate cGMP caused GAF-mediated activation by itself. The other PDE2 GAF ligands identified in the FRET experiments (four analogues with an EC50 < 1 µM; most of the analogues with an EC50 < 10 µM) displayed lower stimulation of PDE2 activity than 5,6-DM-cBIMP and 5,6-DCl-cBIMP.
Figure 2.
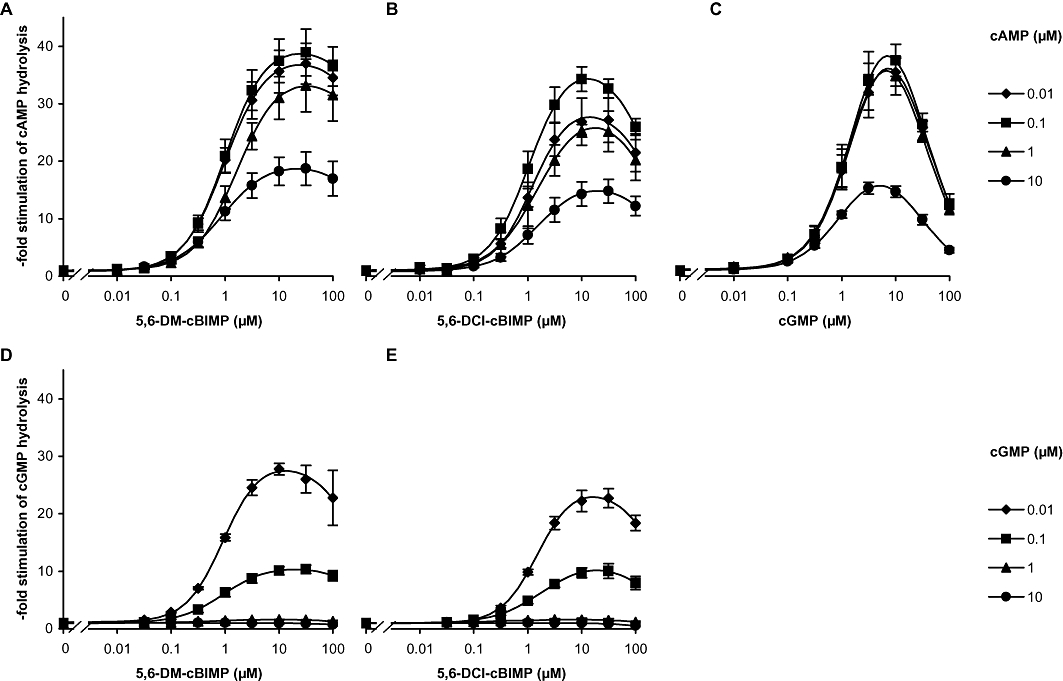
Stimulation of PDE2 by the GAF domain ligands 5,6-DM-cBIMP and 5,6-DCI-cBIMP. PDE activity was measured in cytosolic fractions of PDE2 overexpressing HEK cells in the presence of increasing concentrations of 5,6-DM-cBIMP (A, D), 5,6-DCI-cBIMP (B, E) or cGMP (C) at the indicated concentrations of cAMP (A–C) and cGMP (D–E) as substrate. The reactions were carried out in 5 min incubations as described in detail in Methods. Data shown are mean ± SD of at least three independent experiments performed in duplicate.
Activation of PDE5 by the GAF ligand 8-Br-cGMP
Next, the effects of Sp-8-Br-cGMPS and 8-Br-cGMP on PDE5 were studied. At a low substrate concentration (0.1 µM), Sp-8-Br-cGMPS did not cause a pronounced activation of PDE5 at concentrations up to 100 µM (1.6-fold) indicating insufficient specificity for GAF domains (data not shown). In contrast, as shown in Figure 3, 8-Br-cGMP caused a 20-fold activation at 0.03 µM cGMP, which declined at higher cGMP concentrations (17-fold at 0.1 µM cGMP, sixfold at 1 µM cGMP and no activation at 10 µM cGMP). As 8-Br-cGMP is an effective activator of the cyclic GMP-dependent protein kinases I and II, the PDE5-stimulating properties of almost all other analogues that bound to the FRET construct with an EC50 < 10 µM were determined. However, even though these cGMP analogues elicited conformational changes comparable to cGMP in the FRET constructs, none of the tested compounds caused any relevant PDE5 activation. The lack of GAF domain-induced activation can be attributed to competition of the analogues with the substrate turnover at the catalytic domain, indicating a similar affinity ratio of the analogues and cGMP to the GAF and catalytic domains respectively.
Figure 3.
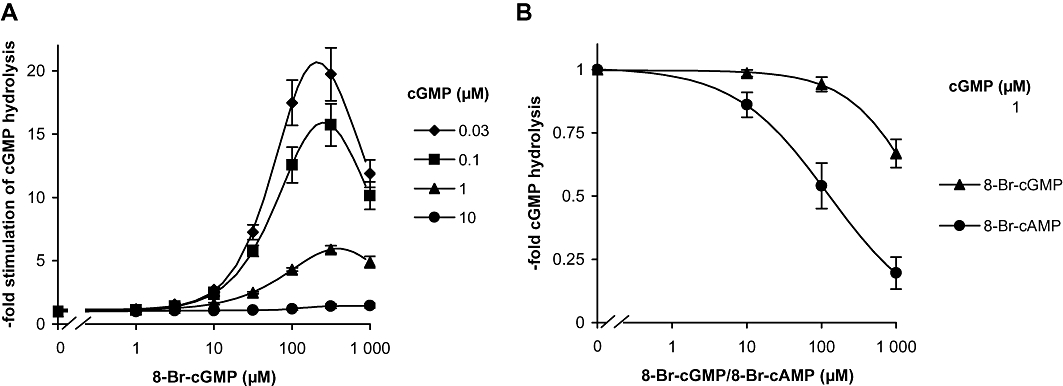
Stimulation of PDE5 by the GAF domain ligand 8-Br-cGMP. (A) PDE5 activity was measured in cytosolic fractions of PDE5 overexpressing HEK cells in the presence of increasing concentrations of 8-Br-cGMP at the indicated concentrations of cGMP as substrate. (B) Activity of a PDE5 construct containing only the catalytic domain was measured at 1 µM cGMP in the presence of increasing concentrations of 8-Br-cGMP or 8-Br-cAMP. The reactions were carried out for 5 min as described in detail in Methods. Data shown are mean ± SD of at least three independent experiments performed in duplicate.
A construct containing only the catalytic domain for PDE5 was used to confirm that inhibition by 8-Br-cGMP observed at the highest concentration (1000 µM) was caused by competition at the catalytic site (Figure 3B, upper curve). As in the holoenzyme, the highest concentration of 8-Br-cGMP (1000 µM) inhibited the catalytic domain construct. For comparison, inhibition of the catalytic domain by 8-Br-cAMP was also determined and was found to be more marked than that by 8-Br-cGMP as described previously (Poppe et al., 2008).
Time course of PDE5 activation
While recording the concentration–response curves for 8-Br-cGMP, we got the impression that cGMP degradation was not linear over time. Therefore, we monitored the time course of PDE5 activation by assessing GMP accumulation at 10 s intervals with low, medium and high cGMP concentrations (0.03, 1, 100 µM; Figure 4A–C; results with additional concentrations shown in Figure S2). At all substrate concentrations, a time-dependent increase in PDE activity was observed. Even at a substrate concentration of 0.03 µM cGMP, at which the GAF domains of PDE5 can be considered ligand-free, maximal activity was not observed until 120 s. The values for the half-life of the activity increase were derived from the product accumulation curves (Figures 5 and S3). Because the scatter was predominantly due to different PDE5 preparations, the curves had to be normalized to the 240 s values for the purpose of statistical analysis. As a control for our assay conditions, PDE2 was measured for comparison. As expected, PDE2 was fully active at the first time point measured (10 s), which shows that the slow time-dependent increase in PDE5 activity was not caused by the experimental conditions (Figure 6A). Moreover, PDE activity in platelet cytosol, which can be attributed almost exclusively to PDE5 under the conditions applied (data not shown), showed a comparable time-dependent increase in activity when measured under identical assay conditions (Figure 6B,C). The activity increase observed is therefore a general feature of PDE5 and not limited to the recombinant enzyme.
Figure 5.
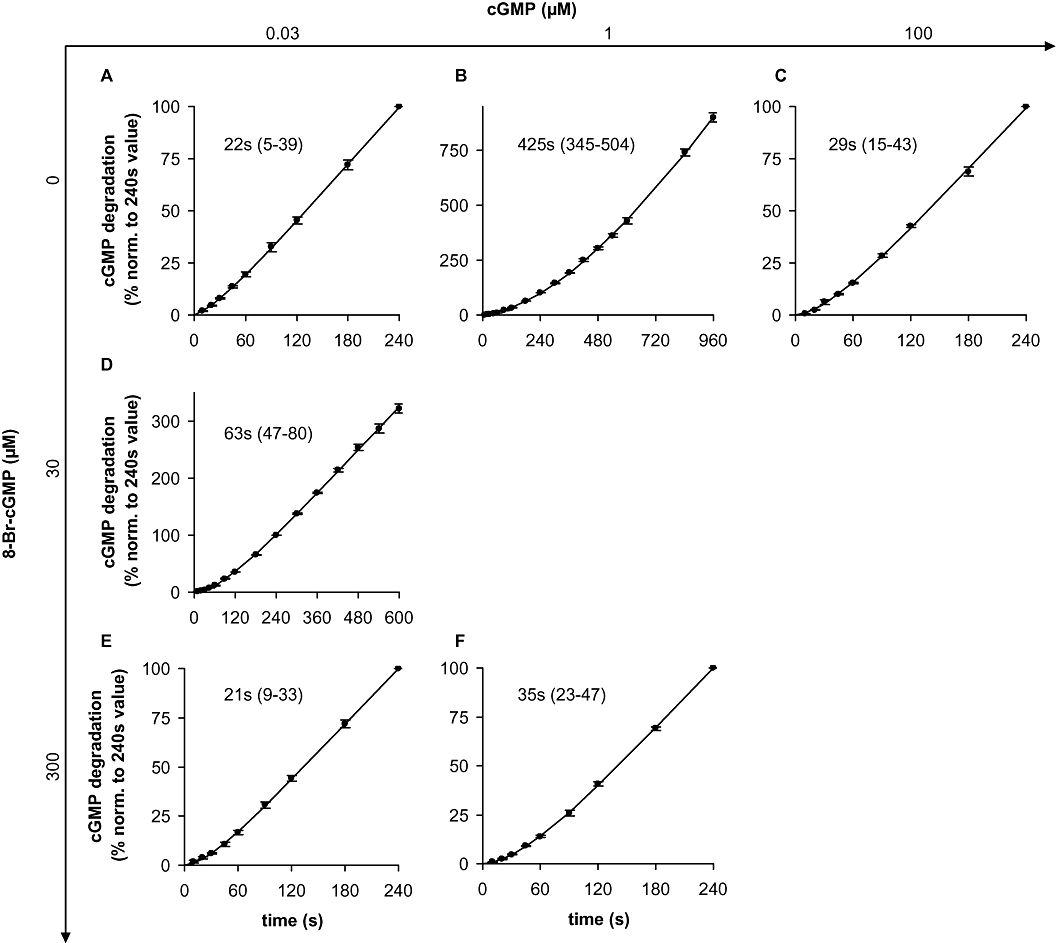
PDE5 activation kinetics: GMP accumulation and statistical analysis. GMP accumulation was measured at the indicated time points in cytosolic fractions of PDE5 overexpressing HEK cells at 0.03 µM (A, D, E), 1 µM (B, F) or 100 µM (C) cGMP as substrate either in the absence (A, B, C) or in the presence of 30 µM (D) or 300 µM (E, F) 8-Br-cGMP. The reactions were carried out as described in detail in Methods. Data are normalized to the GMP value at 240 s to reduce scatter caused by different PDE5 expression levels. Data shown are mean ± SD of at least three independent experiments performed in duplicate. Values for half-life and 95% confidence intervals (in parentheses) were calculated by non-linear regression as described in Methods.
Figure 6.
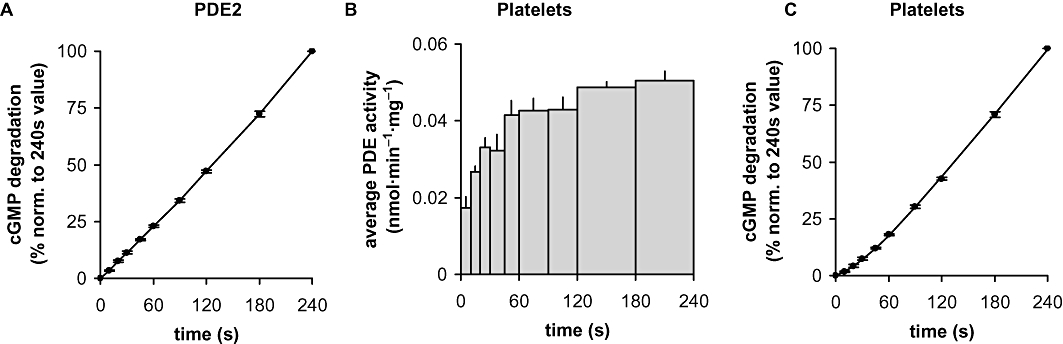
(A) cGMP degradation by PDE2 was measured at the indicated time points in cytosolic fractions of PDE2 overexpressing HEK cells at 1 µM cGMP as substrate. Data were normalized to the GMP value at 240 s. (B) Time course of PDE5 activation was determined in human platelet cytosolic fractions at a substrate concentration of 0.03 µM cGMP as described in Methods and the legend to Figure 4. (C) Statistical analysis of the GMP accumulation determined in platelet cytosolic fractions. Data were normalized to the GMP value at 240 s. Values for half-life and 95% confidence interval (in parentheses) were calculated by non-linear regression as described in Methods. Data shown are mean ± SD of at least three independent experiments performed in duplicate.
Next, we analysed activation of PDE5 at the same substrate concentration (0.03 µM) in the presence of a maximally activating concentration of the GAF ligand 8-Br-cGMP (Figure 4E). Under these conditions, the enzyme showed a 20-fold activation already at the first point measured (10 s) indicating that at a high ligand concentration, GAF-induced activation is fast. However, also in these measurements, an activity increase (threefold) over time also occurred (120 s). At an intermediate ligand concentration (30 µM 8-Br-cGMP, Figure 4D), GAF-induced activation at 10 s was negligible, but 6 min were required to reach final velocity (10-fold activation).
Subsequently, we studied cGMP as a GAF ligand. At a very high cGMP concentration (100 µM cGMP), the GAF domains should be occupied immediately. Accordingly, PDE5 activity at the first time point was 200-fold higher than with 0.03 µM cGMP. The factor 200 is explained by a 10-fold km effect due to the higher substrate concentration and a 20-fold GAF-dependent activation of PDE5. The GAF-dependent activation of PDE5 by cGMP occurs as fast as the one observed with the high 8-Br-cGMP concentration (see Figure 4E), but as in the experiments with a low substrate concentration, a time-dependent increase in activity (sevenfold) within 120 s occurred with 100 µM cGMP. At an intermediate substrate concentration of 1 µM cGMP, a slow (600 s), approximately 20-fold PDE5 activation took place, which is reminiscent of that at the half-maximally active 8-Br-cGMP concentration (30 µM). The 1 µM substrate concentration caused a half-maximal stimulation of the enzyme as a twice as high activity (60 nmol·min−1·mg−1) was obtained in the presence of a maximally activating 8-Br-cGMP concentration (Figure 4F). Under these conditions, GAF-dependent activation was fast and the activity increase took only 120 s. To sum up, in addition to the GAF-dependent activation, we observed a time-dependent increase in PDE5 activity. This time-dependent activity increase occurred within approximately 120 s and was independent of the GAF ligand, whereas the GAF-dependent activation was fast at high ligand concentrations and took minutes at cGMP concentrations around the EC50.
Phosphorylation of PDE5
Cyclic GMP binding to the GAF domain of PDE5 has been reported to be stabilized by phosphorylation at serine-102. To analyse the impact of PDE5 phosphorylation, we performed experiments with phosphorylated PDE5 and used antibodies that specifically recognize the phosphorylated enzyme to check phosphorylation. Our attempt to phosphorylate ligand-free PDE5 was unsuccessful; we were unable to detect any phosphorylation without the substrate or a GAF domain ligand (data not shown) confirming that cGMP bound to the GAF domain is a prerequisite for phosphorylation, as found previously (Turko et al., 1998). Hence, we incubated recombinantly expressed PDE5 in the presence of 8-Br-cGMP with and without the cAMP-dependent kinase to compare activity of the phosphorylated versus the non-phosphorylated PDE5. After incubation, measurement of PDE activity at various substrate concentrations (0.03, 0.1, 1, 10 and 100 µM) showed an approximately 35% higher activity of the phosphorylated species indicative of an increase in Vmax (Figure 7). Phosphorylation has been reported to have an important impact on the dissociation of the GAF ligand. Therefore, subsequent to phosphorylation induced by incubation with kinase and 8-Br-cGMP, samples were diluted to decrease the concentration of the free GAF ligand. Dissociation of the GAF ligand was then monitored by measuring the activity at a low concentration of cGMP (0.03 µM) in 10 s incubations (Figure 7C). With a half-life of 3.4 versus 1.5 min, deactivation of phospho-PDE5 was clearly slower than that of the non-phosphorylated enzyme. We concluded that phosphorylation enhances cGMP-induced PDE5 activation by increasing the activity and slowing down the dissociation of the GAF ligand.
Figure 7.
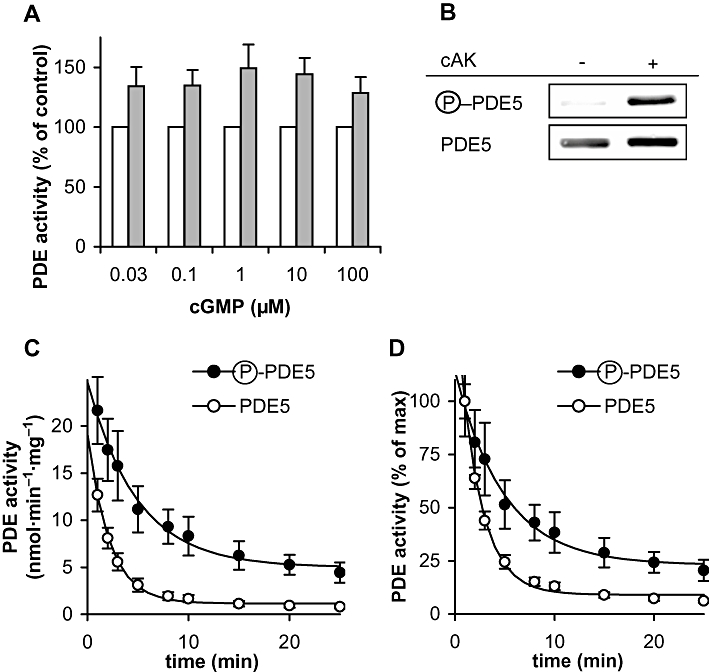
Phosphorylated PDE5 shows higher catalytic rates and a slower deactivation. PDE5 was phosphorylated as described in detail in the Methods. (A) Comparison of activity of phosphorylated versus non-phosphorylated PDE5 at the indicated concentrations of cGMP as substrate. The reactions were carried out as described in detail in Methods. P < 0.05 in paired t-test for phospho- versus non-phospho-PDE5 at all substrate concentrations. (B) Aliquots were analysed in Western blots with antibodies specifically detecting phosphorylated PDE5 and antibodies against PDE5 to check loading. The blot shown is representative of four performed with similar results (full lanes of the blots are depicted in Figure S4). cAK, cAMP-dependent protein kinase (C) PDE5 deactivation was monitored by measuring PDE activity of phosphorylated versus non-phosphorylated PDE5 in 10 s incubations at the indicated time points after a 1000-fold dilution to reduce the GAF ligand 8-Br-cGMP to concentrations below 0.3 µM. (D) Data from (C) normalized to the 1 min value. Data shown are mean ± SD of at least three independent experiments performed in duplicate.
Discussion
GAF domain ligands for PDE2 and PDE5
To enable a better characterization of the allosteric GAF domain-dependent activation of PDE2 and PDE5, we performed a systematic search for GAF domain ligands among 30 nucleotide analogues using GAF domain-containing FRET constructs. Although our screen with PDE2 and PDE5 GAF domain FRET constructs disappointingly did not yield any antagonists, several agonists for the PDE2 and PDE5 GAF domains were identified.
5,6-DM-cBIMP and 5,6-DCl-cBIMP were relatively specific for PDE2 compared with PDE5. Both analogues have been suggested before to act as PDE2 activators (Genieser et al., 1992), cBIMP has been shown to activate PDE from rat liver or bovine adrenal gland twofold (Erneux et al., 1981; Miot et al., 1985). In our study, these analogues caused an almost 40-fold activation of PDE2-catalysed cAMP hydrolysis, which is similar to the activation by cGMP reported here and by others (Yamamoto et al., 1983). Moreover, the analogues allowed us to demonstrate a pronounced GAF-mediated stimulation of PDE2's cGMP-degrading activity (30-fold) at low cGMP concentrations, which is sixfold higher than the reported stimulation by cAMP (Yamamoto et al., 1983). The EC50 for the cGMP-induced activation determined in this study, 1 µM, is in good accordance with the apparent Kd of 2 µM observed by Stroop and Beavo (1991). 5,6-DM-cBIMP and 5,6-DCl-cBIMP competed with substrate turnover only at very high concentrations (see Figure 2), which implies a comparatively low affinity to the catalytic domain. Some metabolism of the GAF ligands at these high concentrations could not be excluded. However, the pronounced stimulation of PDE activity demonstrates a much higher specificity of the analogues to the GAF domain compared with the catalytic domain than cGMP.
For PDE5, two specific GAF ligands (Sp-8-Br-cGMPS, 8-Br-cGMP) were identified with the FRET constructs. Experiments with native PDE revealed that only 8-Br-cGMP can be used as a GAF ligand, because Sp-8-Br-cGMPS even at high concentrations failed to cause relevant PDE5 activation. In earlier studies, 8-Br-cGMP inhibited cGMP binding to PDE5 by only 20%, which led to the conclusion that 8-Br-cGMP does not bind to PDE5 (Francis et al., 1980). However, in our study higher concentrations (up to 300 µM) elicited a substantial stimulation. 8-Br-cGMP caused a 20-fold activation of PDE5 at a very low substrate concentration of cGMP (0.03 µM), which declined to 17-fold at 0.1 µM cGMP and sixfold at 1 µM cGMP. At 10 µM cGMP, no 8-Br-cGMP-dependent activation was observed. These results show that even at a substrate concentration of 0.1 µM cGMP, some cGMP is bound to the GAF domains already, at 1 µM cGMP approximately half of GAF domains are saturated, and at 10 µM cGMP the GAF domain is fully saturated. Due to technical reasons, we were unable to measure PDE activity at a substrate concentration below 0.03 µM cGMP, but as 8-Br-cGMP activation of PDE5 at 0.03 is only slightly higher than at 0.1 µM cGMP (20- vs. 17-fold), the GAF domains can be considered to be largely unoccupied at 0.03 µM cGMP. An EC50 of about 1 µM cGMP at PDE5's GAF domains can also be estimated from the kinetic analysis of PDE5 activation (see below), which is in reasonable agreement with the Kd of 0.2 µM observed by Zoraghi et al. (2005).
In a millimolar concentration, 8-Br-cGMP competed with substrate turnover (see Figure 3), remarkably, 8-Br-cGMP inhibited turnover at these concentrations less effectively than 8-Br-cAMP. In an earlier study that used microcalorimetry to directly assess turnover of cyclic nucleotide analogues, degradation of 8-Br-cAMP but not of 8-Br-cGMP by PDE5 was described (Poppe et al., 2008), an unexpected finding that nevertheless is in line with our observations.
It should be noted that the cGMP affinity was a lot higher in the FRET constructs than in the holoenzymes (as judged by activation of PDE activity) in both PDE5 and PDE2, which is in accordance with results from earlier binding studies (Wu et al., 2004; Zoraghi et al., 2005). The discrepancy implies that the catalytic domain alters the properties of the GAF domain-containing regulatory domain, an observation that is also supported by the finding that sildenafil binding to PDE5's catalytic domain has an impact on cGMP binding to the GAF domain (Turko et al., 1999).
Similarly, the half-maximal effective 8-Br-cGMP concentration for PDE5 stimulation was in the range of 30 µM and far higher than the EC50 observed with the FRET construct (0.6 µM). Yet, because this study used isolated FRET-tagged GAF domains only as tools to identify promising cGMP analogues, a thorough investigation of the molecular basis of this discrepancy was not performed. Furthermore, the principle of reciprocity implies that the catalytic domain in the holoenzyme should affect ligand binding to the GAF domains, if the GAF domains can regulate the catalytic turnover.
Direct assessment of the stimulating effect of cGMP binding on PDE5 cGMP hydrolysis inevitably represents a challenge, because cGMP acts as an activator and substrate. Different approaches have been made to demonstrate the consequences of cGMP binding to the GAF domain on the catalytic domain. In one of these studies, the effect of the physiological GAF ligand, cGMP, on binding of the synthetic PDE5 inhibitor sildenafil was examined (Corbin et al., 2003). An increase in sildenafil affinity in the presence of low cGMP concentrations suggested that cGMP binding to the GAF domains induced a conformational change leading to higher sildenafil affinity and presumably activation of the enzyme. Another study (Rybalkin et al., 2003) took advantage of the relatively slow cGMP dissociation and investigated activation of the enzyme after cGMP pre-incubation. In that study, a 10-fold stimulation of PDE5 and a higher potency of sildenafil to inhibit the stimulated enzyme were found. Our approach to employ a reasonably specific, synthetic GAF domain ligand (8-Br-cGMP) to investigate stimulated turnover of the physiological substrate, cGMP, represents the second possible strategy to solve the problem that cGMP acts as activator and substrate and revealed a 20-fold dynamic range of PDE5 activation.
The almost unchanged 8-Br-cGMP EC50 values for stimulation of the PDE5 holoenzyme at different cGMP concentrations raise the question why the EC50 values were not shifted by increasing cGMP concentrations. The finding can be explained in the case that the Kd values at the GAF domains for 8-Br-cGMP and cGMP are in the range of the observed EC50s. Under the assumption of Kd of 30 and 1 µM for 8-Br-cGMP and cGMP, respectively, the Cheng and Prusoff equation (Cheng and Prusoff, 1973) yields 8-Br-cGMP EC50 values of 31, 33 and 60 µM at increasing cGMP concentrations (0.03, 0.1 and 1 µM). The small difference expected would be barely visible on a logarithmic scale, especially as the stimulation factors by 8-Br-cGMP also vary with the cGMP concentration.
PDE5 activation kinetics
In contrast to the fast GAF domain-dependent activation of PDE2, the increase in PDE5 activity was slow. A non-linear PDE5 activity has been described before and attributed to slow binding of cGMP (0.1 and 1 µM) to the GAF domains because blocking cGMP binding to the GAF domains or saturating the GAF domains with cGMP linearized the kinetics (Rybalkin et al., 2002). In contrast, our study performed on a more detailed time scale revealed a time-dependent but GAF ligand-independent increase in PDE5 activity in addition to the GAF ligand-dependent activation.
GAF ligand-dependent PDE5 activation
The GAF ligand-dependent PDE5 activation was fast at high GAF ligand concentrations (300 µM 8-Br-cGMP or 100 µM cGMP) as stimulated PDE5 activity (20-fold) was already observed after 10 s. However, at intermediate GAF ligand concentrations (30 µM 8-Br-cGMP or 1 µM cGMP), the GAF domain-dependent PDE5 activation was much slower and dominated the time course of PDE5 activity described below (600 s). The ligand concentration-dependence of the activation kinetics indicates cGMP binding to the GAF domains as the rate-limiting step.
cGMP binding experiments with the PDE5 holoenzyme and the isolated GAF domains have been published in the past (McAllister-Lucas et al., 1993; Turko et al., 1996; Francis et al., 2002; Zoraghi et al., 2005). In summary, these studies described a slow and a fast cGMP binding site/component on PDE5 that did not correspond to the GAF-A and GAF-B domains, respectively, but were components of cGMP binding to the GAF-A domain. The slow site displayed a dissociation half-life between 72 and 465 min. Together with a cGMP Kd between 0.1 and 1 µM, an association half-life between 10 and 232 min can be derived at a 1 µM cGMP concentration. For the fast site, the observed dissociation half-life between 29 and 42 min would result in an association half-life between 2.6 and 20 min at 1 µM cGMP. The activation half-life of 7 min (see Figure 5B) determined in our study fits fairly well within this range and suggests that the fast cGMP binding site described previously mediates activation of the enzyme.
Time-dependent PDE5 activity increase
Surprisingly, in addition to the GAF ligand-dependent PDE5 activation, we found a time-dependent increase in activity (threefold activity increase within 120 s), which was independent of a GAF domain ligand as it was observed under conditions at which the GAF domains can be considered ligand-free (0.03 µM cGMP) or fully saturated (addition of 300 µM 8-Br-cGMP or 100 µM cGMP). The molecular basis for the time-dependent increase in PDE5 activity is unknown. As the increase occurred at very low and very high substrate concentrations, cGMP binding to the catalytic domains does not appear to be the limiting step. One can only speculate about the existence of at least two conformations of PDE5's catalytic domain with the transition between the lower and higher activity states occurring within 2 min. Untypically slowly responding, so-called hysteretic behaviour has been described for regulatory as well as metabolic enzymes previously (Frieden, 1979; 2008;).
A recent study described a new ‘super-high’ sensitivity state for sildenafil inhibition (Rybalkina et al., 2010) raising the question whether the different sensitivity states are related to the two conformations described above. As the two sensitivity states were observed only in the non-stimulated enzyme whereas the two conformations described in our study were observed at non-stimulating (0.03 µM cGMP) and stimulating cGMP concentrations (100 µM), the sildenafil sensitivity states appear to be distinct from the two activity states.
Recently, a general model for GAF-mediated PDE activation derived from the structure of full length PDE2A was proposed (Pandit et al., 2009). According to this model, a dimer interface is formed between the catalytic domains if the H-loop occludes the catalytic pocket (closed conformation). PDE2 cycles between the closed and open conformations and cGMP binding to GAF-B stabilizes the open conformation by moving the two catalytic domains apart. This allows the H-loop to swing out thereby enabling substrate binding. It is tempting to speculate that cGMP binding in PDE5 induces an even larger conformational reorganization, because cGMP binds to the more distant GAF-A in this enzyme. These changes could therefore require more time, although the time scale of minutes at cGMP concentrations near the EC50 of 1 µM remains nevertheless unexpected. With regard to the proposed model, it can further be speculated that the two conformations of PDE5's catalytic domain with lower and higher activity correspond to different orientations of the H-loop. Structural analyses of the H-loop of unligated PDE5 and inhibitor PDE5 complexes demonstrated the existence of four different conformations of this loop (Wang et al., 2006), all of which differed from the H-loop conformations in other PDEs. Furthermore, in contrast to PDE2, the H-loop in the unligated PDE5 did not occlude the catalytic pocket. Whether the lower and higher activity states correspond to different conformations of the H-loop therefore awaits further structural analyses.
Consequences of PDE5 activation kinetics for intracellular cGMP signals
PDE5-rich cells respond to NO stimulation with a cGMP spike (Mullershausen et al., 2001). A slow response of cGMP-degrading activity to variations in cGMP is a prerequisite for development of such a spike. Extensive mathematical models have been developed to describe the activity profiles of cGMP-forming and cGMP-degrading activities that underlie these cGMP spikes (Mo et al., 2004). In these models, besides the impact of cGMP concentration on PDE5 activity, rate constants for increases in PDE activity were required to explain the observed cGMP peaks. Thus, without a slowly releasing ‘break’ on PDE5 activity, the observed intracellular cGMP signals in cells containing high PDE5 levels would remain inexplicable.
PDE5 phosphorylation
Phosphorylation analysis of PDE5 confirmed the results by others that only the enzyme with a bound GAF ligand can be phosphorylated efficiently by the cAMP- and cGMP-dependent protein kinases (Thomas et al., 1990). In this context, it is interesting to note that only those FRET constructs, which included PDE5's phosphorylation site, were responsive to cGMP. Phosphorylated PDE5 exhibited approximately 35% higher catalytic rates than the non-phosphorylated species at various substrate concentrations indicating a general Vmax effect. In addition to an increased catalytic rate, dissociation of the GAF ligand was significantly slower in the phosphorylated PDE5 compared with the non-phosphorylated enzyme indicating that phosphorylation conserves PDE5 activation (half-life of 3.4 vs. 1.5 min). In earlier studies, a longer half-life has been suggested (Mullershausen et al., 2004). In those studies, dissociation of the GAF ligand was determined by monitoring PDE5 activity at a substrate concentration of 100 nM cGMP, a concentration at which – as shown in the present study – some PDE5 activation already occurs.
The impact of PDE5 phosphorylation on cGMP binding to the isolated regulatory domain of PDE5 has been described before (Francis et al., 2002). In that study, phosphorylation shifted the 50:50 ratio between the high and a low affinity cGMP binding sites (corresponding to the slow and fast sites discussed above, half-life 39 and 265 min respectively) completely to the high affinity component (slow site, half-life 339 min). Albeit deactivation of both, the non-phosphorylated and phosphorylated holoenzymes, was considerably faster in our study, the data are in accordance with PDE5 phosphorylation slowing down deactivation of the enzyme.
In summary, our data show that PDE5 activation under physiological conditions occurs unexpectedly slow, is long-lasting – in the range of minutes – and greatly depends on the cGMP levels under resting conditions in a specific tissue. Although the cGMP-induced activation of PDE2 and PDE5 are both mediated by GAF domains, the activation kinetics are quite diverse and reflect their different biological functions; PDE2, responsible for the cross-talk between cGMP and cAMP has to respond fast to variations in cGMP levels, whereas PDE5 as a molecular memory mediating feedback inhibition within the cGMP cascade has to act in a delayed but sustained manner.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (Ko1157) and the KOFFER (Kommission für Finanzautonomie und Ergänzungsmittel of the Medical Faculty). The authors gratefully acknowledge the technical assistance of Ulla Krabbe, Arkadius Pacha, Erika Mannheim and Caroline Vollmers.
Glossary
Abbreviations
- FRET
fluorescence resonance energy transfer
- GAF
protein domain first identified in cGMP-specific and cGMP-stimulated phosphodiesterases, Anabaenaadenylyl cyclases, Escherichia coliFhlA
- PDE
phosphodiesterase
Conflicts of interest
H.G.G. is CEO and F.S. is head of Research and Design of BIOLOG Life Science Institute, which sells cyclic nucleotide analogues for research purposes.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Structures of the tested analogues.
Figure S2 Kinetics of PDE5 activation.
Figure S3 PDE5 activation kinetics: GMP accumulation and statistical analysis.
Figure S4 Complete lanes of the Western blot shown in Figure 7B.
Table S1 Full chemical names of the tested analogues.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (4th edn.) 2009;158:S1–S254. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Ponting CP. The GAF domain: an evolutionary link between diverse phototransducing proteins. Trends Biochem Sci. 1997;22:458–459. doi: 10.1016/s0968-0004(97)01148-1. [DOI] [PubMed] [Google Scholar]
- Beavo JA, Hardman JG, Sutherland EW. Stimulation of adenosine 3′,5′-monophosphate hydrolysis by guanosine 3′,5′-monophosphate. J Biol Chem. 1971;246:3841–3846. [PubMed] [Google Scholar]
- Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Corbin JD, Turko IV, Beasley A, Francis SH. Phosphorylation of phosphodiesterase-5 by cyclic nucleotide-dependent protein kinase alters its catalytic and allosteric cGMP-binding activities. Eur J Biochem. 2000;267:2760–2767. doi: 10.1046/j.1432-1327.2000.01297.x. [DOI] [PubMed] [Google Scholar]
- Corbin JD, Blount MA, Weeks JL, 2nd, Beasley A, Kuhn KP, Ho YS, et al. [3H]sildenafil binding to phosphodiesterase-5 is specific, kinetically heterogeneous, and stimulated by cGMP. Mol Pharmacol. 2003;63:1364–1372. doi: 10.1124/mol.63.6.1364. [DOI] [PubMed] [Google Scholar]
- Erneux C, Couchie D, Dumont JE, Baraniak J, Stec WJ, Abbad EG, et al. Specificity of cyclic GMP activation of a multi-substrate cyclic nucleotide phosphodiesterase from rat liver. Eur J Biochem. 1981;115:503–510. doi: 10.1111/j.1432-1033.1981.tb06231.x. [DOI] [PubMed] [Google Scholar]
- Francis SH, Lincoln TM, Corbin JD. Characterization of a novel cGMP binding protein from rat lung. J Biol Chem. 1980;255:620–626. [PubMed] [Google Scholar]
- Francis SH, Bessay EP, Kotera J, Grimes KA, Liu L, Thompson WJ, et al. Phosphorylation of isolated human phosphodiesterase-5 regulatory domain induces an apparent conformational change and increases cGMP binding affinity. J Biol Chem. 2002;277:47581–47587. doi: 10.1074/jbc.M206088200. [DOI] [PubMed] [Google Scholar]
- Friebe A, Mullershausen F, Smolenski A, Walter U, Schultz G, Koesling D. YC-1 potentiates nitric oxide- and carbon monoxide-induced cyclic GMP effects in human platelets. Mol Pharmacol. 1998;54:962–967. doi: 10.1124/mol.54.6.962. [DOI] [PubMed] [Google Scholar]
- Frieden C. Slow transitions and hysteretic behavior in enzymes. Annu Rev Biochem. 1979;48:471–489. doi: 10.1146/annurev.bi.48.070179.002351. [DOI] [PubMed] [Google Scholar]
- Frieden C. A lifetime of kinetics. J Biol Chem. 2008;283:19873–19878. doi: 10.1074/jbc.X800003200. [DOI] [PubMed] [Google Scholar]
- Genieser HG, Winkler E, Butt E, Zorn M, Schulz S, Iwitzki F, et al. Derivatives of 1-beta-D-ribofuranosylbenzimidazole 3′,5′-phosphate that mimic the actions of adenosine 3′,5′-phosphate (cAMP) and guanosine 3′,5′-phosphate (cGMP) Carbohydr Res. 1992;234:217–235. doi: 10.1016/0008-6215(92)85050-a. [DOI] [PubMed] [Google Scholar]
- Gross-Langenhoff M, Hofbauer K, Weber J, Schultz A, Schultz JE. cAMP is a ligand for the tandem GAF domain of human phosphodiesterase 10 and cGMP for the tandem GAF domain of phosphodiesterase 11. J Biol Chem. 2006;281:2841–2846. doi: 10.1074/jbc.M511468200. [DOI] [PubMed] [Google Scholar]
- McAllister-Lucas LM, Sonnenburg WK, Kadlecek A, Seger D, Trong HL, Colbran JL, et al. The structure of a bovine lung cGMP-binding, cGMP-specific phosphodiesterase deduced from a cDNA clone. J Biol Chem. 1993;268:22863–22873. [PubMed] [Google Scholar]
- Manganiello VC, Tanaka T, Murashima S. Cyclic GMP-stimulated cyclic nucleotide phosphodiesterases. In: Beavo J, Houslay MD, editors. Cyclic Nucleotide Phosphodiesterases: Structure, Regulation and Drug Action. New York: John Wiley and Sons Ltd; 1990. pp. 61–85. [Google Scholar]
- Martinez SE, Wu AY, Glavas NA, Tang XB, Turley S, Hol WG, et al. The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding. Proc Natl Acad Sci USA. 2002;99:13260–13265. doi: 10.1073/pnas.192374899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miot F, Van Haastert PJ, Erneux C. Specificity of cGMP binding to a purified cGMP-stimulated phosphodiesterase from bovine adrenal tissue. Eur J Biochem. 1985;149:59–65. doi: 10.1111/j.1432-1033.1985.tb08893.x. [DOI] [PubMed] [Google Scholar]
- Mo E, Amin H, Bianco IH, Garthwaite J. Kinetics of a cellular nitric oxide/cGMP/phosphodiesterase-5 pathway. J Biol Chem. 2004;279:26149–26158. doi: 10.1074/jbc.M400916200. [DOI] [PubMed] [Google Scholar]
- Moss J, Manganiello VC, Vaughan M. Substrate and effector specificity of a guanosine 3′:5′-monophosphate phosphodiesterase from rat liver. J Biol Chem. 1977;252:5211–5215. [PubMed] [Google Scholar]
- Mullershausen F, Russwurm M, Thompson WJ, Liu L, Koesling D, Friebe A. Rapid nitric oxide-induced desensitization of the cGMP response is caused by increased activity of phosphodiesterase type 5 paralleled by phosphorylation of the enzyme. J Cell Biol. 2001;155:271–278. doi: 10.1083/jcb.200107001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullershausen F, Friebe A, Feil R, Thompson WJ, Hofmann F, Koesling D. Direct activation of PDE5 by cGMP: long-term effects within NO/cGMP signaling. J Cell Biol. 2003;160:719–727. doi: 10.1083/jcb.200211041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullershausen F, Russwurm M, Koesling D, Friebe A. In vivo reconstitution of the negative feedback in nitric oxide/cGMP signaling: role of phosphodiesterase type 5 phosphorylation. Mol Biol Cell. 2004;15:4023–4030. doi: 10.1091/mbc.E03-12-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit J, Forman MD, Fennell KF, Dillman KS, Menniti FS. Mechanism for the allosteric regulation of phosphodiesterase 2A deduced from the X-ray structure of a near full-length construct. Proc Natl Acad Sci USA. 2009;106:18225–18230. doi: 10.1073/pnas.0907635106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppe H, Rybalkin SD, Rehmann H, Hinds TR, Tang XB, Christensen AE, et al. Cyclic nucleotide analogs as probes of signaling pathways. Nat Methods. 2008;5:277–278. doi: 10.1038/nmeth0408-277. [DOI] [PubMed] [Google Scholar]
- Russell TR, Terasaki WL, Appleman MM. Separate phosphodiesterases for the hydrolysis of cyclic adenosine 3′,5′-monophosphate and cyclic guanosine 3′,5′-monophosphate in rat liver. J Biol Chem. 1973;248:1334–1340. [PubMed] [Google Scholar]
- Russwurm M, Mullershausen F, Friebe A, Jager R, Russwurm C, Koesling D. Design of fluorescence resonance energy transfer (FRET)-based cGMP indicators: a systematic approach. Biochem J. 2007;407:69–77. doi: 10.1042/BJ20070348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russwurm C, Zoidl G, Koesling D, Russwurm M. Dual acylation of PDE2A splice variant 3: targeting to synaptic membranes. J Biol Chem. 2009;284:25782–25790. doi: 10.1074/jbc.M109.017194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybalkin SD, Rybalkina IG, Feil R, Hofmann F, Beavo JA. Regulation of cGMP-specific phosphodiesterase (PDE5) phosphorylation in smooth muscle cells. J Biol Chem. 2002;277:3310–3317. doi: 10.1074/jbc.M106562200. [DOI] [PubMed] [Google Scholar]
- Rybalkin SD, Rybalkina IG, Shimizu-Albergine M, Tang XB, Beavo JA. PDE5 is converted to an activated state upon cGMP binding to the GAF A domain. EMBO J. 2003;22:469–478. doi: 10.1093/emboj/cdg051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybalkina IG, Tang XB, Rybalkin SD. Multiple affinity states of cGMP-specific phosphodiesterase for sildenafil inhibition defined by cGMP-dependent and cGMP-independent mechanisms. Mol Pharmacol. 2010;77:670–677. doi: 10.1124/mol.109.062299. [DOI] [PubMed] [Google Scholar]
- Stroop SD, Beavo JA. Structure and function studies of the cGMP-stimulated phosphodiesterase. J Biol Chem. 1991;266:23802–23809. [PubMed] [Google Scholar]
- Thomas MK, Francis SH, Corbin JD. Substrate- and kinase-directed regulation of phosphorylation of a cGMP-binding phosphodiesterase by cGMP. J Biol Chem. 1990;265:14971–14978. [PubMed] [Google Scholar]
- Turko IV, Haik TL, McAllister-Lucas LM, Burns F, Francis SH, Corbin JD. Identification of key amino acids in a conserved cGMP-binding site of cGMP-binding phosphodiesterases. A putative NKXnD motif for cGMP binding. J Biol Chem. 1996;271:22240–22244. doi: 10.1074/jbc.271.36.22240. [DOI] [PubMed] [Google Scholar]
- Turko IV, Francis SH, Corbin JD. Binding of cGMP to both allosteric sites of cGMP-binding cGMP-specific phosphodiesterase (PDE5) is required for its phosphorylation. Biochem J. 1998;329:505–510. doi: 10.1042/bj3290505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turko IV, Ballard SA, Francis SH, Corbin JD. Inhibition of cyclic GMP-binding cyclic GMP-specific phosphodiesterase (Type 5) by sildenafil and related compounds. Mol Pharmacol. 1999;56:124–130. doi: 10.1124/mol.56.1.124. [DOI] [PubMed] [Google Scholar]
- Wang H, Liu Y, Huai Q, Cai J, Zoraghi R, Francis SH, et al. Multiple conformations of phosphodiesterase-5: implications for enzyme function and drug development. J Biol Chem. 2006;281:21469–21479. doi: 10.1074/jbc.M512527200. [DOI] [PubMed] [Google Scholar]
- Wu AY, Tang XB, Martinez SE, Ikeda K, Beavo JA. Molecular determinants for cyclic nucleotide binding to the regulatory domains of phosphodiesterase 2A. J Biol Chem. 2004;279:37928–37938. doi: 10.1074/jbc.M404287200. [DOI] [PubMed] [Google Scholar]
- Wyatt TA, Naftilan AJ, Francis SH, Corbin JD. ANF elicits phosphorylation of the cGMP phosphodiesterase in vascular smooth muscle cells. Am J Physiol. 1998;274:H448–H455. doi: 10.1152/ajpheart.1998.274.2.H448. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Manganiello VC, Vaughan M. Purification and characterization of cyclic GMP-stimulated cyclic nucleotide phosphodiesterase from calf liver. Effects of divalent cations on activity. J Biol Chem. 1983;258:12526–12533. [PubMed] [Google Scholar]
- Zoraghi R, Bessay EP, Corbin JD, Francis SH. Structural and functional features in human PDE5A1 regulatory domain that provide for allosteric cGMP binding, dimerization, and regulation. J Biol Chem. 2005;280:12051–12063. doi: 10.1074/jbc.M413611200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.