

Abstract
BACKGROUND AND PURPOSE
Although carboplatin is currently used as a therapeutic drug for ovarian, breast, and non-small cell lung cancers, it has serious side effects including renal and cardiac toxicity. Herein, we examined the effect of carboplatin on murine renal tubular cell (RTC) apoptosis both in vivo and in vitro and the underlying molecular mechanisms associated with its activation of the nuclear factor of activated T-lymphocytes-3 (NFAT3).
EXPERIMENTAL APPROACH
Mechanisms of carboplatin-mediated renal apoptosis were examined using NFAT-reporter transgenic mice and RTCs with NFAT3 overexpression or knockdown.
KEY RESULTS
We demonstrated that carboplatin initiated an intrinsic apoptotic pathway of activating caspase-3 and -9, accompanied by a decrease in the ratio of Bcl-XL/Bax and a significant increase in Bcl-XS. Carboplatin increased NFAT activation in NFAT-luciferase reporter transgenic mice, RTCs and cells exogenously overexpressing NFAT3 that exacerbated cell death. Furthermore, the addition of either N-acetylcysteine (NAC, an antioxidant) or NFAT inhibitors, including FK-506 (tacrolimus), cyclosporin A (CsA, a calcineurin inhibitor), and BAPTA-AM (a calcium chelator) successfully reversed carboplatin-mediated cell apoptosis, which was further confirmed using siNFAT3. Additionally, NAC blocked NFAT3 activation by inhibition of NADPH oxidase activation, and ERK/JNK and PKC pathways, resulting in a decrease in cell apoptosis; the therapeutic effect of NAC was verified in vivo.
CONCLUSION AND IMPLICATIONS
The results presented herein show that carboplatin-mediated reactive oxygen species might signal calcineurin and NFAT3 activation in RTCs, whereas NAC and NFAT inhibitors reversed carboplatin-mediated RTC apoptosis, suggesting that oxidative stress-mediated NFAT3 activation is essential for carboplatin-mediated RTC apoptosis.
Keywords: carboplatin, nuclear factor of activated T-lymphocytes, apoptosis, reactive oxygen species, renal tubular cells
Introduction
Nuclear factor of activated T-lymphocytes (NFAT) family proteins contain three functional domains: (i) a Rel-similarity domain (DNA-binding activity and interactions with AP-1); (ii) an NFAT-homology region (intracellular localization); and (iii) a transcriptional activation domain (Masuda et al., 1998). Calcineurin (PP2B; serine-threonine phosphatase) is a calcium/calmodulin-activated enzyme that transmits signals to the nucleus through the dephosphorylation and translocation of the NFAT. Hyperphosphorylated NFAT transcription factors are sequestered in the cytoplasm, but are immediately translocated to the nucleus after calcineurin-mediated dephosphorylation.
Five NFAT genes, nfatc1–4 and nfat5, have been identified, each with distinct temporally and spatially regulated expression patterns (Rao et al., 1997; Bushdid et al., 2003). NFAT 1–4 activate gene transcription by integrating inputs from the calcium/calcineurin and protein kinase C/mitogen-activated protein kinase signalling pathways. NFAT3 is mainly expressed outside the immune system with high levels in the kidneys, lungs, placenta, etc.; it has also been shown to be essential for cardiac development and mitochondrial function (Bushdid et al., 2003). Furthermore, it was previously shown that NFAT activation is mediated by the generation of reactive oxygen species (ROS) by asbestos, nickel subsulphide, and vanadium, and that scavenging of these compound-mediated ROS with N-acetylcysteine (NAC) resulted in inhibition of NFAT activation (Huang et al., 2001a,b; Li et al., 2002).
Carboplatin (cis-diammine-1,1-cyclobutanedicarboxylateplatinum II), a second-generation platinum-containing anti-cancer drug, is currently being used in the clinic against lung, ovarian, head and neck cancers (Pivot et al., 2001; Fujiwara et al., 2003). The antitumour action of carboplatin is mediated by alkylation of DNA followed by the killing of cancerous cells. Carboplatin is more water-soluble and has fewer adverse effects than its analogue, cisplatin, and has equivalent DNA-damaging activity to cisplatin at similar toxic doses (Alberts, 1995). Because carboplatin has fewer toxic adverse effects than cisplatin, increased doses of carboplatin are commonly used in the clinic in order to achieve optimal antitumour effects. The predominant dose-limiting toxicities of carboplatin are bone marrow suppression and ototoxicity caused by free radical oxidative injury to those organs (Husain et al., 2001). An increasing interest in factors regulating cellular accumulation of platinum may be one of the influences controlling the cytotoxicity of platinum derivatives. Recently, the involvement of copper transporters, hCtrl, ATP7A and ATP7B, and organic cation transporters (OCTs) in the kinetics or cytotoxicity of platinum derivatives was reported, although OCTs contribute less to carboplatin transportation than other platinum compounds (Burger et al., 2010). Additionally, it was shown that a cannabinoid-2 (CB2) receptor agonist attenuated the cisplatin-induced inflammatory response, oxidative/nitrosative stress, and the cell death in the kidneys and improved renal function (Mukhopadhyay et al., 2010a), whereas CB1 receptors promoted enhanced inflammation and tissue injury (Mukhopadhyay et al., 2010b). We previously showed that treatment of rat cultured cardiomyocytes with carboplatin caused marked apoptosis, which was inhibited by the ROS scavenger, N-acetylcysteine (Husain et al., 2001; Cheng et al., 2008). However, the molecular mechanisms of carboplatin-mediated renal tubular cell (RTC) apoptosis have not been elucidated.
Calcineurin/NFAT-induced upregulation of the Fas ligand/Fas death pathway was shown to be involved in methamphetamine-induced neuronal apoptosis (Jayanthi et al., 2005). There are two major pathways for caspase activation: (i) the death receptor-mediated pathway (extrinsic pathway) through the death-inducing signalling complex as the activating complex for procaspase-8; and (ii) the mitochondrion-mediated pathway (intrinsic pathway) through apoptosomes as the activating complex for procaspase-9 (Chowdhury et al., 2008). Caspase-3 is the major caspase that amplifies signals from intrinsic or extrinsic pathways to cleave cellular substrates in apoptotic cells (Lakhani et al., 2006).
In this study, we examined whether carboplatin-induced renal tubular cell apoptosis also results from oxidative stress and what the underlying molecular mechanism is in relation to NFAT3 activation both in vivo and in vitro.
Methods
Animals
Animal care and treatment were conducted in conformity with the Principles of Laboratory Animal Care formulated by the National Society for Medical Research. Male NFAT-luciferase reporter transgenic mice (8 weeks old, 25–30 g) from a FVB/N background were used in the study. The transgene containing nine concatamerized high-affinity NFAT binding sites from the interleukin-4 promoter and a minimal promoter was a gift from Dr Jeffery D. Molkentin (Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA) (Wilkins et al., 2004). These mice were fed a regular chow diet and maintained under conventional housing conditions in our animal facility.
Histological and biochemical assessments of mice treated with carboplatin
Carboplatin (Sigma, St. Louis, MO, USA) was dissolved in double-distilled water (ddH2O) just before use. Age-matched wild-type and transgenic mice at 8 weeks old were injected (i.p.) with a single dose of carboplatin (50, 75, or 100 mg·kg−1) for 6 days to determine the optimal working concentration based upon the mouse survival rate, whereas control mice were injected with an equal amount of ddH2O (n = 11 per group). The blood urea nitrogen (BUN) and serum creatinine levels were measured using Fuji Dri-Chem Slides (Fujifilm, Kanagawa-ken, Japan) at the indicated time. Serial paraffin-embedded sections (5 µm) from kidney samples of both the control and carboplatin-treated groups were stained with haematoxylin and eosin (H&E) for histological morphological analysis and frozen sections (10 µm) of those were prepared for terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) assay at day 3 (n = 8). For NAC therapeutic intervention, NAC and carboplatin at a single dose of 500 mg·kg−1 and 75 mg·kg−1, respectively, were sequentially given to mice and NAC was continuously given for the other 2 days. At day 4, mice were killed and samples obtained for the analysis of ROS-mediated lipid peroxidation, cell apoptosis and Western blot analysis of NFAT3 and caspase-3 activation (n = 6).
Immunohistochemistry
Tissue sections were pretreated with 3% H2O2 for 10 min at room temperature to exhaust endogenous peroxidase activities. After incubation in PBS containing 1% BSA and 1% goat serum at 37°C for 30 min, sections were treated with, firstly, anti-NFAT3 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and anti-hydroxy-2-nonenal antibody (HNE) (Abcam Inc., Cambridge, MA, USA) for 30 min at 37°C followed by 3 washes in PBS. Sections were then incubated with horseradish peroxidase – conjugated goat/mouse secondary antibody at 37°C for 30 min. After colour had been developed with 0.1% DAB/0.01% H2O2, sections were postfixed with 2.5% glutaraldehyde in PBS for 30 min, counterstained in Contrast Green Solution for 3 min, mounted, and then observed and photographed with an Olympus BX51 light microscope.
Cell culture
We used rat renal tubular cells (RTCs), NRK-52E, in this study. We purchased rat proximal tubular (NRK-52E) cells from the Bioresource Collection and Research Center (Hsinchu, Taiwan), and cultured them in DMEM supplemented with an antibiotic/antifungal solution and 10% FBS (pH 7.2). They were grown until the monolayer became confluent. The medium for the cultured cells was then changed to serum-free medium, and cells were incubated overnight before the experiment.
Caspase-3 activity assay
Cells were seeded at 2 × 104 cells per well in a 24-well plate, and treated with the indicated concentrations of carboplatin for 20 h. An Apo-ONE™ homogeneous caspase-3 assay was performed following the manufacturer's instructions (Promega) as previously described (Cao et al., 2003). To confirm caspase-9 and -3 activation, similar experiments were performed after 3 h of pretreatment with 10 µM of the respective caspase-9 and -3 inhibitors, Z-LEHD-FMK and Z-DEVD-FMK (R&D Systems, Minneapolis, MN, USA), followed by carboplatin treatment. The assay used a profluorescent-specific caspase-3 substrate, Z-DEVDR110 (rhodamine 110). The luminescence of each sample was measured at 485/527 nm in a Varioskan Flash microplate spectrophotometer with ScanIt software (Thermo Fisher Scientific, Rochester, NY, USA).
Analysis of gene regulation by luciferase assay and gene expression by reverse-transcription polymerase chain reaction (RT-PCR) and Western blot analysis
For the reporter activity assay, cells were seeded in 24-well plates at a density of 1 × 105 cells per well. In brief, cells were transiently transfected with 1.02 µg of plasmid DNA containing 0.02 µg of the Renilla luciferase construct, pRL-TK (Promega, Madison, WI, USA), to control transfection efficiency, and 1 µg of the pGL4.10-NFAT using LipofectAMINE 2000™ (Invitrogen, Carlsbad, CA, USA). Subsequent procedures were as described previously (Lee et al., 2010).
The method to obtain total RNA for the RT-PCR analysis was as described previously (Pang et al., 2008) with minor modifications. Sequences of the primer pairs for amplification of each gene were 5′-TCTTCAGGACCTCTGCCCTA-3′ and 5′-AGCCTAGGAGCTTGACCACA -3′ for the NFAT3 gene (180 bp); and 5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′ for the GAPDH gene (451 bp). Total RNA, at 5 µg, of extracts from RTCs was used. The level of the housekeeping gene, GAPDH, was analysed and used to demonstrate the presence of the same amount of total complementary (c)DNA in each RNA sample.
Antibodies for NFAT3, caspase-8, Bcl-XL, BAX, lamin A/C, total and phosphorylated MAPKs, and PKC (Santa Cruz Biotechnology, Santa Cruz, CA, USA), caspase-9 (Cell Signaling, Beverly, MA, USA), cleaved caspase-3 (Cayman Chemical, Ann Arbor, MI, USA), GAPDH (Ab Frontier, Seoul, Korea), were included in the assay. RTCs in 10-cm2 dishes were harvested after treatment with 200 µM of carboplatin for the indicated time points and partitioned into cytosol and nuclear fractions using NE-PERTM nuclear extraction reagents (Pierce, Rockford, IL, USA) with the addition of protease inhibitors according to the manufacturer's instructions. The indicated cellular fractions (50 µg) were electrophoresed on a 10% sodium dodecylsulphate (SDS)-polyacrylamide gel and then transblotted onto a Hybond-P membrane (GE Healthcare, Hong Kong, China). Subsequent procedures were as described previously (Lin et al., 2008).
Establishment of NFAT3-Flag and HO-1 transfectants, NFAT3 and Bax small interfering (si)RNA preparation, and transient transfection
Human NFAT3 (pCMV-SPORT6-NFAT3) was purchased from Invitrogen and subcloned into pcDNA3.1 Flag using the BamHI/NotI cutting sites. Sequences of the primer pairs for amplification of NFAT3 were 5′-TTGGATCCGCCGCCGCCATGGGGGCGGCCAGCTGCGAGGAT-3′ and 5′-ATTTGCGGCCGCAAGGCAGGAGGCTCTTC-3′. Human NFAT3 cDNA containing the entire coding sequence was inserted into the BamHI-NotI site of the pcDNA-Flag vector. To obtain Bcl-XL cDNA, total RNA was prepared from cultures of primary rat aortic smooth muscle cells by directly lysing cells in TRIzol buffer (Life Technologies, Gaithersburg, MD, USA), and mRNAs were reversed-transcribed into cDNA using an oligo-dT primer by reverse transcriptase (Invitrogen). After first-strand cDNA synthesis, the cDNA was used as a template and amplified by pairs of primers derived from Bcl-XL for the RT-PCR. Sequences of the primer pairs for amplification of Bcl-XL were 5′-TTGGACAATGGACTGGTTGAGCCC-3′ and 5′-TGTCTGGTCACTTCCGACTG-3′. Rat Bcl-XL cDNA containing the entire coding sequence was inserted into the T&A cloning vector (RBC Bioscience), followed by subcloning into the EcoRI-XhoI site of pcDNA3.1. The identities of the sequences were confirmed using an ABI PRISM 377 DNA Analysis System (Perkin-Elmer, North Point, Hong Kong).
An NFAT3 siRNA (CCAAGGUGGUGUUCAUUGATT) duplex was chemically synthesized by Ambion (Austin, TX, USA) and Bax siRNA (CUAGCAAACUGGUGCUCAATT, GCUCUGAACAGUUCAUGAATT, and CAGCUGACAUGUUUGCUGATT) duplexes were chemically synthesized by MDBio (Taipei, Taiwan). RTCs were seeded in a 6-well plate and transfected with either 100 pmol of NFAT3 siRNA (nos. s9483 and s9484, Ambion), Bax siRNAs, or scrambled control siRNA (no. 4611, Ambion) in a 100-µl volume with siPORT NeoFX. The efficiency of siRNA silencing was analysed by Western blotting after transfection for 48 h, followed by carboplatin treatment for the indicated time periods.
The NADPH oxidase activity and protein oxidation by measuring the amount of protein carbonyls
RTCs were treated with 200 µM of carboplatin for various time points and NADPH oxidase was measured as previously described (Clark et al., 2000; Sue et al., 2009). To measure protein carbonyls, cells pretreated with NAC followed by carboplatin challenge for 24 h were extracted in RIPA buffer containing 2% 2-mercaptoethanol to inhibit oxidation. The formation of carbonyl groups as a result of oxidative stress was derived with 2,4-dinitrophenylhydrazine (DNPH) that was carried out on 20 µg of protein for 15 min using an OxyBlot kit following the manufacturer's instructions (Millipore, Bedford, MA, USA).
Determination of ROS generation, apoptosis in RTC cells, and immunofluorescence staining
RTCs grown on a coverglass from each treatment group were loaded with 10 µM of the non-fluorescent dye, 20,70-dichlorodihydrofluorescein diacetate (H2DCFDA, Molecular Probes, Eugene, OR, USA) at 37°C for 30 min in the dark followed by 200 µM of carboplatin challenge for 30 min. Subsequent procedures were as described previously (Cheng et al., 2008). Apoptosis of RTCs with carboplatin challenge for 24 h was identified by a TUNEL assay with an in situ Cell Death Detection kit (Roche, Mannheim, Germany) according to the manufacturer's instructions. For immunofluorescence staining, RTCs were cultured on 0.17-mm poly-L-lysine-coated coverslips, and numbers of cells which had been transfected with pcDNA3.1 or pcDNA3.1-Flag-NFAT3 for 24 h followed by 200 µM carboplatin administration for 1 h were counted at 50∼80% confluence. Then cells were fixed in 4% formaldehyde for 15 min. Cells were permeabilized with 0.2% Triton and 0.1% Tween 20 in blocking buffer (3% BSA in PBS) for 2 h. Antibodies used for staining included mouse anti-Flag (1:1000, Sigma) followed by Texas red-conjugated goat anti-mouse immunoglobulin G (IgG) (1:200, Jackson Immunoresearch, West Grove, PA, USA). Subsequent procedures were as described previously (Chang et al., 2009).
Statistical analysis
Values are expressed as the mean ± SEM of at least three experiments. The significance of the difference from the control groups was analysed by Student's t-test or one-way anova. A value of P < 0.05 was considered statistically significant.
Results
Carboplatin caused morphological and functional renal injury with increased NFAT-driven luciferase activity in kidneys of transgenic mice
An animal study was performed to examine the effect of carboplatin in renal integrity and function, and its molecular action. The 6-day carboplatin treatment was to determine the optimal working concentration based upon the mouse survival rate. We observed that the levels of BUN at days 1 and 2 did not differ in mice receiving 50 mg·kg−1 carboplatin, whereas those receiving 100 mg·kg−1 showed significantly increased mortality (data not shown), but the mouse survival rate after 6-day treatment with 75 mg·kg−1 carboplatin was 100% for wild-type and 82% for transgenic mice (n = 11) with no statistically significant difference. The results in Figure 1A, obtained by renal histological examination with H&E staining and a TUNEL assay, demonstrate that in mice subjected to 75 mg·kg−1 carboplatin challenge renal tubular epithelial cells in the intermediate segment were flattened and had cell apoptotic injury to a greater extent than the control group; these effects were slightly more severe in transgenic than in the wild-type mice. An increase in BUN showed that a concomitant decrease in the renal function also occurred (Figure 1B). Interestingly, transgenic mice demonstrated a significant increase in BUN after 2 days of carboplatin treatment compared to wild-type FVB/N mice, suggesting that the former group was more susceptible to carboplatin-mediated renal injury. Additionally, carboplatin (75 mg·kg−1) given i.p. at day 2 significantly increased NFAT enhancer-driven luciferase activity in fractions of homogenized kidneys of NFAT-luciferase reporter transgenic mice (Figure 1C) and immunohistochemical staining revealed nuclear translocation of NFAT3 at day 1 (Figure 1D), suggesting that carboplatin increased renal NFAT(3) activation in the kidneys of transgenic mice. Additionally, the extent of nuclear translocation of NFAT3, as measured by immunohistochemical staining, in transgenic mice was greater than in the wild-type, which might be due to their greater susceptibility to carboplatin toxicity than their wild-type littermates.
Figure 1.
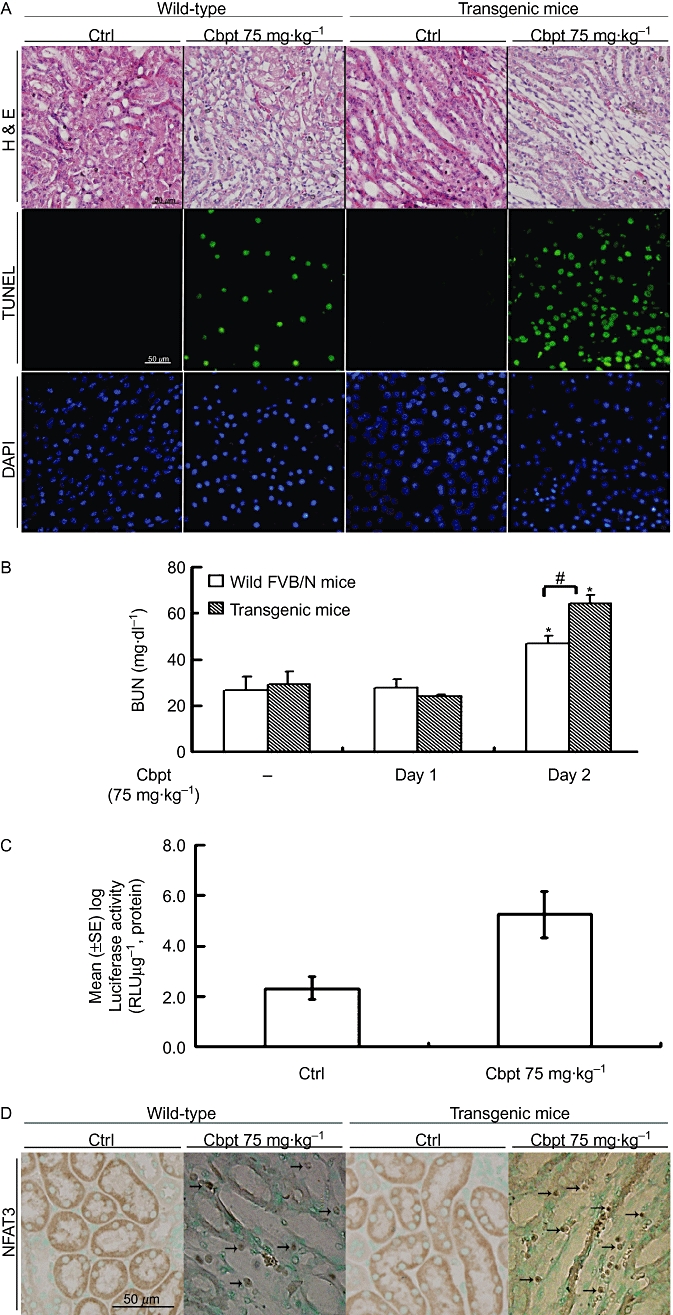
Effects of carboplatin (Cbpt) on the renal histology and function of the wild-type and nuclear factor of activated T-lymphocytes (NFAT)-reporter transgenic mice. Kidneys from mice given Cbpt (75 mg·kg−1 i.p., for 3 days) as the experimental group or those with double-distilled H2O as the control were dissected and sectioned for histological and apoptosis evaluation (A). Representative photomicrographs of haematoxylin and eosin (H&E) staining for morphology, and terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) for apoptosis with DAPI as a counterstaining are shown. Micrographs of representative fields were recorded. Concentration of blood urea nitrogen (BUN) in the aforementioned two groups was measured at day 1 and day 2 of Cbpt treatment (B). Results are the mean ± SD (n = 8). *P < 0.05 versus each respective group on day 1, #P < 0.05 versus wild-type FVB/N mice on day 2. Transgenic mice with kidneys carrying the NFAT-reporter gene were obtained as described in Methods. Mice were treated as described above, and kidneys were extracted on day 2 to determine the luciferase activity assay (C). Results are the mean ± SD (n = 8). *P < 0.05 versus the control. Kidney sections of the wild-type and transgenic mice were treated with Cbpt for 1 day, and then immunostained with an anti-NFAT3 antibody followed by incubation with Contrast Green Solution for nuclear counterstaining to reveal the position of cell nuclei (D). The arrows indicate the positive NFAT3 staining in the nuclei in mice challenged with Cbpt. Micrographs of representative fields were recorded. Representative results of three separate experiments are shown.
Carboplatin-mediated RTC apoptosis occurs through an intrinsic apoptotic pathway associated with ROS
The molecular mechanism of carboplatin-mediated renal injury was further investigated using a culture of rat RTCs. Cells treated with an increasing concentration of carboplatin, 0–400 µM, showed proportionally decreased cell viability (Figure 2A). Toxic effects of carboplatin in RTCs were examined by Western blot analysis of the downstream executioners of extrinsic and intrinsic apoptotic molecules including caspase-3, -8, and -9. The results in Figure 2B show that the activated forms of caspase-9 and -3, but not caspase-8, of RTCs challenged with 0–400 µM of carboplatin for 18 h concentration-dependently increased, suggesting that an intrinsic apoptotic pathway may be involved in carboplatin-mediated RTC injury. To prove the essential roles of caspase-9 and -3 in carboplatin-mediated RTC apoptosis, respective inhibitors of caspase-9 and -3 were added to prevent cell apoptosis. The results in Figure 2C,D show that the additional treatment with caspase-9 and -3 inhibitors reversed carboplatin-mediated caspase-3 activation and apoptosis. RTC apoptotic cell death caused by 200 µM carboplatin was associated with a time-dependent decrease in the ratio of Bcl-XL/Bax and a marked increase in a proapoptotic molecule, Bcl-XS, as shown in Figure 2E. Furthermore, the roles of increased Bax and decreased Bcl-XL expressions were further investigated using siRNA and overexpression approaches. The results in Figure 2F show that cells overexpressing Bcl-XL or knocking-down Bax by siBax alleviated carboplatin-mediated caspase-3 activation, suggesting the importance of Bcl-XL/Bax in carboplatin-mediated RTC apoptosis.
Figure 2.
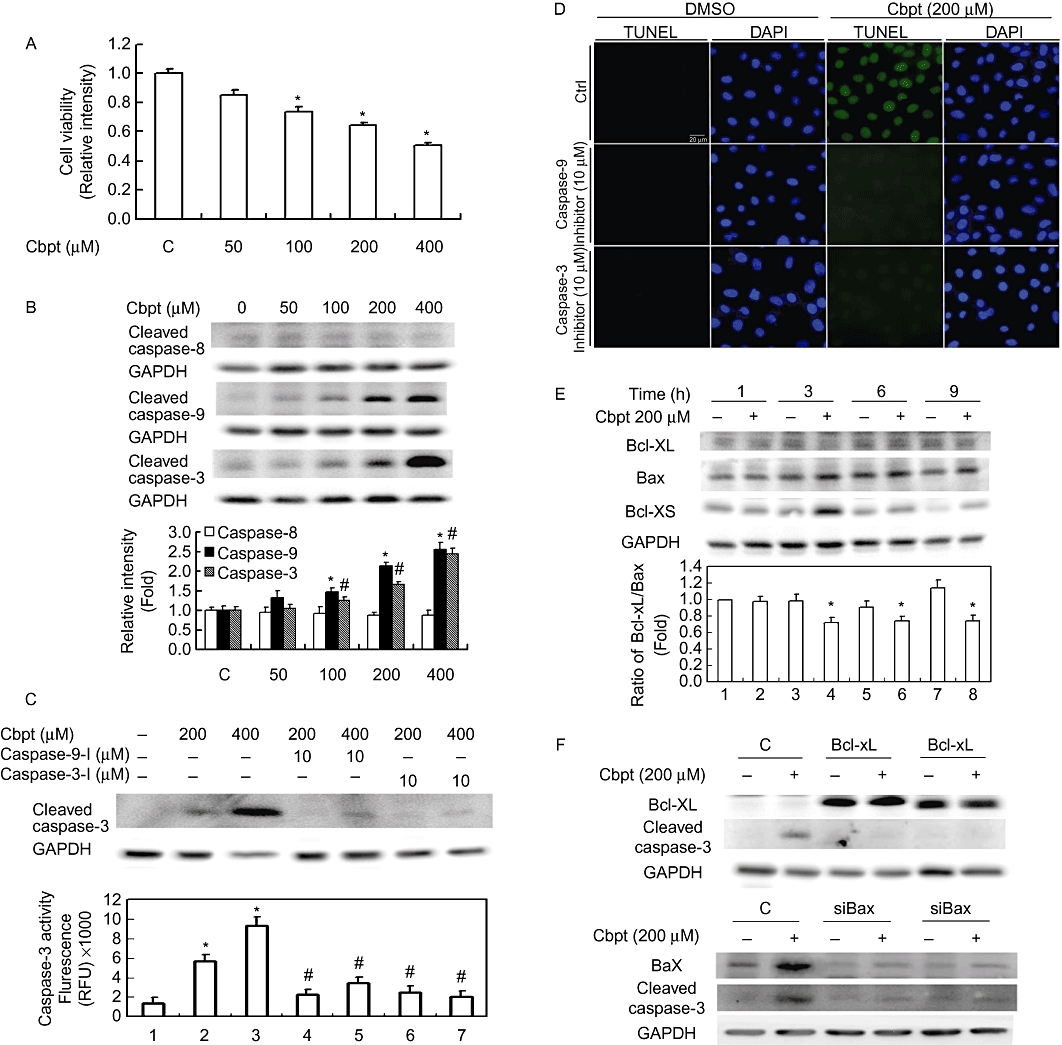
Carboplatin (Cpbt) induced renal tubular cell (RTC) apoptosis through an intrinsic apoptotic pathway. (A) Carboplatin inhibited cell viability in a concentration-dependent manner. Cells were challenged with an increasing concentration of Cbpt from 0–400 µM for 24 h, and cell viability was assessed by a 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. (B) Western blot analysis of active forms of caspase-3, -8, and -9 in cells challenged with various concentrations of Cbpt for 18 h. Equal loading in each lane or transfer was confirmed by incubation with an anti-GAPDH antibody. Representative results of three separate experiments are shown. Histograms in the lower panel show the band intensities of the indicated molecules by densitometry. Data were derived from three independent experiments and are presented as the mean ± SEM. Effects of caspase-3 and -9 inhibitors on Cbpt-mediated caspase-3 activation (C) and RTC apoptosis (D). Cells were pretreated with inhibitors of caspase-3 and -9 for 3 h followed by 18 or 24 h of Cbpt treatment to examine the roles of caspase-3 and -9 on Cbpt-mediated caspase-3 activation (18 h) and cell apoptosis (24 h). (E) Western blot analysis of anti- and proapoptotic molecules of Bcl-XL and Bax/Bcl-XS. Cells were treated with 200 µM of Cbpt for the indicated time points. Cells were transfected with pcDNA3.1-Bcl-XL or siBax for 24 h followed by 18 h of Cbpt treatment to evaluate the effect of Bcl-XL overexpression or Bax knockdown on Cbpt-mediated caspase-3 activation by a Western blot analysis (F). Representative results of three separate experiments are shown, and data are presented as the mean ± SEM (*P < 0.05 vs. the control; #P < 0.05 vs. the Cbpt-treated group).
Carboplatin increased NFAT enhancer-driven luciferase activity, its nuclear translocation, and exogenous overexpression of Flag-NFAT3 exacerbated carboplatin-mediated renal cell death
We showed above in Figure 1C that carboplatin increased renal luciferase activity in NFAT-transgenic mice. Whether activation of NFAT also occurs in vitro after carboplatin challenge was also examined. The results in Figure 3A demonstrate that 4 h of carboplatin treatment significantly increased NFAT-driven luciferase activity in RTCs transfected with pGL4.10-NFAT as compared to those with empty vector alone (Figure 3A). Likewise, RTCs treated with 200 µM carboplatin for 30 and 60 min showed a significant increase in nuclear translocation of NFAT3, suggesting that NFAT3 is activated in RTCs challenged with carboplatin (Figure 3B). The effect of activated NFAT3 on carboplatin-mediated RTC apoptosis was further examined using an NFAT3 overexpression system. The results in Figure 4A, immunofluorescent staining, show that carboplatin increased the nuclear translocation of exogenous Flag-NFAT3 in RTCs transfected with pcDNA3.1-Flag-NFAT3. Additionally, Western blot analysis demonstrated that the extent of caspase-3 activation by carboplatin was exacerbated in cells overexpressing Flag-NFAT3 with a concomitant increase in pro-apoptotic molecules, Bcl-XS and Bax, and a decrease in an anti-apoptotic molecule, Bcl-XL, as compared to cells transfected with the pcDNA3.1 empty vector alone (Figure 4B). Furthermore, apoptotic cells stained by a TUNEL assay (Figure 4C) and cell viability by a 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Figure 4D) produced results that agreed with the findings presented in Figure 4B, indicated that exogenous NFAT3 overexpression in RTCs mimicked the effect of carboplatin in RTC apoptosis, and the addition of carboplatin exacerbated the extent of cell death.
Figure 3.
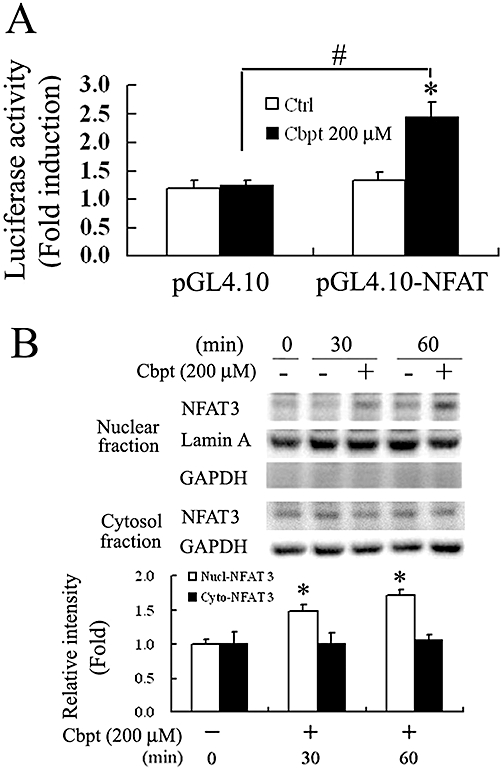
Carboplatin (Cbpt) increased nuclear factor of activated T-lymphocytes (NFAT)-driven luciferase activity and nuclear translocation in renal tubular cells (RTCs). (A) Cbpt-mediated increase in NFAT-driven luciferase activity in RTCs. Cells were transiently transfected with pGL4.10/NFAT and pRL-TK for 24 h, followed by 200 µM Cbpt for 4 h. Details are given in Methods. Results are presented as the mean ± SEM of four independent experiments. *P < 0.05 versus the control, #P < 0.05 versus the control with additional Cbpt treatment. (B) Increases in the nuclear translocation of NFAT3 in RTCs treated with Cbpt. Cells challenged with 200 µM Cbpt for 1 h were harvested or partitioned into the cytosolic and nuclear fractions of NFAT3. Representative results of three separate experiments are shown. GAPDH and lamin A were, respectively, used as the internal controls for the cytosolic and nucleus fractions. Histograms in the lower panel show the band intensities of the indicated molecules by densitometry. Data were derived from three independent experiments and are presented as the mean ± SEM. Significantly different (*P < 0.05 vs. the control; Nucl-NFAT3; #P < 0.05 vs. the control treated with Cbpt).
Figure 4.
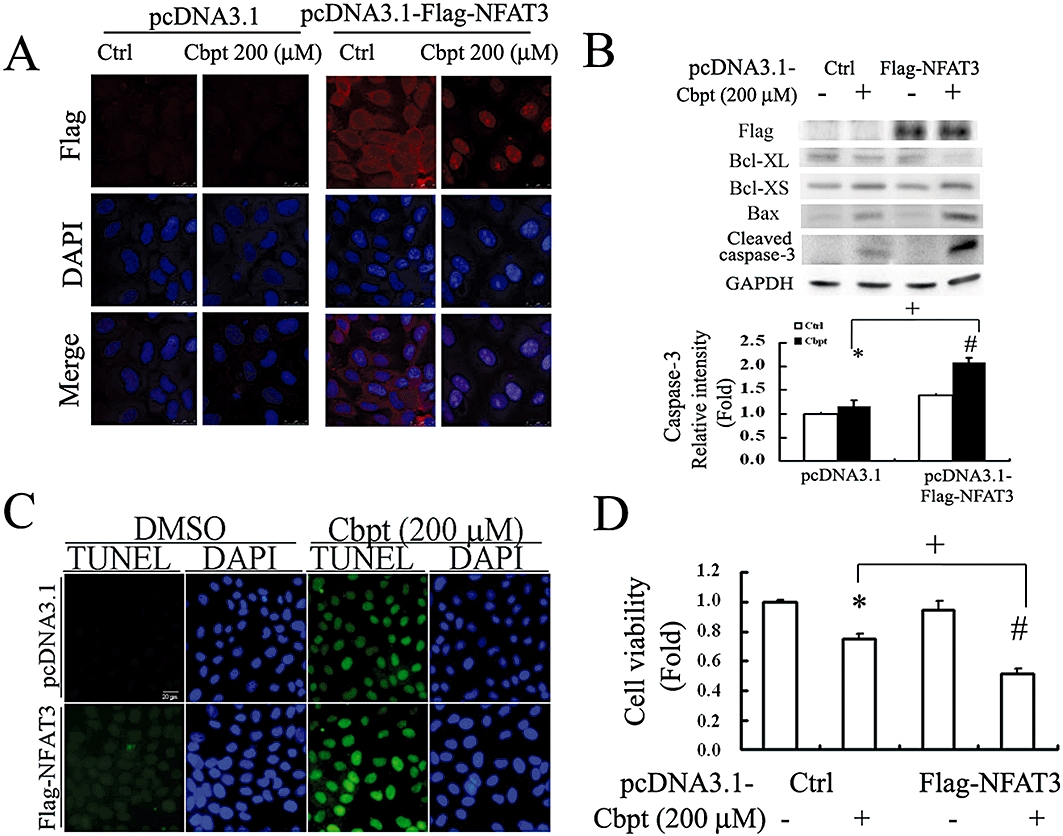
Overexpression of exogenous Flag-nuclear factor of activated T-lymphocyte-3 (NFAT3) exacerbates the effects of carboplatin (Cbpt) in renal tubular cells (RTCs). Cells transfected with pcDNA3.1-Flag-NFAT3 for 24 h were challenged with 200 µM Cbpt treatment for 1 h for nuclear translocation of exogenous Flag-NFAT3 by immunofluorescence staining (A), for 6 h or 18 h as analysed by Western blotting for apoptotic-related molecules (Bcl-XL, Bcl-XS and BAX) or the active form of caspase-3 (B), respectively, or for 24 h for RTC apoptosis by a terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) assay (C) and cell viability by a 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (D). Details are given in Methods. Representative results of three separate experiments are shown, and data are presented as the mean ± SEM (*P < 0.05 vs. pcDNA3.1 alone; #P < 0.05 vs. pcDNA3.1-Flag-NFAT3 alone; +P < 0.05 vs. the Cbpt-treated group).
Inhibition of NFAT activation by calcineurin inhibitors and knockdown of NFAT3 by siRNA reversed NFAT3 activation and subsequent caspase-3 activation caused by carboplatin
NFAT activation has been reported to be regulated by calcineurin, which can be inhibited by FK-506 (tacrolimus), CsA and BAPTA-AM (Huang et al., 2001b). The addition of FK-506 (Figure 5A), CsA (Figure 5B) and BAPTA-AM (Figure 5C) significantly reversed the carboplatin-mediated nuclear translocation of NFAT and subsequent caspase-3 activation. Additionally, knockdown of NFAT3 by siNFAT3 decreased messenger (m)RNA and protein levels of NFAT3 and the consequent active form of caspase-3 with a concomitant reversal of carboplatin-mediated alterations in the anti-apoptotic molecule of Bcl-XL and the pro-apoptotic molecules of Bax and Bcl-XS (Western blot analysis) and, hence, decreased apoptotic cell death (TUNEL assay). These results indicate the essential role of NFAT3 activation in carboplatin-mediated RTC apoptotic signalling (Figure 5D).
Figure 5.
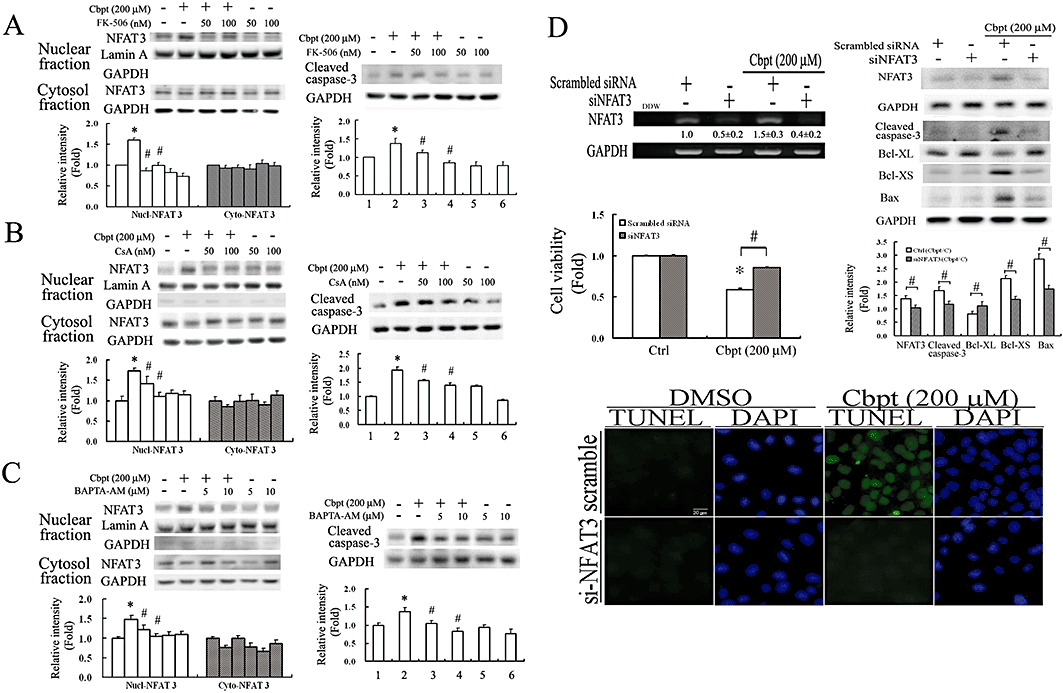
Carboplatin-mediated caspase-3 activation was reversed by inactivation of NFAT by calcineurin inhibitors including FK-506, cyclosporin A (CsA) and BAPTA-AM, or knockdown of nuclear factor of activated T-lymphocyte-3 (NFAT3) by small interfering (si)NFAT3. Cells were pretreated with 30 or 40 min of the indicated concentrations of FK-506 (A), CsA (B) and BAPTA-AM (C), respectively, followed by challenge with 200 µM Cbpt for 1 or 18 h and then examined by Western blot analysis for the nuclear translocation of NFAT3 or the active form of caspase-3. Representative results of three separate experiments are shown. (D) Effect of siNFAT3 on Cbpt-mediated apoptotic molecules and cell apoptosis. Cells were transfected with siNFAT3 for 24 h, followed by 1 h or 18 h of Cbpt treatment to examine the efficiency of siRNA knockdown by an RT-PCR analysis and the effect of NFAT3 knockdown on Cbpt-mediated alterations of apoptotic-related molecules including Bcl-XL, Bax, and Bcl-XS, and caspase-3 activation by Western blots, RTC viability, and apoptosis by 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) assays. Representative results of three separate experiments are shown, and data are presented as the mean ± SEM (*P < 0.05 vs. the control; #P < 0.05 vs. Cbpt-treated group).
Carboplatin-mediated ROS generation was involved in the signalling pathways of NFAT3 activation, which was attenuated by NAC both in vitro and in vivo
Given the findings that carboplatin causes ROS generation (Cheng et al., 2008) and ROS stimulates NFAT activation to trigger apoptosis in various cell types (Li et al., 2002; Cheng et al., 2008), we examined whether carboplatin-mediated NFAT3 activation is associated with ROS and whether scavenging of ROS reverses NFAT3-mediated RTC apoptosis. The result in Figure 6A show that carboplatin induced ROS in RTCs through by increasing the active form of p47PHOX (a) and then enhancing NADPH oxidase activity (b), which can be reversed by the antioxidant and the glutathione precursor, NAC. Additionally, the results in panels (c) and (d) of Figure 6A show that NAC effectively reversed carboplatin-mediated ROS generation and protein oxidation. Furthermore, the signalling molecules involved in the rescue of carboplatin-mediated NFAT3 activation by NAC were examined. We showed that carboplatin-mediated ERK, JNK and PKC activation was eliminated by the ROS scavenger, NAC, and their respective inhibitors, U0126, SP600125 and RO318220, also reversed carboplatin-mediated NFAT3 activation (Figure 6B). Moreover, the results in Figure 6C show that NAC markedly alleviated carboplatin-mediated NFAT3 activation, and subsequent caspase-3 activation, suggesting that ROS is essential for carboplatin-mediated RTC apoptosis through NFAT3 activation. The results in Figure 6D show that the respective inhibitors of ERK/JNK/PKC/ROS decreased RTC apoptosis, confirming the causal effect of NFAT3 activation and subsequent caspase-3 activation depicted in Figure 6B,C. Furthermore, the therapeutic effect of NAC in preventing carboplatin-mediated renal damage was examined in vivo using the wild-type littermates. The results in Figure 7A show that NAC reversed ROS-mediated lipid peroxidation induced by carboplatin; this was assayed by measuring protein bound 4-hydroxy-2-nonenal (HNE) and immunohistochemical staining. Additionally, NAC reversed carboplatin-mediated NFAT3 and caspase-3 activation (Figure 7B), increase in creatinine and BUN (Figure 7C), and cell apoptosis (Figure 7D) in vivo, suggesting the importance of ROS's involvement in NFAT3 activation and subsequent renal apoptosis induced by carboplatin.
Figure 6.
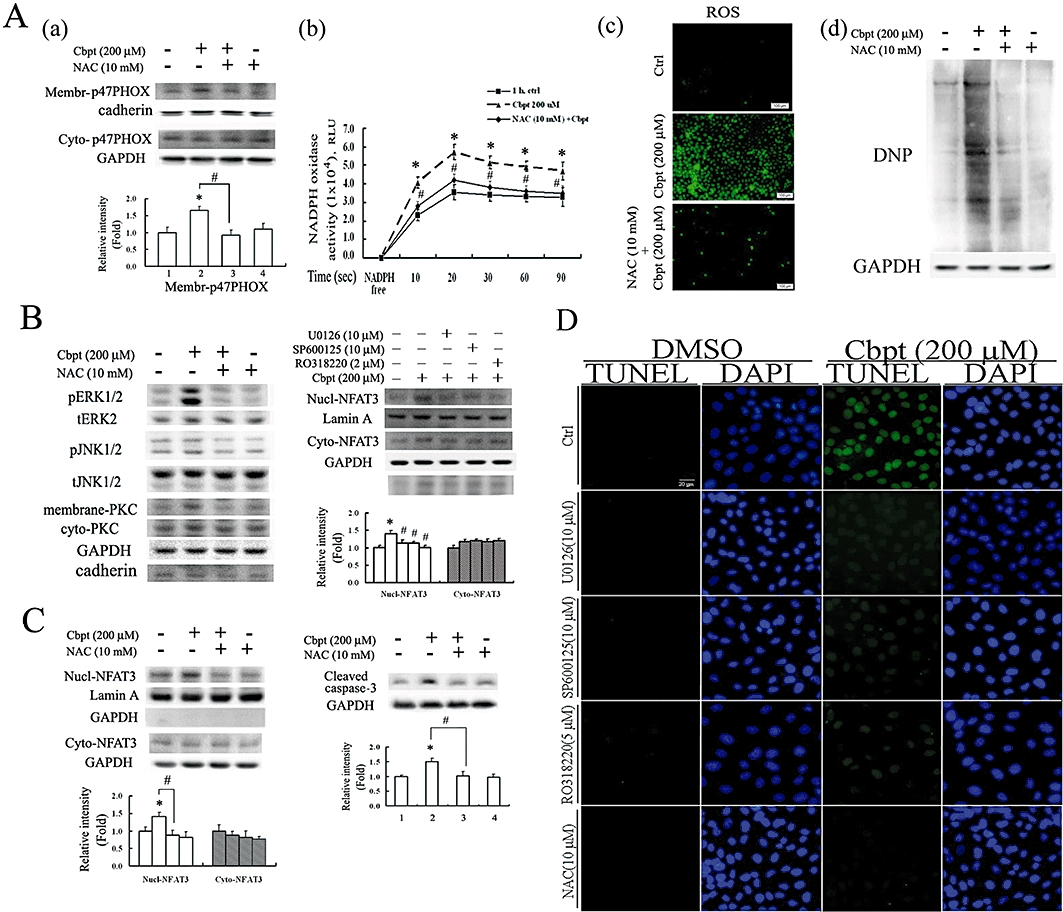
Oxidative stress involved in the signalling pathways of NFAT3 activation and rat tubular cells (RTCs) apoptosis by carboplatin (Cbpt), and attenuated by N-acetylcysteine (NAC). (A) Alleviation of the Cbpt-mediated increase in NADPH oxidase activity by eliminating membranous recruitment of p47PHOX by NAC, with a concomitant decrease in ROS generation. Cells with or without 30 min of NAC pretreatment followed by 1 h of Cbpt challenge were harvested for cytosolic-membranous fractions of p47PHOX (a), a component of NADPH oxidase, and analysed by Western blotting. NADPH oxidase activity (b) in cells with various treatments was detected by a luminometer, as described in Methods. Representative CM-H2DCFDA fluorescent photomicrographs of Cbpt-mediated ROS production and Western blot analysis of carbonyl group formation induced by Cbpt in RTCs with the above-mentioned treatments are shown in panels (c) and (d) of (A). (B) Elimination of Cbpt-mediated NFAT3 activation by NAC through an inhibition in ERK, JNK and PKC activation. Cells were pretreated with NAC prior to 30 min of Cbpt treatment for Western blot analysis of activated ERK/JNK/PKC. Additionally, cells were pretreated with their respective inhibitors for 1 h prior to Cbpt challenge for another hour and partitioned into nuclear-cytosolic fractions for analysis of activated NFAT3 by Western blots. (C) Elimination of Cbpt-induced caspase-3 activation by NAC-mediated NFAT3 inactivation. Cells were treated as described previously, except that cell lysates were harvested for Western blot analysis of cytosolic-nuclear fractions of NFAT3 and caspase-3 at 1 and 18 h of Cbpt treatment, respectively. Results are representative data from three separate experiments. Histograms in the lower panels of (A) (B) and (C) show the band intensities of the indicated molecules by densitometry. Data were derived from three independent experiments and are presented as the mean ± SEM. Significantly different (*P < 0.05 vs. the control; #P < 0.05 vs. Cbpt-treated group). (D) Attenuation of Cbpt-mediated RTC apoptosis by inhibitors of ERK/JNK/PKC/ROS as observed by terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) assays. Results are representative data from three separate experiments.
Figure 7.
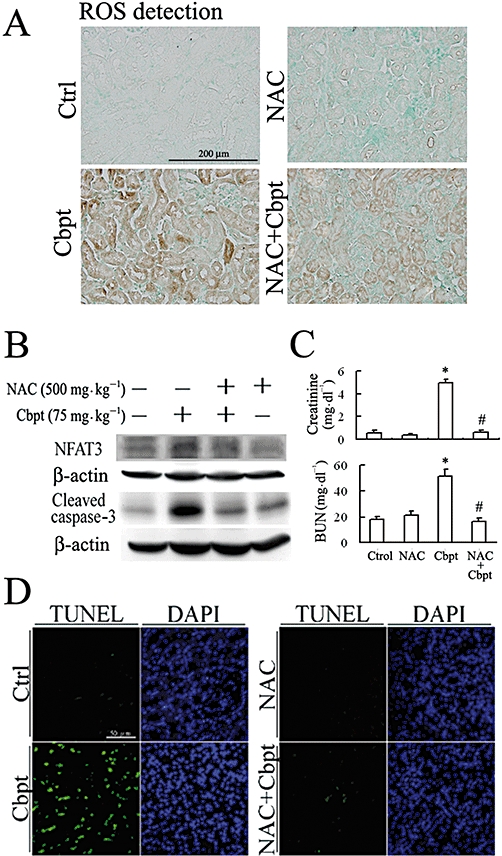
The therapeutic effect of NAC and its attenuation of Cbpt-mediated renal injury. The wild-type mice were treated with a single dose of 75 mg·kg−1 Cbpt for 3 days or with 500 mg·kg−1 NAC as a therapeutic intervention for 3 consecutive days. On day 4, murine kidneys were harvested and sectioned for immunochemical staining by an anti-HNE antibody for ROS-mediated lipid peroxidation, followed by incubation with a second antibody conjugated with horseradish peroxidase. Contrast Green Solution was used for nuclear counterstaining to reveal the position of cell nuclei (A). Micrographs of representative fields were recorded. Representative results of three separate experiments are shown. The therapeutic effects of NAC on Cbpt-mediated renal injury were evaluated by Western blot analysis of NFAT3 and subsequent caspase-3 activation (B), renal function indicators of creatinine and BUN (C) and cell apoptosis (D). Results are the mean ± SD (n = 6). *P < 0.05 versus the control; #P < 0.05 versus the wild-type mice with Cbpt treatment.
Discussion
Carboplatin is generally and widely used for treating various tumours, and we had previously shown that carboplatin-mediated cardiomyocyte apoptosis is ascribable to ROS generation. However, the effect of carboplatin in renal toxicity is not clear. Herein, our findings demonstrate its deleterious effects in renal integrity, function and apoptosis in vivo, as well as its potential molecular mechanisms in eliciting this toxic effect in vitro. Our present data show that nuclear translocation of NFAT3 is responsible for carboplatin-mediated apoptosis through an ROS-dependent signalling pathway, as inhibition of ROS generation by NAC reversed NFAT3 activation and caspase-3 activation both in vitro and in vivo. Additionally, inactivation of calcineurin by FK-506, CsA and BAPTA-AM decreased NFAT3 activation and the ensuing carboplatin-mediated renal apoptosis. Therefore, carboplatin-induced renal apoptosis is mediated by ROS-mediated NFAT3 activation through a pathway dependent on calcineurin and calcium.
The results of our present study demonstrate that carboplatin-mediated ROS generation is involved in the increased NADPH oxidase activity which is accompanied by an increase in p47PHOX membranous assembling. Interestingly, our in vitro results, similar to those of Fujii et al. (2007), show that NAC, an antioxidant, decreased the p47PHOX membranous recruitment as well as ROS production. The underlying mechanism was found to be activation of PI3K that elicits p47PHOX phosphorylation and the membranous recruitment of p47PHOX to increase NADPH oxidase activity, which is critical for α-platelet-derived growth factor receptor-induced production of ROS in fibroblasts and HIV Nef-mediated rapid superoxide anion release from the U937 human monoblastic cell line (Baumer et al., 2008; Olivetta et al., 2009). However, the signalling pathway through which carboplatin increases p47PHOX membranous recruitment remains to be determined.
The mechanism of cisplatin-induced nephrotoxicity is complex and involves numerous interconnected processes, such as formation of ROS, DNA damage, increased Bax translocation into mitochondria through ERK activation, and caspase activation (Lee et al., 2001; Mirowski et al., 2003; Kim et al., 2005). Additionally, it has been suggested that nucleus translocation of p53 correlated with activation of caspase-3, -8 and -9 in renal cell apoptosis through intrinsic and extrinsic apoptotic pathways, although 50% of cisplatin-induced renal proximal tubular cell apoptosis has been shown to be mediated by additional mechanisms independent of p53 and caspase-3, -8, and -9 (Cummings and Schnellmann, 2002). By contrast, based on our findings, carboplatin, a second-generation platinum compound, did not cause RTC apoptosis through an extrinsic apoptotic pathway (Figure 2B) and there was no significant alteration in the p53 protein level in RTC treated with carboplatin (data not shown). However, our findings demonstrate that, in addition to ERK1/2, JNK/PKC signalling pathways are also involved in the activation of NFAT3 and by decreasing the ratio of Bcl-XL/Bax and significantly increasing the pro-apoptotic molecule, Bcl-XS, subsequently lead to RTC apoptosis. Whether cisplatin-mediated RTC apoptosis is also dependent on NFAT3 activation is not clear. Importantly, the results presented in this study suggest the essential role of activated NFAT3 in carboplatin-induced Bcl-X alternative splicing in terms of the findings in Figures 4B and 5D; the mechanism for this remains to be further investigated.
In the present study, our findings demonstrate that carboplatin causes renal apoptosis both in vitro and in vivo, by NFAT3 activation, which is further linked with oxidative stress in vitro. Using a cell culture system, we revealed that carboplatin-mediated ROS production by increasing the activity of NADPH oxidase, which activates NFAT3 and consequently RTC apoptosis through the ERK/JNK/PKC-dependent pathways (summarized in Figure 8). The activation of NFAT through the ERK/JNK/PKC pathways is agreed by recent studies showing that PKC affects Ca2+ mobilization and NFAT activation in primary mouse T Cells (Pfeifhofer et al., 2003) and calcineurin-NFAT signalling regulates the cardiac hypertrophic response in coordination with the MAPKs (Molkentin, 2004). Moreover, our in vitro findings show that NFAT3 overexpression mimicked the effect of carboplatin in RTCs, and this is exacerbated by carboplatin treatment. Conversely, elimination of NFAT3 by siRNA knockdown effectively reversed carboplatin-mediated caspase-3 activation and cell viability. This result agrees with methamphetamine-induced neuronal apoptosis and cardiac glycoside-induced U937 cell (histiocytic lymphoma) apoptosis, although they occur through calcineurin/NFAT-induced upregulation of the Fas ligand (FasL)/Fas and then caspase-8 activation (an extrinsic apoptotic process) (Jayanthi et al., 2005; Raghavendra et al., 2007). In contrast, we did not observe apparent induction of FasL (data not shown) and activation of caspase-8 (Figure 2B); instead the intrinsic apoptosis executioners, caspase-9 and -3, were activated by carboplatin. However, the precise physiological roles of NFAT3 remain unclear, even though it has been extensively studied in the cardiovascular system. Additionally, the role of NFAT varies at different physiological stages. For instance, simultaneous disruption of NFAT3 and NFAT4 causes embryonic lethality on embryonic day 11 due to cardiovascular defects (Graef et al., 2001a), whereas overexpression of NFAT3 induces cardiac hypertrophy in adult mice (Molkentin, 2000; Graef et al., 2001b). Determining whether the pathological role of NFAT3 in renal toxicity is to elicit programmed cell apoptosis to prevent further deterioration remains to be investigated.
Figure 8.
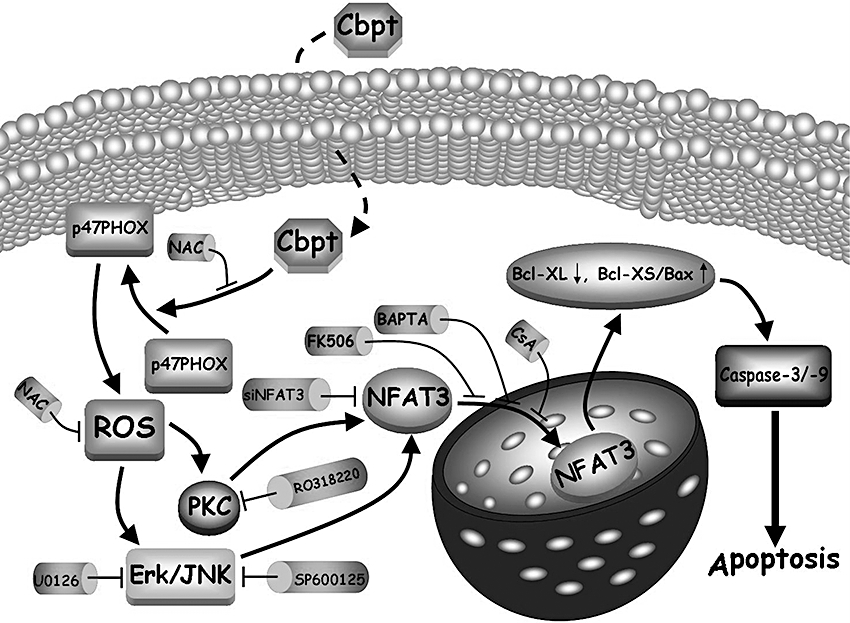
Summary of signal pathways of carboplatin (Cbpt) on NFAT3 activation attributed to RTC apoptosis. In the proposed signal pathways, Cbpt-mediated ROS production activates ERK/JNK/PKC signalling pathways, followed by the activation of NFAT3 with a concomitant increase in the ratio of Bax/Bcl-XL, a pro-apoptotic molecule, Bcl-XS, and caspase-3/-9 activation, and the subsequent RTC apoptosis. Various pharmacological inhibitors such as NAC, U0126, SP600125 and RO318220 or siNFAT3 can block its respective target molecule to inhibit RTC apoptosis caused by Cbpt.
Intriguingly, although our results herein demonstrate that activation of NFAT3 in response to carboplatin-mediated oxidative stress triggered an RTC apoptotic signalling pathway, activation of NFAT3 by arsenite in human bronchial epithelial cells has been shown to exert an antiapoptotic effect through induction of Cox-2 expression (Ding et al., 2006). Additionally, mice lacking NFATc2 developed more and larger solid tumours than wild-type littermates in a murine model of bronchoalveolar adenocarcinoma. The extent of central tumour necrosis decreased in tumours in NFAT2 (−/−) mice, and this finding was associated with reduced tumour necrosis factor-α and interleukin-2 (IL)-2 production by CD8+ T cells (Maxeiner et al., 2009). However, one of the gene expression profiles, NFAT, was shown to identify patients with a higher risk of recurrence (Rosell et al., 2007), which was supported by the finding demonstrating that high NFAT expression was present in Chinese NSCLCs and that NFAT expression might be involved in the development of human lung cancer (Zhang et al., 2007).
An i.v. infusion of carboplatin, as a single dose of 300–500 mg·m−2, is commonly used to treat patients with solid tumours. The dose/concentration of carboplatin used (in vivo or in vitro) in the present study (200 µM; 75 mg·kg−1) was equivalent to 225 mg·m−2 and is near the lower range used clinically, but was based upon previous studies where 120 or 50 mg·kg−1 of carboplatin was used for in vivo i.p. injections in mice (Boughattas et al., 1990; Wang et al., 2004). The lower range of carboplatin used clinically causes renal injury but a higher dose (600 mg·m−2 or 200 mg·kg−1) has also been used to induce bone marrow suppression but this markedly reduced the survival rate (20% at day 1 post-treatment) in mice (Wang et al., 2004). This might have been due to the fact that the kidneys as excretory organs are relatively susceptible to the toxicity of carboplatin chemotherapy. Additionally, it has been shown that carboplatin produces less nephrotoxicity than other platinum-containing drugs such as cisplatin, nedaplatin and even the third-generation oxaliplatin (Kitada et al., 2008); however, the concentration of carboplatin used in our investigation was twice as much as their highest concentration used (100 µM). Additionally, they demonstrated that concentrations of carboplatin as low as 1 µM induced apoptotic injury in LLC-PK1 cells, as evident from DNA ladder formation. The results presented in our study also demonstrate that transgenic mice are more susceptible to carboplatin-mediated renal damage compared to the wild-type mice. This might have been due to random insertion of a transgene in the mouse chromosome and NFAT-luciferase transgenic mice displaying detectable activity in most tissues, although the NFAT enhancer is positioned 5′ to the α-myosin heavy chain gene promoter, a cardiac muscle-specific promoter, to create NFAT-luc. Other organs (i.e. the heart and brain) overexpressing this transgene might somehow contribute to the more severe renal injury observed in the transgenic mice; the mechanism for this remains to be investigated. Furthermore, we also observed that the extent of NFAT3 activation was greater in the transgenic mice than in the wild-type, which might have accounted for the transgenic mice having more severe renal injury after carboplatin challenge. Most importantly using the transgenic mice model, we proved that, in agreement with our in vitro findings, mice administered carboplatin exhibited increased NFAT3 activation and apoptosis in the kidneys. Additionally, the attenuation of carboplatin-mediated murine renal injury, including NFAT3 activity, ROS, renal function, and cell apoptosis, by the antioxidant, NAC, suggests the causal effect of oxidative stress on NFAT3 activation and subsequent RTC apoptosis.
This is the first study to report that carboplatin-mediated renal apoptosis is associated with NFAT3 activation, signalled by ROS generation and resulting in cell apoptosis. In summary, the data presented herein provide evidence to support the essential role of NFAT3 in carboplatin-mediated renal apoptosis. Identifying the transcription factor of NFAT3 and its underlying mechanisms of activation will provide an important molecular basis for the design of new therapeutic strategies to treat the complications (side effects) associated with carboplatin chemotherapy. The NAC, an antioxidant, or other antioxidants might be potential targets for protecting cells and tissues against carboplatin-mediated oxidative injuries. Further studies using NAC or other antioxidants will clarify the possibility of developing this new ‘therapy’ for the prevention or treatment of renal tubular apoptosis associated with carboplatin chemotherapy for tumours
Acknowledgments
We are grateful to Dr Jeffery D. Molkentin (Cincinnati Children's Hospital Medical Center, Cincinnati, OH, USA) for the gift of the NFAT-reporter transgenic mice. This study was sponsored by a grant from Taipei Medical University-associated Wan Fang Hospital (98TMU-WFH-09).
Glossary
Abbreviations
- BAPTA-AM
1,2-bis-(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid-acetoxymethyl ester
- CsA
cyclosporin A
- NAC
N-acetylcysteine
- NFAT3
nuclear factor of activated T-lymphocyte-3
- RTC
renal tubular cell
Conflict of interest
The authors state no conflict of interest.
References
- Alberts DS. Carboplatin versus cisplatin in ovarian cancer. Semin Oncol. 1995;22:88–90. [PubMed] [Google Scholar]
- Baumer AT, Ten Freyhaus H, Sauer H, Wartenberg M, Kappert K, Schnabel P, et al. Phosphatidylinositol 3-kinase-dependent membrane recruitment of Rac-1 and p47phox is critical for alpha-platelet-derived growth factor receptor-induced production of reactive oxygen species. J Biol Chem. 2008;283:7864–7876. doi: 10.1074/jbc.M704997200. [DOI] [PubMed] [Google Scholar]
- Boughattas NA, Levi F, Fournier C, Hecquet B, Lemaigre G, Roulon A, et al. Stable circadian mechanisms of toxicity of two platinum analogs (cisplatin and carboplatin) despite repeated dosages in mice. J Pharmacol Exp Ther. 1990;255:672–679. [PubMed] [Google Scholar]
- Burger H, Zoumaro-Djayoon A, Boersma AW, Helleman J, Berns EM, Mathijssen RH, et al. Differential transport of platinum compounds by the human organic cation transporter hOCT2 (hSLC22A2) Br J Pharmacol. 2010;159:898–908. doi: 10.1111/j.1476-5381.2009.00569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushdid PB, Osinska H, Waclaw RR, Molkentin JD, Yutzey KE. NFATc3 and NFATc4 are required for cardiac development and mitochondrial function. Circ Res. 2003;92:1305–1313. doi: 10.1161/01.RES.0000077045.84609.9F. [DOI] [PubMed] [Google Scholar]
- Cao X, Deng X, May WS. Cleavage of Bax to p18 Bax accelerates stress-induced apoptosis, and a cathepsin-like protease may rapidly degrade p18 Bax. Blood. 2003;102:2605–2614. doi: 10.1182/blood-2003-01-0211. [DOI] [PubMed] [Google Scholar]
- Chang CC, Tsai SY, Lin H, Li HF, Lee YH, Chou Y, et al. Aryl-hydrocarbon receptor-dependent alteration of FAK/RhoA in the inhibition of HUVEC motility by 3-methylcholanthrene. Cell Mol Life Sci. 2009;66:3193–3205. doi: 10.1007/s00018-009-0102-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CF, Juan SH, Chen JJ, Chao YC, Chen HH, Lian WS, et al. Pravastatin attenuates carboplatin-induced cardiotoxicity via inhibition of oxidative stress associated apoptosis. Apoptosis. 2008;13:883–894. doi: 10.1007/s10495-008-0214-9. [DOI] [PubMed] [Google Scholar]
- Chowdhury I, Tharakan B, Bhat GK. Caspases – an update. Comp Biochem Physiol B Biochem Mol Biol. 2008;151:10–27. doi: 10.1016/j.cbpb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Clark JE, Foresti R, Green CJ, Motterlini R. Dynamics of haem oxygenase-1 expression and bilirubin production in cellular protection against oxidative stress. Biochem J. 2000;348(Pt 3):615–619. [PMC free article] [PubMed] [Google Scholar]
- Cummings BS, Schnellmann RG. Cisplatin-induced renal cell apoptosis: caspase 3-dependent and -independent pathways. J Pharmacol Exp Ther. 2002;302:8–17. doi: 10.1124/jpet.302.1.8. [DOI] [PubMed] [Google Scholar]
- Ding J, Li J, Xue C, Wu K, Ouyang W, Zhang D, et al. Cyclooxygenase-2 induction by arsenite is through a nuclear factor of activated T-cell-dependent pathway and plays an antiapoptotic role in Beas-2B cells. J Biol Chem. 2006;281:24405–24413. doi: 10.1074/jbc.M600751200. [DOI] [PubMed] [Google Scholar]
- Fujii S, Zhang L, Kosaka H. Albuminuria, expression of nicotinamide adenine dinucleotide phosphate oxidase and monocyte chemoattractant protein-1 in the renal tubules of hypertensive Dahl salt-sensitive rats. Hypertens Res. 2007;30:991–998. doi: 10.1291/hypres.30.991. [DOI] [PubMed] [Google Scholar]
- Fujiwara K, Sakuragi N, Suzuki S, Yoshida N, Maehata K, Nishiya M, et al. First-line intraperitoneal carboplatin-based chemotherapy for 165 patients with epithelial ovarian carcinoma: results of long-term follow-up. Gynecol Oncol. 2003;90:637–643. doi: 10.1016/s0090-8258(03)00377-9. [DOI] [PubMed] [Google Scholar]
- Graef IA, Chen F, Chen L, Kuo A, Crabtree GR. Signals transduced by Ca(2+)/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell. 2001a;105:863–875. doi: 10.1016/s0092-8674(01)00396-8. [DOI] [PubMed] [Google Scholar]
- Graef IA, Chen F, Crabtree GR. NFAT signaling in vertebrate development. Curr Opin Genet Dev. 2001b;11:505–512. doi: 10.1016/s0959-437x(00)00225-2. [DOI] [PubMed] [Google Scholar]
- Huang C, Ding M, Li J, Leonard SS, Rojanasakul Y, Castranova V, et al. Vanadium-induced nuclear factor of activated T cells activation through hydrogen peroxide. J Biol Chem. 2001a;276:22397–22403. doi: 10.1074/jbc.M010828200. [DOI] [PubMed] [Google Scholar]
- Huang C, Li J, Costa M, Zhang Z, Leonard SS, Castranova V, et al. Hydrogen peroxide mediates activation of nuclear factor of activated T cells (NFAT) by nickel subsulfide. Cancer Res. 2001b;61:8051–8057. [PubMed] [Google Scholar]
- Husain K, Scott RB, Whitworth C, Somani SM, Rybak LP. Dose-response of carboplatin-induced hearing loss in rats: antioxidant defense system. Hear Res. 2001;151:71–78. doi: 10.1016/s0300-2977(00)00081-4. [DOI] [PubMed] [Google Scholar]
- Jayanthi S, Deng X, Ladenheim B, McCoy MT, Cluster A, Cai NS, et al. Calcineurin/NFAT-induced up-regulation of the Fas ligand/Fas death pathway is involved in methamphetamine-induced neuronal apoptosis. Proc Natl Acad Sci USA. 2005;102:868–873. doi: 10.1073/pnas.0404990102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Kim HJ, Kwon CH, Kim JH, Woo JS, Jung JS, et al. Role of ERK activation in cisplatin-induced apoptosis in OK renal epithelial cells. J Appl Toxicol. 2005;25:374–382. doi: 10.1002/jat.1081. [DOI] [PubMed] [Google Scholar]
- Kitada N, Takara K, Itoh C, Minegaki T, Tsujimoto M, Sakaeda T, et al. Comparative analysis of cell injury after exposure to antitumor platinum derivatives in kidney tubular epithelial cells. Chemotherapy. 2008;54:217–223. doi: 10.1159/000140465. [DOI] [PubMed] [Google Scholar]
- Lakhani SA, Masud A, Kuida K, Porter GA, Jr, Booth CJ, Mehal WZ, et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FP, Jen CY, Chang CC, Chou Y, Lin H, Chou CM, et al. Mechanisms of adiponectin-mediated COX-2 induction and protection against iron injury in mouse hepatocytes. J Cell Physiol. 2010;224:837–847. doi: 10.1002/jcp.22192. [DOI] [PubMed] [Google Scholar]
- Lee RH, Song JM, Park MY, Kang SK, Kim YK, Jung JS. Cisplatin-induced apoptosis by translocation of endogenous Bax in mouse collecting duct cells. Biochem Pharmacol. 2001;62:1013–1023. doi: 10.1016/s0006-2952(01)00748-1. [DOI] [PubMed] [Google Scholar]
- Li J, Huang B, Shi X, Castranova V, Vallyathan V, Huang C. Involvement of hydrogen peroxide in asbestos-induced NFAT activation. Mol Cell Biochem. 2002;234–235:161–168. [PubMed] [Google Scholar]
- Lin H, Lee JL, Hou HH, Chung CP, Hsu SP, Juan SH. Molecular mechanisms of the antiproliferative effect of beraprost, a prostacyclin agonist, in murine vascular smooth muscle cells. J Cell Physiol. 2008;214:434–441. doi: 10.1002/jcp.21214. [DOI] [PubMed] [Google Scholar]
- Masuda ES, Imamura R, Amasaki Y, Arai K, Arai N. Signalling into the T-cell nucleus: NFAT regulation. Cell Signal. 1998;10:599–611. doi: 10.1016/s0898-6568(98)00019-9. [DOI] [PubMed] [Google Scholar]
- Maxeiner JH, Karwot R, Sauer K, Scholtes P, Boross I, Koslowski M, et al. A key regulatory role of the transcription factor NFATc2 in bronchial adenocarcinoma via CD8+ T lymphocytes. Cancer Res. 2009;69:3069–3076. doi: 10.1158/0008-5472.CAN-08-1678. [DOI] [PubMed] [Google Scholar]
- Mirowski M, Rozalski M, Krajewska U, Balcerczak E, Mlynarski W, Wierzbicki R. Induction of caspase 3 and modulation of some apoptotic genes in human acute promyelocytic leukemia HL-60 cells by carboplatin with amifostine. Pol J Pharmacol. 2003;55:227–234. [PubMed] [Google Scholar]
- Molkentin JD. Calcineurin and beyond: cardiac hypertrophic signaling. Circ Res. 2000;87:731–738. doi: 10.1161/01.res.87.9.731. [DOI] [PubMed] [Google Scholar]
- Molkentin JD. Calcineurin-NFAT signaling regulates the cardiac hypertrophic response in coordination with the MAPKs. Cardiovasc Res. 2004;63:467–475. doi: 10.1016/j.cardiores.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay P, Pan H, Rajesh M, Batkai S, Patel V, Harvey-White J, et al. CB1 cannabinoid receptors promote oxidative/nitrosative stress, inflammation and cell death in a murine nephropathy model. Br J Pharmacol. 2010a;160:657–668. doi: 10.1111/j.1476-5381.2010.00769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay P, Rajesh M, Pan H, Patel V, Mukhopadhyay B, Batkai S, et al. Cannabinoid-2 receptor limits inflammation, oxidative/nitrosative stress, and cell death in nephropathy. Free Radic Biol Med. 2010b;48:457–467. doi: 10.1016/j.freeradbiomed.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivetta E, Mallozzi C, Ruggieri V, Pietraforte D, Federico M, Sanchez M. HIV-1 Nef induces p47(phox) phosphorylation leading to a rapid superoxide anion release from the U937 human monoblastic cell line. J Cell Biochem. 2009;106:812–822. doi: 10.1002/jcb.22041. [DOI] [PubMed] [Google Scholar]
- Pang PH, Lin YH, Lee YH, Hou HH, Hsu SP, Juan SH. Molecular mechanisms of p21 and p27 induction by 3-methylcholanthrene, an aryl-hydrocarbon receptor agonist, involved in antiproliferation of human umbilical vascular endothelial cells. J Cell Physiol. 2008;215:161–171. doi: 10.1002/jcp.21299. [DOI] [PubMed] [Google Scholar]
- Pfeifhofer C, Kofler K, Gruber T, Tabrizi NG, Lutz C, Maly K, et al. Protein kinase C theta affects Ca2+ mobilization and NFAT cell activation in primary mouse T cells. J Exp Med. 2003;197:1525–1535. doi: 10.1084/jem.20020234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pivot X, Cals L, Cupissol D, Guardiola E, Tchiknavorian X, Guerrier P, et al. Phase II trial of a paclitaxel-carboplatin combination in recurrent squamous cell carcinoma of the head and neck. Oncology. 2001;60:66–71. doi: 10.1159/000055299. [DOI] [PubMed] [Google Scholar]
- Raghavendra PB, Sreenivasan Y, Ramesh GT, Manna SK. Cardiac glycoside induces cell death via FasL by activating calcineurin and NF-AT, but apoptosis initially proceeds through activation of caspases. Apoptosis. 2007;12:307–318. doi: 10.1007/s10495-006-0626-3. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Rosell R, Skrzypski M, Jassem E, Taron M, Bartolucci R, Sanchez JJ, et al. BRCA1: a novel prognostic factor in resected non-small-cell lung cancer. PLoS ONE. 2007;2:e1129. doi: 10.1371/journal.pone.0001129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sue YM, Cheng CF, Chang CC, Chou Y, Chen CH, Juan SH. Antioxidation and anti-inflammation by haem oxygenase-1 contribute to protection by tetramethylpyrazine against gentamicin-induced apoptosis in murine renal tubular cells. Nephrol Dial Transplant. 2009;24:769–777. doi: 10.1093/ndt/gfn545. [DOI] [PubMed] [Google Scholar]
- Wang H, Li M, Rinehart JJ, Zhang R. Pretreatment with dexamethasone increases antitumor activity of carboplatin and gemcitabine in mice bearing human cancer xenografts: in vivo activity, pharmacokinetics, and clinical implications for cancer chemotherapy. Clin Cancer Res. 2004;10:1633–1644. doi: 10.1158/1078-0432.ccr-0829-3. [DOI] [PubMed] [Google Scholar]
- Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, et al. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- Zhang K, Li N, Chen Z, Shao K, Zhou F, Zhang C, et al. High expression of nuclear factor of activated T cells in Chinese primary non-small cell lung cancer tissues. Int J Biol Markers. 2007;22:221–225. doi: 10.1177/172460080702200310. [DOI] [PubMed] [Google Scholar]