

Abstract
BACKGROUND AND PURPOSE
The Ca2+ paradox is an important phenomenon associated with Ca2+ overload-mediated cellular injury in myocardium. The present study was undertaken to elucidate molecular and cellular mechanisms for the development of the Ca2+ paradox.
EXPERIMENTAL APPROACH
Fluorescence imaging was performed on fluo-3 loaded quiescent mouse ventricular myocytes using confocal laser scanning microscope.
KEY RESULTS
The Ca2+ paradox was readily evoked by restoration of the extracellular Ca2+ following 10–20 min of nominally Ca2+-free superfusion. The Ca2+ paradox was significantly reduced by blockers of transient receptor potential canonical (TRPC) channels (2-aminoethoxydiphenyl borate, Gd3+, La3+) and anti-TRPC1 antibody. The sarcoplasmic reticulum (SR) Ca2+ content, assessed by caffeine application, gradually declined during Ca2+-free superfusion, which was further accelerated by metabolic inhibition. Block of SR Ca2+ leak by tetracaine prevented Ca2+ paradox. The Na+/Ca2+ exchange (NCX) blocker KB-R7943 significantly inhibited Ca2+ paradox when applied throughout superfusion period, but had little effect when added for a period of 3 min before and during Ca2+ restoration. The SR Ca2+ content was better preserved during Ca2+ depletion by KB-R7943. Immunocytochemistry confirmed the expression of TRPC1, in addition to TRPC3 and TRPC4, in mouse ventricular myocytes.
CONCLUSIONS AND IMPLICATIONS
These results provide evidence that (i) the Ca2+ paradox is primarily mediated by Ca2+ entry through TRPC (probably TRPC1) channels that are presumably activated by SR Ca2+ depletion; and (ii) reverse mode NCX contributes little to the Ca2+ paradox, whereas inhibition of NCX during Ca2+ depletion improves SR Ca2+ loading, and is associated with reduced incidence of Ca2+ paradox in mouse ventricular myocytes.
Keywords: Ca2+ paradox, transient receptor potential canonical channel, sarcoplasmic reticulum, Na+/Ca2+ exchange
Introduction
The Ca2+ paradox (Zimmerman and Hülsmann, 1966), which rapidly develops upon restoration of extracellular Ca2+ following Ca2+-free superfusion, has many features in common with cellular damage associated with reperfusion of ischaemic myocardium, including the elevation of intracellular Ca2+, development of contracture, loss of mechanical and electrical activity, depletion of high-energy phosphate stores, and release of intracellular enzymes (Chapman and Tunstall, 1987; Piper, 2000). The Ca2+ paradox has therefore been regarded as an important experimental model for studying the morphological, electrophysiological and biochemical basis of myocardial injury associated with Ca2+ overload. However, it has also been noted that there are some differences in the mechanisms of cellular injury due to Ca2+ paradox and those associated with ischaemia-reperfusion (Piper, 2000). Several structural and functional disorders have been suggested to mediate the Ca2+ paradox, such as a weakening of the cell membrane, incomplete mechanical uncoupling between myocytes and intracellular Na+ accumulation leading to the reverse-mode activation of the Na+/Ca2+ exchange (NCX) (Chapman and Tunstall, 1987; Chatamra and Chapman, 1996; Piper, 2000). However, there is still considerable controversy as to the precise ionic and cellular basis for the development of Ca2+ overload during the Ca2+ paradox (Busselen, 1987; Chapman and Tunstall, 1987; Chatamra and Chapman, 1996; Jansen et al., 1998; Van Echteld et al., 1998; Piper, 2000).
The transient receptor potential canonical (TRPC) channels are Ca2+-permeable non-selective cation channels widely expressed in diverse cell types (Nilius et al., 2007; Vassort and Alvarez, 2009). TRPC channels comprise seven isoforms (TRPC1-7; Alexander et al., 2009) and all isoforms except TRPC2 have been found in mammalian heart at mRNA and/or protein levels (Ju et al., 2007; Ohba et al., 2007; Seth et al., 2009; Vassort and Alvarez, 2009). While some of the TRPC channels can be activated by several stimuli, such as diacyl glycerol, mechanical stretch and redox processes (Poteser et al., 2006), TRPC channels are typically activated following depletion of endoplasmic/sarcoplasmic reticulum (ER/SR) Ca2+ stores caused by stimulation of Ca2+ release or inhibition of Ca2+ uptake. TRPC channels are therefore implicated in the Ca2+ entry across the plasma membrane known as store-operated Ca2+ entry (SOCE; Xu and Beech, 2001; Rosado et al., 2002; Vazquez et al., 2004; Beech, 2005; Nilius et al., 2007; Vassort and Alvarez, 2009). There is accumulating evidence that TRPC channels mediate many physiological and pathological processes, including the activation of transcription factors, vascular contractility, platelet activation, apoptosis and cardiac automaticity, hypertrophy, and arrhythmias (Rosado et al., 2002; Beech, 2005; Ju et al., 2007; Nilius et al., 2007; Ohba et al., 2007; Seth et al., 2009; Vassort and Alvarez, 2009).
The present study was undertaken to elucidate the molecular and cellular mechanisms underlying the development of the Ca2+ paradox in mouse ventricular myocytes. Our results show for the first time that TRPC channels, presumably activated through the SR Ca2+ depletion that occurs during Ca2+-free superfusion, contribute to the development of the Ca2+ paradox.
Methods
Preparation of mouse ventricular myocytes
The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no. 85-23, revised 1996) and all protocols were approved by the institution's Animal Care and Use Committee (2008-11-7). Ventricular myocytes were isolated from hearts of adult C57BL/6J mice (Charles River Japan) using an enzymatic dissociation procedure, as described previously (Shioya, 2007). Briefly, 7- to 10-week-old mice (20–25 g body weight) were killed by sodium pentobarbital overdose (300 mg·kg−1, i.p.) with heparin (8000 U·kg−1, i.p.). The hearts were rapidly excised, cannulated via the ascending aorta and perfused in a retrograde manner at 37°C, initially with normal Tyrode solution for 3 min and then with cell isolation buffer (CIB) supplemented with 0.4 mmol·L−1 EGTA, for 2–3 min. This was followed by 8–10 min of perfusion with enzyme I solution (CIB supplemented with 1 mg·mL−1 collagenase, 0.06 mg·mL−1 trypsin, 0.06 mg·mL−1 protease and 0.3 mmol·L−1 CaCl2). After perfusion, the ventricles were removed, chopped into small pieces and further digested at 37°C for 10 min in enzyme II solution [CIB supplemented with 1 mg·mL−1 collagenase, 0.06 mg·mL−1 trypsin, 0.06 mg·mL−1 protease, 2 mg·mL−1 bovine serum albumin (BSA) and 0.7 mmol·L−1 CaCl2]. The supernatant was centrifuged (3 min at 14×g) and the myocyte pellet was resuspended in 15 mL of CIB supplemented with 2 mg·mL−1 BSA and 1.2 mmol·L−1 CaCl2. The myocytes were incubated for 10 min, centrifuged (3 min at 14×g) and resuspended in normal Tyrode solution supplemented with 2 mg·mL−1 BSA and antibiotics (penicillin/streptomycin). Isolations were excluded if the fraction of rod-shaped viable myocytes was below 50%. Myocytes were used for experiments within 8 h after dissociation. A previous study has confirmed that mouse ventricular myocytes isolated in this way have normal electrophysiological and contractile properties (Shioya, 2007).
Solutions and chemicals
Normal Tyrode solution contained (in mmol·L−1) 140 NaCl, 5.4 KCl, 1.8 CaCl2, 0.5 MgCl2, 0.33 NaH2PO4, 5.5 glucose and 5 HEPES (pH adjusted to 7.4 with NaOH). The nominally Ca2+-free Tyrode solution was prepared by simply omitting CaCl2 (no added EGTA) from the normal Tyrode solution. CIB contained (in mmol·L−1) 130 NaCl, 5.4 KCl, 0.5 MgCl2, 0.33 NaH2PO4, 22 glucose, 25 HEPES (pH adjusted to 7.4 with NaOH) and 50 U·mL−1 bovine insulin. The extracellular solution used to measure whole-cell TRPC currents was K+-free Tyrode solution supplemented with nisoldipine, which contained (in mmol·L−1) 140 NaCl, 1.8 CaCl2, 0.5 MgCl2, 0.33 NaH2PO4, 5.5 glucose, 0.001 nisoldipine and 5 HEPES (pH adjusted to 7.4 with NaOH). The pipette solution contained (in mmol·L−1) 90 Cs-aspartate, 30 CsCl, 20 tetraethylammonium chloride (TEA-Cl), 2 MgCl2, 5 Tris-ATP, 0.1 Li2-GTP, 5 EGTA, 2 CaCl2 and 5 HEPES (pH adjusted to 7.2 with CsOH). The concentration of free Ca2+ in the pipette solution was calculated to be approximately 0.1 µmol·L−1 (Fabiato and Fabiato, 1979; Tsien and Rink, 1980).
Collagenase (Type 2) was obtained from Worthington Biochemical Corporation (Lakewood, NJ, USA), and trypsin, protease, BSA and bovine insulin were from Sigma (St. Louis, MO, USA). Fluo-3 acetoxymethyl ester (fluo-3 AM) was from Dojin Chemicals (Kumamoto, Japan). Rabbit anti-TRPC1 antibody directed against an extracellular epitope of human TRPC1 (ACC-010), rabbit anti-TRPC3 antibody directed against an intracellular C-terminal epitope of mouse TRPC3 (ACC-016), rabbit anti-TRPC4 antibody directed against an intracellular C-terminal epitope of mouse TRPC4 (ACC-018), rabbit anti-TRPC5 antibody directed against an intracellular epitope of human TRPC5 (ACC-020) were from Alomone Laboratories (Jerusalem, Israel), and normal rabbit IgG was from Santa Cruz Biotechnology (Santa Cruz, CA, USA). AlexaFluor® 488-conjugated anti-rabbit IgG was from Molecular Probes (Eugene, OR, USA).
Various test compounds were added to normal Tyrode and/or nominally Ca2+-free Tyrode solutions, as indicated. These included: thapsigargin (Wako Pure Chemical Industries, Osaka, Japan), 2-aminoethoxydiphenyl borate (2-APB; Tocris Cookson Inc., Ellisville, MO, USA), GdCl3 (Sigma), LaCl3 (Nacalai tesque, Kyoto, Japan), SKF-96365 (Sigma), verapamil (Sigma), KB-R7943 (Tocris), 2,4-dinitrophenol (DNP, Wako Pure Chemical Industries), Na2-ATP (ATP, Sigma), uridine 5′-triphosphate (trisodium salt, UTP, Sigma), caffeine (Sigma) and tetracaine (Sigma). Concentrated stock solution was made for thapsigargin (10 mmol·L−1), 2-APB (20 mmol·L−1) and KB-R7943 (5 mmol·L−1) in dimethyl sulphoxide, verapamil (20 mmol·L−1) in ethanol, and GdCl3 (100 mmol·L−1), LaCl3 (100 mmol·L−1) and SKF-96365 (10 mmol·L−1) in distilled water. These chemicals were stored in aliquots at −20°C. DNP, ATP, UTP, caffeine and tetracaine were directly added to the bathing solutions.
Fluo-3 fluorescence imaging with laser scanning confocal microscope
Fluo-3 fluorescence images were obtained from quiescent (not paced) mouse ventricular myocytes because myocytes failed to respond to electrical stimulation during superfusion with nominally Ca2+-free Tyrode solution. Ventricular myocytes were loaded with 5 µmol·L−1 fluo-3 AM for 20 min at 37°C and were washed to remove excess extracellular dye in normal Tyrode solution supplemented with BSA. Fluo-3 loaded myocytes were then resuspended in normal Tyrode solution supplemented with 2 mg·mL−1 BSA for an additional 30 min to allow for the intracellular hydrolysis of fluo-3 AM before experiments. An aliquot of fluo-3 loaded myocytes was allowed to settle onto the glass bottom of a recording chamber (0.5 mL in volume) mounted on the stage of an Eclipse TE2000-E inverted microscope (Nikon, Tokyo, Japan), equipped with a C1si spectral imaging confocal laser scanning system (Nikon). The chamber was continuously perfused with bath solution at a constant rate of 2–3 mL·min−1 at room temperature (23–25°C). The myocytes were excited with an argon laser beam (wavelength 488 nm) at 0.4 or 30 s intervals, and data were collected for emission intensity at wavelength of 515 nm. Fluo-3 fluorescence images were analysed frame by frame using a Nikon EZ-C1 software to calculate average intensity in each myocyte, which was used as an estimate of intracellular Ca2+ levels. Fluo-3 fluorescence intensity (F) was expressed either as arbitrary units (a.u., Figures 1, 3, 4 and 6) or relative value (F/F0) compared with initial value obtained just before application of caffeine (F0, Figures 7 and 9). All intensity values were calculated by subtracting the background fluorescence. The ratio between the length and width was also measured in each myocyte image, and a decrease in length/width ratio of <2 was defined as injured or dead.
Figure 1.
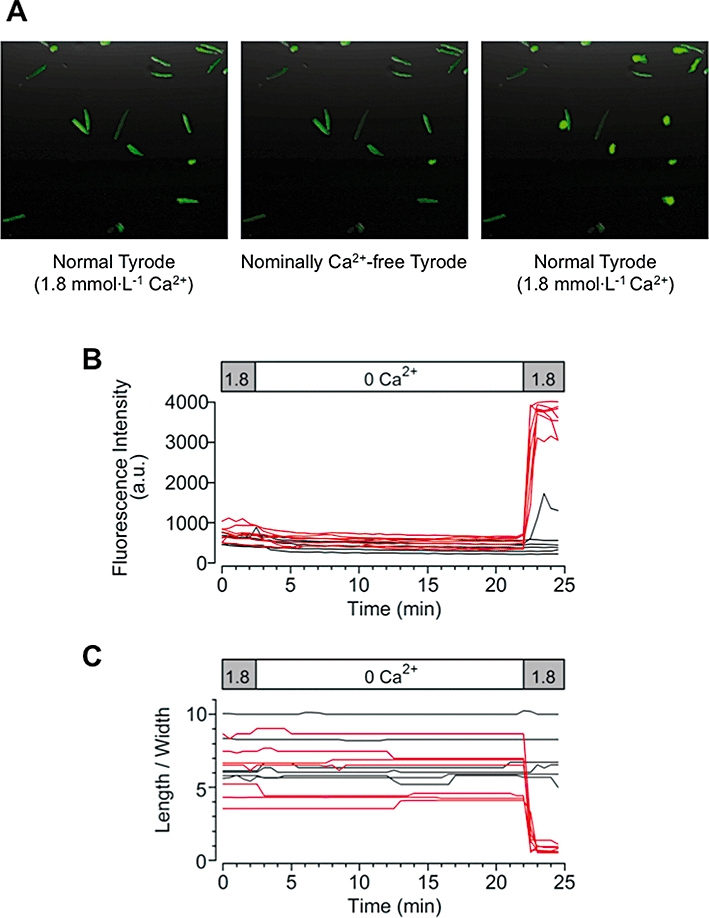
Ca2+ paradox detected in mouse ventricular myocytes. (A) Ventricular myocytes loaded with fluo-3 were successively superfused, initially with normal Tyrode solution for 5 min, then with nominally Ca2+-free Tyrode solution for 20 min, and again with normal Tyrode solution. Fluo-3 fluorescence images within the same field of view that were collected during the respective superfusion, as indicated. Time courses of changes in fluo-3 fluorescence intensity (B) and cell morphology (C) plotted individually for each of the 13 rod-shaped viable myocytes, obtained from the experiment shown in (A).
Figure 3.

Effects of transient receptor potential canonical (TRPC) channel blockers on the occurrence of Ca2+ paradox. Time course of changes in fluo-3 fluorescence intensity (A) and length/width ratio (B), calculated from fluorescence image obtained every 30 s for each of 15 myocytes, during the presence of 2-APB (10 µmol·L−1) throughout the superfusion period. (C) Concentration-dependent inhibition of the Ca2+ paradox by 2-APB, fitted with a Hill equation yielding an IC50 of 3.6 µmol·L−1 and nH of 2.6. (D) Effects of various blockers of TRPC channels and L-type Ca2+ channel on the occurrence of Ca2+ paradox. **P < 0.01 compared with control. There were no significant differences between the effects of Gd3+ and La3+ at 10 or 100 µmol·L−1.
Figure 4.
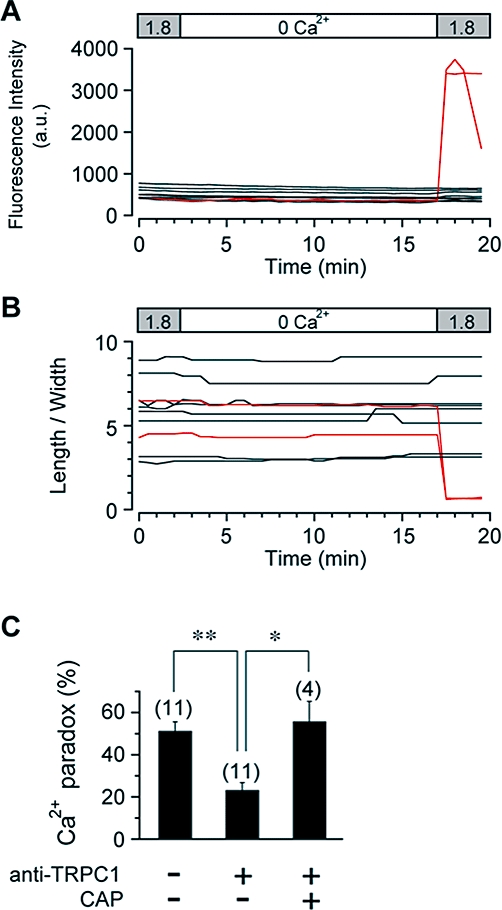
Prevention of Ca2+ paradox by anti-TRPC1 antibody. Ventricular myocytes were preincubated with anti-TRPC1 antibody (15 µg·mL−1) without or with control antigen peptide (CAP) for 15 min. Time courses of changes in fluo-3 fluorescence (A) and length/width ratio (B) were recorded every 30 s from each of 10 anti-TRPC1-pretreated myocytes within the same field of view during superfusion as indicated. (C) Occurrence of Ca2+ paradox in ventricular myocytes in control and after preincubation with anti-TRPC1 antibody without or with CAP. *P < 0.05 and **P < 0.01 compared with anti-TRPC1-pretreated myocytes without CAP.
Figure 6.
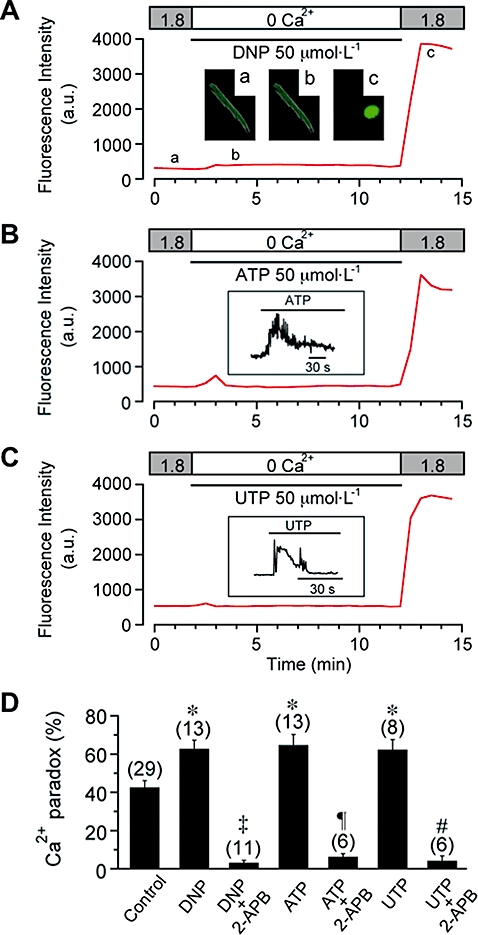
Potentiation of Ca2+ paradox by metabolic inhibition or by the presence of extracellular ATP and UTP. (A) Effect of metabolic inhibition during Ca2+ depletion on the Ca2+ paradox. DNP (50 µmol·L−1) was added (and glucose was removed) during Ca2+ depletion, as indicated. Inset shows fluorescence images at time points (a, b, c) indicated in (A). (B) and (C) Effect of extracellular ATP (50 µmol·L−1, B) and UTP (50 µmol·L−1, C) during Ca2+ depletion on the Ca2+ paradox. Inset shows fluorescence signals (acquired every 0.4 s) displaying Ca2+ transient evoked by ATP (B) and UTP (C) on an expanded time scale. Fluorescence intensity was measured every 30 s in experiments shown in panels (A) (B) and (C). (D) Potentiation of Ca2+ paradox by DNP (62.8 ± 4.5%, n = 13, N = 4), ATP (64.8 ± 5.5%, n = 13, N = 5) and UTP (62.4 ± 5.2%, n = 8, N = 3), and its inhibition by 2-APB (3.2 ± 1.2%, n = 11, N = 2; 6.3 ± 1.6%, n = 6, N = 2; and 4.2 ± 2.5%, n = 6, N = 3 respectively). *P < 0.05 compared with control (42.6 ± 3.5%, n = 29, N = 9); ‡P < 0.01 compared with DNP without 2-APB; ¶P < 0.01 compared with ATP without 2-APB; #P < 0.01 compared with UTP without 2-APB. There was no significant difference between the ATP and UTP groups.
Figure 7.
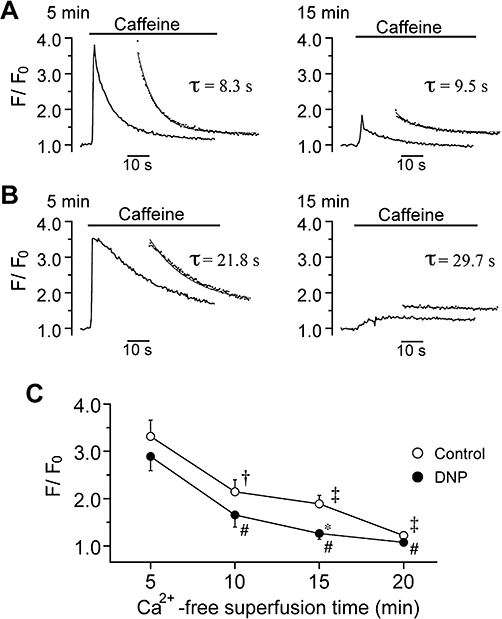
Depletion of sarcoplasmic reticulum Ca2+ content during Ca2+-free superfusion. Ca2+ transient evoked by bath application of caffeine (10 mmol·L−1) after 5 (left panel) and 15 min (right) of Ca2+-free superfusion without (A) and with (B) 50 µmol·L−1 DNP. Fluorescence signals were continuously acquired every 0.4 s. The inset in each panel shows single exponential fit (continuous line) to the decay of caffeine-induced Ca2+ transient (dotted points), yielding τ as indicated. (C) Peak amplitude of caffeine-induced Ca2+ transient was measured with reference to the baseline value prior to caffeine application (F/F0) and plotted as a function of Ca2+-free superfusion time, in control and in the presence of DNP. †P < 0.05 and ‡P < 0.01 compared with 5 min of superfusion in control. #P < 0.05 compared with 5 min of superfusion with DNP. *P < 0.05 compared with control (at 15 min).
Figure 9.
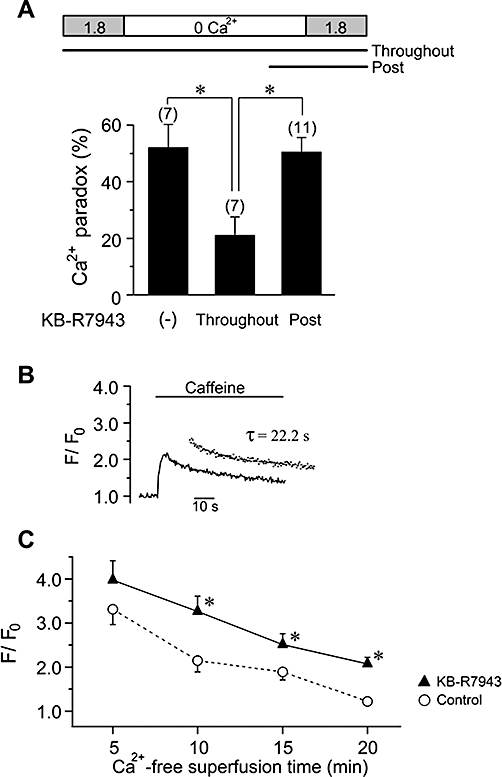
Functional linkage of NCX and Ca2+ paradox. (A) The Ca2+ paradox evoked by Ca2+ restoration after 15 min of Ca2+-free superfusion, in the absence and presence of KB-R7943 (5 µmol·L−1) applied throughout the superfusion period (Throughout) or for a period of 3 min before and during Ca2+ restoration (Post), as indicated. *P < 0.05, compared with Throughout. (B) Caffeine-induced Ca2+ transient after 15 min of Ca2+-free superfusion with KB-R7943 (5 µmol·L−1). Inset shows single exponential fit to the decay, yielding τ of 22.2 s. (C) Peak amplitude of Ca2+ transient with reference to the baseline value (F/F0) plotted against Ca2+-free superfusion time, in control (the data are the same as in Figure 7C) and in the presence of KB-R7943. *P < 0.05 compared with control at each time.
Fluo-3 loaded ventricular myocytes were successively superfused, initially with normal Tyrode solution for 5 min, then with nominally Ca2+-free Tyrode solution for 10–20 min, and again with normal Tyrode solution. In most experiments, 10–20 rod-shaped viable myocytes were observed within a single field of view during the initial superfusion with normal Tyrode solution. Ventricular myocytes that generated spontaneous Ca2+ waves during initial superfusion with normal Tyrode and/or subsequent superfusion with nominally Ca2+-free Tyrode solution were excluded from the analysis (less than approximately 2% of total viable myocytes). The data (fluo-3 fluorescence and length/width ratio) for initial superfusion with normal Tyrode solution were shown for the latter 2.5 min, and those for ventricular myocytes that developed the Ca2+ paradox were marked red in the Figures. The periods of exposure to various reagents and changes in extracellular Ca2+ concentrations between 0 (nominally Ca2+-free Tyrode solution) and 1.8 mmol·L−1 (normal Tyrode solution) are denoted by horizontal bars or boxes in the Figures. The concentration-response curve for inhibitory action of 2-APB or tetracaine on the Ca2+ paradox was drawn by a least-squares fit of a Hill equation: percentage incidence of Ca2+ paradox = 1/(1 + ([D]/IC50)nH), where [D] is the drug concentration, IC50 is the concentration of the drug causing a half-maximal inhibition and nH is the Hill coefficient.
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde in phosphate buffered saline (PBS) for 30 min at room temperature and were washed three times with PBS. Fixed cells were treated with 0.2% Triton X-100 and 10% BSA in PBS for 1 h and then incubated with primary antibodies for 15–17 h at 4°C. The cells were then washed with PBS and incubated with secondary antibodies for 3 h at room temperature. The primary antibodies were: human anti-TRPC1 (1:50 dilution), mouse anti-TRPC3 (1:50 dilution) and mouse anti-TRPC4 (1:50 dilution). Secondary antibody was AlexaFluor® 488-conjugated anti-rabbit IgG. Fluorescence images were acquired using a Nikon C1si confocal laser scanning system on an inverted microscope (TE2000-E, Nikon).
Whole-cell patch-clamp recordings
Whole-cell membrane currents (Hamill et al., 1981) were recorded from isolated mouse ventricular myocytes using an EPC-8 patch-clamp amplifier (HEKA, Lambrecht, Germany). Fire-polished pipettes pulled from borosilicate glass capillaries (Narishige Scientific Instrument Lab., Tokyo, Japan) had a resistance of 2.0–3.5 MΩ when filled with the pipette solution. An aliquot of cell (ventricular myocyte) suspension was transferred to a recording chamber (0.5 mL in volume) mounted on the stage of a Nikon TMD-300 inverted microscope and was allowed to adhere lightly to the glass bottom for at least 1–2 min. The chamber was continuously perfused at a constant rate of 2 mL·min−1 with bath solutions at 34–36°C. The voltage ramp protocol (dV·dt−1=±0.25 V·s−1) was repeated every 8 s and consisted of three phases: an initial +90 mV depolarizing phase from a holding potential of −40 mV, a second hyperpolarizing phase of −160 mV and then a third phase returning to the holding potential. The current-voltage (I-V) relationship was measured during the second hyperpolarizing phase. Voltage-clamp protocols and data acquisition were controlled with PATCHMASTER software (Version 1.03, HEKA), and current records were filtered at 1 kHz, digitized at 5 kHz through an LIH-1600 interface (HEKA), and stored on a Macintosh computer. Cell membrane capacitance (Cm) was calculated from the capacitive transients elicited by 20-ms voltage-clamp steps (±5 mV) from a holding potential of −40 mV, using the following relationship (Bénitah et al., 1993): Cm=τcI0/ΔVm (1 −I∞/I0), where τc is the time constant of the capacitive transient, I0 is the initial peak current amplitude, ΔVm is the amplitude of voltage step (5 mV) and I∞ is the steady-state current value. The sampling rate for these measurements of Cm was 50 kHz with a low-pass 10 kHz filter. The average Cm for mouse ventricular myocytes used in the present study was 153.8 ± 8.5 pF (n = 28, N = 10). To account for differences in cell size, the current amplitude was normalized to Cm in each cell and presented as current density (in pA·pF−1).
Statistical analysis
Data values are expressed as mean ± SEM, with the number of animals (cell isolations) and experiments indicated by N and n respectively. On the bar graphs, the number of experiments is shown in parentheses. Statistical comparisons between two groups were evaluated by Mann–Whitney U-test and comparisons among multiple groups were performed by Kruskal–Wallis test followed by Mann–Whitney U-test. A value of P < 0.05 was considered statistically significant.
Results
Ca2+ paradox observed in mouse ventricular myocytes
Figure 1 demonstrates a typical experiment showing the effects of re-addition of extracellular Ca2+ on ventricular myocytes, obtained by examining fluo-3 fluorescence images collected at 30 s intervals. Fluo-3-loaded ventricular myocytes were initially stabilized by superfusion with normal (Ca2+-containing) Tyrode solution for 5 min, and then were successively superfused with nominally Ca2+-free Tyrode solution for 20 min and subsequently with normal Tyrode solution. Figure 1A illustrates fluo-3 fluorescence images of ventricular myocytes within the same field of view, and 13 of the rod-shaped viable myocytes were detected during the initial superfusion with normal Tyrode solution (left panel). After the superfusate was switched to nominally Ca2+-free Tyrode solution (middle panel), fluorescence intensity of ventricular myocytes gradually declined to 73.2 ± 3.1% of baseline level (in a.u.; Figure 1B), while cell morphology as measured by the length/width ratio was not appreciably affected (100.6 ± 2.0% of baseline; Figure 1C), during the 20 min of superfusion. These results indicate that intracellular free Ca2+ levels decreased to some extent during the Ca2+-free superfusion. However, on return to normal Tyrode solution, the fluorescent intensity was abruptly elevated in 7 out of 13 myocytes (53.8%), accompanied by hypercontracture as determined by a decrease in length/width ratio of <2 (Figure 1A, right panel; Figure 1B,C). There was no recovery from hypercontracture during a 10 min period of Ca2+ restoration (data not shown). In the present study, an irreversible hypercontracture due to elevated cytosolic Ca2+ was defined as the Ca2+ paradox. The occurrence of the Ca2+ paradox upon re-addition of extracellular Ca2+ was progressively but insignificantly increased by prolonging the duration of the Ca2+-free superfusion time from 10 to 20 min (48.3 ± 6.6%, 55.4 ± 6.1% and 64.3 ± 5.9% at 10, 15 and 20 min, respectively, n = 11–18, N = 11–18).
Functional expression of TRPC channels in mouse ventricular myocytes
The possibility that Ca2+ entry through TRPC channels contributes to the Ca2+ paradox was examined in the following experiments. The functional expression of TRPC channels was initially tested by whole-cell patch-clamp experiments. The sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA) inhibitor thapsigargin has been shown to activate TRPC channels by passively depleting the SR Ca2+ stores in various cell types (Xu and Beech, 2001; Rosado et al., 2002; Vazquez et al., 2004; Beech, 2005; Nilius et al., 2007; Vassort and Alvarez, 2009). As demonstrated in Figure 2A,B, bath application of thapsigargin increased the membrane current during the voltage-ramp protocol from +50 to −110 mV, which exhibited an almost linear I-V relationship with a reversal potential of ∼0 mV (Figure 2C, b-a). This increase in membrane current was completely abolished by subsequent addition of the TRPC channel blocker 2-APB (Figure 2A–C; Bootman et al., 2002; Flemming et al., 2003; Liu et al., 2007; Zhou et al., 2007). Based on these electrophysiological and pharmacological properties, it seems reasonable to suggest that this thapsigargin-activated current probably represents TRPC channel currents.
Figure 2.
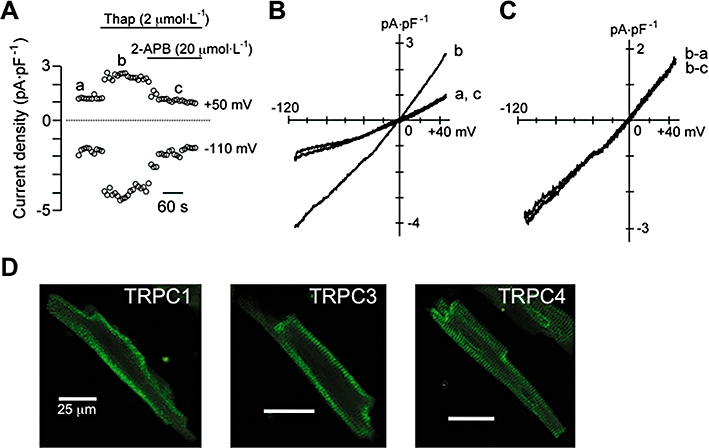
Functional expression of transient receptor potential canonical (TRPC) channels in mouse ventricular myocytes. (A–C) Activation of TRPC current by thapsigargin recorded under conditions where Na+, Ca2+ and K+ channel currents were minimized. (A) Time course of changes in membrane current measured at +50 and −110 mV during the voltage-ramp protocol (from +50 to −110 mV), before and during exposure to thapsigargin (Thap, 2 µmol·L−1), without and then with 2-APB (20 µmol·L−1). (B) I-V relationships measured at time points (a, b, c) indicated in (A). (C) Difference currents obtained by digital subtraction as indicated (b-a: thapsigargin-activated current; b-c: 2-APB-sensitive current). (D) Immunostaining of TRPC1, TRPC3 and TRPC4. Scale bar in all panels, 25 µm.
Immunocytochemistry experiments using anti-TRPC1, TRPC3 and TRPC4 antibodies detected immunofluorescence signals predominantly in the peripheral region of the myocytes (Figure 2D), thus supporting the expression of TRPC1, TRPC3 and TRPC4 channel proteins in mouse ventricular myocytes, consistent with previous observations on these cells (Fauconnier et al., 2007; Williams and Allen, 2007; Seth et al., 2009).
Contribution of TRPC channels to the Ca2+ paradox
To elucidate whether Ca2+ entry through TRPC channels mediates the Ca2+ paradox, we examined the effects of various TRPC channel blockers (2-APB, Gd3+, La3+ and SKF-96365) on changes in fluo-3 fluorescence images of ventricular myocytes during re-addition of extracellular Ca2+. In each experiment, 10–20 rod-shaped viable myocytes were typically observed within a single field of view during the initial superfusion with normal Tyrode solution, and intracellular Ca2+ levels (assessed by fluo-3 fluorescence) and cell morphology (length/width ratio) were examined in the absence and presence of each of these compounds in the superfusion media throughout the superfusion period.
Figure 3A,B shows the results of a representative experiment examining the effect of 2-APB (10 µmol·L−1) on the occurrence of the Ca2+ paradox. In the presence of 2-APB, the fluorescence intensity slightly declined during superfusion with nominally Ca2+-free Tyrode solution (79.1 ± 3.9% of baseline levels, 51 myocytes from four experiments) to an extent similar to control (72.1 ± 4.3%; 40 myocytes from four experiments, N.S.), indicating that there was little, if any, effect of 2-APB on intracellular Ca2+ levels during Ca2+-free superfusion. However, 2-APB markedly prevented the occurrence of the Ca2+ paradox upon restoration of extracellular Ca2+; only 1 out of 15 myocytes (6.7%) was judged to undergo the Ca2+ paradox, by an abrupt elevation of intracellular Ca2+ levels (Figure 3A) accompanied by a reduction of length/width ratio (Figure 3B).
The occurrence of Ca2+ paradox, measured in each experiment as percentage of hypercontracted myocytes compared with the total number of rod-shaped viable myocytes during initial superfusion with normal Tyrode solution, averaged 48.1 ± 3.2% (n = 38, N = 12) under control conditions (Figure 3D). Figure 3C illustrates the inhibitory action of 2-APB at concentrations between 1 and 10 µmol·L−1; the Ca2+ paradox was almost completely abolished by 10 µmol·L−1 2-APB (3.6 ± 2.5%, n = 9, N = 4). The data are well fitted by a Hill equation with a half-maximal inhibitory concentration (IC50) of 3.6 µmol·L−1 and closely resemble previous results obtained for the inhibitory action of 2-APB on SOCE (IC50, 5 µmol·L−1; Zhou et al., 2007). It should be noted that 2-APB also blocks the Ca2+ release from inositol-1,4,5-trisphosphate (IP3) receptors; however, this effect is only seen at a much higher concentration range with an IC50 of 42 µmol·L−1 (Zhou et al., 2007). The occurrence of the Ca2+ paradox was also almost totally blocked by Gd3+ and La3+ at 100 µmol·L−1 (5.5 ± 2.2%, n = 14, N = 6 and 3.4 ± 1.4%, n = 7, N = 4, respectively) but was only minimally affected by SKF-96365 (SKF) at 10 µmol·L−1 (39.6 ± 6.1%, n = 11, N = 4; Figure 3D). It should be added that both Gd3+ and La3+ at 10 µmol·L−1 significantly suppressed the Ca2+ paradox (11.4 ± 2.0%, n = 6, N = 2, and 14.1 ± 2.7%, n = 6, N = 2, respectively; data not shown). The L-type Ca2+ channel blocker verapamil (10 µmol·L−1) had no effect (50.2 ± 10.5%, n = 5, N = 2; Figure 3D). We also confirmed that the Ca2+ paradox was markedly prevented by the addition of La3+ (100 µmol·L−1) for a period of 3 min before and during Ca2+ re-addition (data not shown, 7.1 ± 2.6%, n = 8, N = 2), which is similar to the incidence of the Ca2+ paradox during the presence of La3+ (100 µmol·L−1) throughout the superfusion (3.4 ± 1.4%, n = 7, N = 4). These results are thus consistent with the view that Ca2+ entry through TRPC channels during Ca2+ re-addition primarily contributes to the Ca2+ paradox.
It has been shown that La3+ and Gd3+ at concentrations of 10–100 µmol·L−1 inhibit TRPC1 but potentiate TRPC4 and TRPC5 (Schaefer et al., 2000; Strübing et al., 2001; Flemming et al., 2003; Jung et al., 2003). Furthermore, SKF-96365 blocks multiple TRPC isoforms including TRPC3 and TRPC6, but has less of an inhibitory effect on TRPC1 (Halaszovich et al., 2000; Inoue et al., 2001; Flemming et al., 2003). As judged from these pharmacological profiles of TRPC isoforms, it seems likely that TRPC1 is predominantly involved in mediating the Ca2+ paradox in mouse ventricular myocytes.
Previous studies have demonstrated that the ability of TRPC1 to mediate SOCE is selectively blocked by external application of anti-TRPC1 antibody raised against the extracellular amino acid sequence 557–571, which is predicted to lie in the pore region of the channel. This antibody has therefore been used as a powerful tool to prove mammalian TRPC1 function (Xu and Beech, 2001; Rosado et al., 2002; Ahmmed et al., 2004; Beech, 2005). For example, store-operated currents through TRPC1 channels heterologously expressed in human microvessel endothelial cells are partially but significantly suppressed by pretreatment with 15 µg·mL−1 anti-TRPC1 antibody for 15 min (Ahmmed et al., 2004). The same pretreatment protocol was used in the present experiments. Figure 4A,B illustrates the effect of pretreatment with anti-TRPC1 antibody, where only 20% (2 out of 10) of ventricular myocytes exhibited the Ca2+ paradox upon re-addition of extracellular Ca2+ following a 15 min of Ca2+-free superfusion. As summarized in Figure 4C, the occurrence of the Ca2+ paradox was significantly suppressed by pretreatment with anti-TRPC1 antibody, which was completely reversed by pre-absorption with the control antigen peptide (CAP). These results further support the involvement of TRPC1 in Ca2+ entry associated with the Ca2+ paradox.
We also examined the effect of pretreatment with normal IgG as well as anti-TRPC4 and anti-TRPC5 antibodies raised against intracellular epitopes on the occurrence of the Ca2+ paradox evoked upon Ca2+ restoration following 15 min of Ca2+-free superfusion, as negative control experiments. As expected, there was no significant effect of IgG and these antibodies (Figure S1). These data, however, do not necessarily rule out the possible involvement of TRPC4 and TRPC5 channels in the Ca2+ paradox, because it seems unlikely that these anti-TRPC4 and anti-TRPC5 antibodies cross the cell membranes, react with their intracellular epitopes and block the function of TRPC4 and TRPC5 channels.
It is important to detect the presence of TRPC1 channel currents in mouse ventricular myocytes. We therefore examined the effect of pretreatment with anti-TRPC1 antibody on the thapsigargin (2 µmol·L−1)-induced TRPC currents using the whole-cell patch-clamp method. TRPC currents, as determined by the thapsigargin-activated current, were significantly reduced by pretreatment with anti-TRPC1 antibody, which was largely reversed by pre-absorption with CAP (Figure 5A,B). These observations indicate that the TRPC1 channel, which is thought to mediate the Ca2+ paradox (Figure 4), is actually functional in mouse ventricular myocytes.
Figure 5.

Inhibition of thapsigargin-induced transient receptor potential canonical (TRPC) current by anti-TRPC1 antibody. Ventricular myocytes were preincubated with anti-TRPC1 antibody without or with control antigen peptide (CAP) in a way similar to that of Figure 4. Membrane currents were measured using voltage ramps applied every 8 s before and during exposure to thapsigargin (2 µmol·L−1). (A) Superimposed I-V relationships of thapsigargin-induced TRPC currents, obtained by digital subtraction of current traces before and during exposure to thapsigargin, in ventricular myocytes in control and after preincubation with anti-TRPC1 antibody without or with CAP. (B) Current density of thapsigargin-induced TRPC current measured at −110 mV in ventricular myocytes in control and after preincubation with anti-TRPC1 antibody without or with CAP. *P < 0.05 compared with anti-TRPC1-pretreated myocytes without CAP (−0.81 ± 0.25 pA·pF−1; n = 11, N = 3). There was no significant difference between myocytes in control (−2.70 ± 0.55 pA·pF−1; n = 5, N = 3) and those pretreated with anti-TRPC1 antibody with CAP (−2.10 ± 0.57 pA·pF−1; n = 4, N = 3).
Enhancement of the Ca2+ paradox by metabolic inhibition or by the presence of extracellular ATP and UTP
Next, we examined the effect of metabolic inhibition during Ca2+-free superfusion on the occurrence of Ca2+ paradox. In the experiment of Figure 6A, ventricular myocytes were exposed to DNP (50 µmol·L−1)-containing, glucose-free solution during Ca2+ depletion, which was followed by superfusion with DNP-free, glucose-containing solution during Ca2+ restoration. In contrast to the control experiments (Figure 1B), the intracellular Ca2+ level was slightly but noticeably elevated during Ca2+-free superfusion under metabolic inhibition (Figure 6A), which may be ascribable to a reduction of Ca2+ extrusion via NCX associated with intracellular Na+ accumulation (Donoso et al., 1992) and a decreased uptake of Ca2+ to SR via SERCA (Kaplan et al., 1992). The re-addition of extracellular Ca2+ also evoked a rapid onset of hypercontracture due to elevated cytosolic Ca2+ levels (Figure 6A, c). As summarized in Figure 6D, the occurrence of the Ca2+ paradox was significantly potentiated by metabolic inhibition during Ca2+ depletion.
A number of reports have shown that extracellular ATP and UTP stimulate Gq protein-coupled, metabotropic P2Y receptors and thereby evoke Ca2+ release from SR through the formation of IP3, with a subsequent activation of SOCE in various cell types, including cardiac myocytes (Vassort and Alvarez, 2009). Furthermore, when cardiac myocytes are subjected to hypoxic or chemically ischaemic conditions, intracellular ATP and UTP have been suggested to permeate the cell membrane and to act on its plasma membrane receptors through autocrine/paracrine mechanisms (Dutta et al., 2004; Wihlborg et al., 2006; Alvarez et al., 2008). We have therefore examined the Ca2+ paradox when ATP or UTP was present during Ca2+ depletion (Figure 6B,C respectively). As expected, the addition of ATP or UTP (50 µmol·L−1) to nominally Ca2+-free Tyrode solution caused a transient elevation of intracellular Ca2+ levels (Figure 6B,C, inset), which may reflect IP3-induced Ca2+ release from SR. The occurrence of the Ca2+ paradox in the presence of ATP or UTP during Ca2+ depletion was significantly higher than the control (Figure 6D), thus showing that the Ca2+ paradox is enhanced by the presence of ATP or UTP during Ca2+ depletion. This enhancement of the Ca2+ paradox induced by metabolic inhibition or extracellular ATP/UTP was also almost totally abolished in the presence of 10 µmol·L−1 2-APB (Figure 6D), thus supporting the hypothesis that TRPC channels primarily mediate the Ca2+ paradox under these experimental conditions.
In addition to P2Y receptors, extracellular ATP also acts at ionotropic P2X receptors comprising ligand-gated cation channels (North and Barnard, 1997; Ralevic and Burnstock, 1998). However, extracellular ATP enhanced the Ca2+ paradox to an extent similar to extracellular UTP (Figure 6D) that does not activate P2X receptors. This observation therefore suggests that the potentiation of the Ca2+ paradox by extracellular ATP (Figure 6B) is mediated primarily through G protein-coupled P2Y receptors rather than through ionotropic P2X receptors.
The involvement of SR Ca2+ depletion in the development of the Ca2+ paradox
We next examined the possibility that the TRPC channel activation associated with the Ca2+ paradox arises from the depletion of SR Ca2+ content that may occur during the Ca2+-free superfusion period. To assess SR Ca2+ content, ventricular myocytes were exposed to caffeine (10 mmol·L−1) after 5, 10, 15 and 20 min of superfusion with nominally Ca2+-free Tyrode without or with DNP. As demonstrated in Figure 7A,B, bath application of caffeine evoked a transient elevation of intracellular Ca2+ levels; its peak amplitude provides an estimate of the SR Ca2+ content, while the decaying time course reflects Ca2+ extrusion via NCX (Callewaert et al., 1989). The peak amplitude of the Ca2+ transient was decreased by the prolongation of the Ca2+-free superfusion time, both in the absence (Figure 7A,C) and presence (Figure 7B,C) of DNP. In addition, peak Ca2+ amplitude was significantly reduced in the presence of DNP compared with control for a test duration of 15 min (Figure 7C). The SR Ca2+ content was thus found to decrease during Ca2+-free superfusion, consistent with previous observations in rat ventricular myocytes (Díaz et al., 1997), and this was further enhanced by metabolic inhibition.
The decay of the caffeine-induced Ca2+ transient was evaluated by fitting to a single exponential function to obtain a time constant (τ), and was found to be significantly decelerated by the presence of DNP at any of the test durations, compared with control (Figure 7A,B, 20.6 ± 2.2 vs. 9.3 ± 0.7 s after 5 min of Ca2+-free superfusion, P < 0.05; 31.8 ± 4.6 vs. 10.2 ± 0.9 s after 15 min of Ca2+-free superfusion, P < 0.05). A similar decelerated relaxation of the caffeine-induced Ca2+ transient during metabolic inhibition has been shown in guinea pig ventricular myocytes (Seki and MacLeod, 1995) and may reflect a compromised Ca2+ extrusion via NCX primarily due to elevated intracellular Na+ concentrations (Donoso et al., 1992). It should also be noted that, when the Ca2+-free superfusion time was prolonged, the decaying time course remained unchanged in control (Figure 7A), but became slower in the presence of DNP (Figure 7B), suggesting that NCX operating in its forward mode to extrude Ca2+ was not appreciably affected in control but was compromised under metabolic inhibition presumably by a progressive elevation of intracellular Na+ (Donoso et al., 1992).
Evidence has been provided for the presence of spontaneous diastolic Ca2+ leak from the SR through the cardiac ryanodine receptor (RyR2), even in intact ventricular myocytes (Györke et al., 1997; Shannon et al., 2002). It is probable that SR Ca2+ leak during Ca2+-free superfusion is responsible for SR Ca2+ depletion, with a subsequent activation of TRPC channels and development of the Ca2+ paradox. We therefore examined the effect of tetracaine, which potently blocks the Ca2+ leak through RyR2 and thereby preserves SR Ca2+ content, at concentrations of ≥ approximately 0.2 mmol·L−1 (Györke et al., 1997). Figure 8 illustrates the occurrence of the Ca2+ paradox in the absence and presence of tetracaine at concentrations between 0.1 and 1.0 mmol·L−1. The relationship was best fitted with a Hill equation with an IC50 of 0.29 mmol·L−1, which was close to the previous observation for the block of SR Ca2+ release channels by tetracaine (IC50 of 0.26 mmol·L−1; Györke et al., 1997); this suggests that tetracaine prevented the Ca2+ paradox by blocking SR Ca2+ leakage. Overall, the results shown in Figures 7 and 8 are all consistent with the view that the occurrence of the Ca2+ paradox is closely linked to the depletion of the SR Ca2+ content mediated by spontaneous Ca2+ leak from SR via RyR2.
Figure 8.
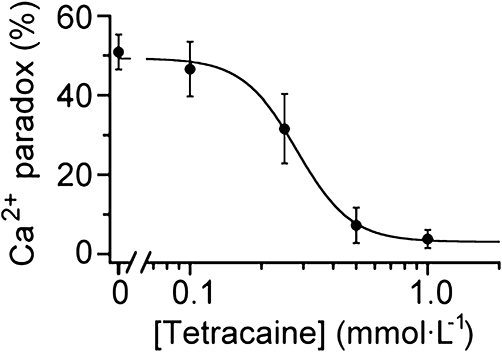
Prevention of Ca2+ paradox by tetracaine. Ventricular myocytes were exposed to tetracaine throughout the superfusion period. The incidence of Ca2+ paradox was plotted as a function of tetracaine concentration and was fitted with a Hill equation, yielding IC50 of 0.29 mmol·L−1 and nH of 3.0. Each data point represents mean ± SEM of 3–8 experiments from 2–4 cell isolations.
Functional linkage of NCX to the development of the Ca2+ paradox
We next addressed the question as to whether and how NCX is involved in the development of the Ca2+ paradox by using its inhibitor KB-R7943 (Kimura et al., 1999). The occurrence of the Ca2+ paradox was examined upon Ca2+ restoration following a 15 min period of Ca2+-free superfusion. When KB-R7943 (5 µmol·L−1) was added to the bath throughout the superfusion period, there was a significant decrease in the occurrence of the Ca2+ paradox (Control, 52.1 ± 8.1% n = 7, N = 5; Throughout, 21.1 ± 6.4%, n = 7, N = 4; P < 0.05; Figure 9A). To examine the possible contribution of Ca2+ entry via reverse mode activation of NCX to the Ca2+ paradox, KB-R7943 was added to the bath for a period of 3 min before and during the restoration of extracellular Ca2+. KB-R7943 applied in this way (Post) did not protect ventricular myocytes from the Ca2+ paradox (Post, 50.5 ± 5.1%, n = 11, N = 5; Figure 9A), which suggests that the Ca2+ paradox is not directly mediated by Ca2+ influx via reverse mode NCX activity.
To elucidate the mechanism for suppression of the Ca2+ paradox by KB-R7943 applied throughout, SR Ca2+ content was measured by caffeine application following 5, 10, 15 and 20 min of Ca2+-free superfusion with KB-R7943. Figure 9B illustrates a typical example of the caffeine-induced Ca2+ transient following a 15 min period of Ca2+-free superfusion in the presence of KB-R7943, and its peak amplitude and decaying time course were measured to estimate the SR Ca2+ content and NCX activity, respectively. As summarized in Figure 9C, SR Ca2+ content was better preserved by the presence of KB-R7943, after 10, 15 and 20 min of Ca2+-free superfusion, compared with control. It should also be noted that the Ca2+ transient decayed more slowly in the presence of KB-R7943, compared with its absence (τ, 28.9 ± 7.5 vs. 10.2 ± 0.9 s after 15 min of Ca2+-free superfusion, P < 0.05), which reflects partial inhibition of the forward mode NCX by KB-R7943. Thus, the inhibition of NCX during the Ca2+-free superfusion period improves SR Ca2+ loading, which may contribute at least partly to reduced incidence of the Ca2+ paradox observed when KB-R7943 was present throughout.
Discussion
Contribution of TRPC channels to the Ca2+ paradox
The present study examines the fluo-3 fluorescence images of quiescent mouse ventricular myocytes obtained using a confocal laser scanning microscope system, and demonstrates that a rapid onset of hypercontracture due to abrupt elevation of intracellular Ca2+ (Ca2+ paradox) can be readily evoked by the re-addition of extracellular Ca2+ following 10–20 min of superfusion with nominally Ca2+-free medium (Figure 1). The cellular events involved in the development of the Ca2+ paradox, as revealed by the present experiments, are illustrated in Figure 10 and are discussed individually. The Ca2+ paradox was prevented by 2-APB in a concentration-dependent manner with an IC50 of 3.6 µmol·L−1 (Figure 3C). As judged from the difference in concentration range at which 2-APB blocks TRPC channels and IP3 receptors (IC50 of 5 and 42 µmol·L−1, respectively; Zhou et al., 2007), it is reasonable to speculate that 2-APB affects TRPC channels rather than IP3 receptors to prevent the Ca2+ paradox. This notion is also supported by the suppression of the Ca2+ paradox by the other blockers of TRPC channels (Gd3+ and La3+; Figures 3D and 10).
Figure 10.
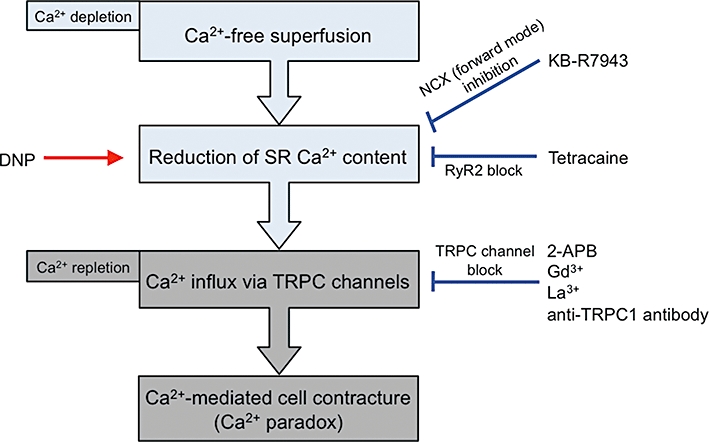
Diagram illustrating the cellular events that lead to the Ca2+ paradox in mouse ventricular myocytes. Reduction of sarcoplasmic reticulum (SR) Ca2+ content during Ca2+-free superfusion (Ca2+ depletion) is facilitated by DNP but is prevented by KB-R7943 (via inhibition of forward mode NCX) and by tetracaine (via RyR2 block). Reduction of SR Ca2+ content triggers Ca2+ influx through transient receptor potential canonical (TRPC) channels upon Ca2+ repletion. Ca2+ paradox is evoked by Ca2+ influx via TRPC channels, which is blocked by TRPC channel blockers (2-APB, Gd3+ and La3+) and anti-TRPC1 antibody. The Ca2+ paradox can be prevented by preserving the SR Ca2+ content and/or by blocking TRPC channels.
Furthermore, functional block of the TRPC1 with anti-TRPC1 antibody resulted in a partial but significant decrease in the Ca2+ paradox (Figure 4). Previous studies have demonstrated that due to its large molecule size, this anti-TRPC1 antibody partially (but not totally) blocks TRPC1 channel functions, as judged from the changes in intracellular Ca2+ concentrations (Ca2+ fluorescence studies; Xu and Beech, 2001; Rosado et al., 2002) or membrane currents (patch-clamp experiments; Ahmmed et al., 2004). It should be noted, however, that this anti-TRPC1 antibody displays a high degree of selectivity (Xu and Beech, 2001; Rosado et al., 2002; Ahmmed et al., 2004; Beech, 2005). The present data therefore suggest that the TRPC channels, probably TRPC1, predominantly mediate Ca2+ entry associated with the Ca2+ paradox in mouse ventricular myocytes. We also provide electrophysiological evidence to suggest the presence of TRPC current by whole-cell patch-clamp experiments with thapsigargin and 2-APB (Figure 2A–C). Furthermore, this thapsigargin-activated current was significantly (but not totally) blocked by pretreatment with anti-TRPC1 antibody, which was largely reversed by pre-absorption of the antibody with CAP (Figure 5). These findings support the presence and function of TRPC1 channels in mouse ventricular myocytes, consistent with a recent report (Seth et al., 2009). However, the functional role of TRPC1 and also other TRPC isoforms in the development of the Ca2+ paradox in cardiac myocytes should be fully examined by further work with genetic approaches, such as the use of dominant-negative TRPC channel isoforms and/or RNA interference.
In the present study, the Ca2+ paradox was almost totally abolished by the presence of the TRPC channel blocker (2-APB, Gd3+ or La3+) under control conditions (Figure 3C,D) and during metabolic inhibition (Figure 6D), which suggests a minimal, if any, contribution of non-specific membrane damage to the development of the Ca2+ paradox in mouse ventricular myocytes.
The present immunocytochemistry experiments confirm the expression of TRPC1, in addition to TRPC3 and TRPC4, in mouse ventricular myocytes (Figure 2D), consistent with previous studies (Fauconnier et al., 2007; Williams and Allen, 2007; Seth et al., 2009). It has been demonstrated that in aortic-banded animal models, TRPC1 is up-regulated and mediates the development of cardiac hypertrophy (Ohba et al., 2007; Seth et al., 2009). It is thus of interest to examine whether or not the Ca2+ paradox injury occurs more severely and/or more extensively in pressure overload-induced hypertrophied myocardium with TRPC1 overexpression.
It is generally accepted that TRPC channels are typically activated following the depletion of Ca2+ in SR (Xu and Beech, 2001; Rosado et al., 2002; Nilius et al., 2007; Vassort and Alvarez, 2009). The present experiments, using caffeine application, revealed that SR Ca2+ content was substantially decreased during Ca2+-free superfusion (Figure 7). Several studies have shown that there is a spontaneous Ca2+ leak from SR through RyR2 even in intact ventricular myocytes and that tetracaine blocks RyR2 in a concentration-dependent manner (IC50 of 0.26 mmol·L−1) with a subsequent increase of SR Ca2+ content (Györke et al., 1997; Shannon et al., 2002). We found that tetracaine was effective at preventing the Ca2+ paradox in a similar concentration range (IC50 of 0.29 mmol·L−1, Figure 8). Taken together, these results suggest that the reduction of SR Ca2+ content (probably via the RyR2-mediated Ca2+ leak) during Ca2+ depletion is critical in predisposing ventricular myocytes to the Ca2+ paradox upon Ca2+ repletion (Figure 10).
The present study further examined the Ca2+ paradox in some pathological conditions where the SR Ca2+ content is affected. Previous work has shown that under metabolic inhibition with sodium cyanide, SR Ca2+ loading is decreased in guinea pig ventricular myocytes (Seki and MacLeod, 1995), which appears to be largely due to an inhibition of Ca2+ uptake via SERCA (Kaplan et al., 1992). We found that the Ca2+ paradox was potentiated by metabolic inhibition during Ca2+-free superfusion (Figure 6D), which may be accounted for, at least partly, by a further reduction of SR Ca2+ content (Figures 7C and 10).
In recent years, evidence has been presented that stromal interacting molecule 1 (STIM1) that resides in ER/SR membranes senses internal Ca2+ store depletion and transmits it to the plasma membrane Orai1 (Liou et al., 2005; Roos et al., 2005; Feske et al., 2006), which constitutes all or part of the Ca2+ release-activated Ca2+ channels (Parekh, 2006). Although functional interaction of TRPC channels with STIM1 and/or Orai1 is currently still unresolved (Huang et al., 2006a; López et al., 2006; DeHaven et al., 2009), it will be interesting to examine whether and how STIM1 and/or Orai 1 play some role in the development of the Ca2+ paradox in cardiac myocytes.
Functional linkage of NCX to Ca2+ paradox
Previous workers have shown that other Ca2+ entry pathways, such as reverse mode NCX and L-type Ca2+ channels, mediate the Ca2+ paradox in the heart (Chapman and Tunstall, 1987; Chatamra and Chapman, 1996). It is generally accepted that intracellular Na+ accumulation during Ca2+ depletion is a prerequisite for the activation of reverse mode NCX. One of the main mechanisms for intracellular Na+ accumulation during Ca2+ depletion is a sustained Na+ entry via the L-type Ca2+ channels, which is pronounced when Ca2+ depletion is combined with Mg2+ depletion with divalent cation-chelating agent (Chapman and Tunstall, 1987; Van Echteld et al., 1998). However, the Ca2+ paradox in mouse ventricular myocytes appears to be independent of reverse mode activation of NCX arising from intracellular Na+ accumulation. Whereas verapamil (10 µmol·L−1) almost completely blocks Na+ entry through the L-type Ca2+ channels under Ca2+ depleted conditions in isolated ventricular myocytes (Imoto et al., 1985), the Ca2+ paradox was unaffected by this blocker at the same concentration (Figure 3D). A previous NMR study demonstrated that intracellular Na+ elevates only when Mg2+ as well as Ca2+ is omitted from the perfusate (Van Echteld et al., 1998), which is different from the present Ca2+-free conditions where 0.5 mmol·L−1 Mg2+ was consistently present with no added EGTA.
We further confirmed that the Ca2+ paradox is not prevented by KB-R7943 (5 µmol·L−1) added for a period of 3 min before and during Ca2+ restoration (Figure 9A), which is long enough for KB-R7943 to produce a steady block of the reverse mode NCX (Kimura et al., 1999). Previous workers have presented evidence that there is no clear relationship between intracellular Na+ levels during Ca2+ depletion and the subsequent occurrence of the Ca2+ paradox upon Ca2+ repletion (Busselen, 1987; Jansen et al., 1998; Van Echteld et al., 1998). These observations appear to be consistent with our results that suggest that the Ca2+ paradox takes place without reverse mode activation of NCX. The present results, however, do not necessarily rule out the possibility that reverse mode NCX contributes to the cellular Ca2+ overload during reperfusion following a period of ischaemia where the intracellular Na+ level is substantially elevated (Donoso et al., 1992; Carmeliet, 1999). It will be interesting to examine the relative contribution and functional role of these two Ca2+ entry pathways (TRPC channels and reverse mode NCX) in mediating ischaemia/reperfusion-induced Ca2+ overload.
Importance of SR Ca2+ loading for prevention of Ca2+ paradox
Previous studies have demonstrated that partial inhibition of the forward mode NCX leads to an elevation of SR Ca2+ content in cardiac myocytes from normal and failing hearts (Hobai et al., 2004; Ozdemir et al., 2008). It is interesting to note that the enhancement of SR Ca2+ loading during inhibition of forward mode NCX is found to be dependent on SOCE activity in neonatal rabbit ventricular myocytes (Huang et al., 2006b). Although KB-R7943 has previously been shown to preferentially block the reverse mode NCX, a subsequent study has confirmed that this compound exerts a much less or even absent mode-dependent action with a virtually identical IC50 of approximately 1 µmol·L−1 for forward and reverse mode inhibition (Kimura et al., 1999). Consistent with this view, the decay of caffeine-induced Ca2+ transient, which reflects Ca2+ extrusion via forward mode NCX, was significantly slowed down by the presence of KB-R7943 (Figure 9B). The addition of KB-R7943 (5 µmol·L−1) during the Ca2+-free superfusion significantly improved SR Ca2+ loading (Figure 9C), which may account at least partly for reduced incidence of the Ca2+ paradox in the presence of this compound throughout the superfusion period. The present study thus reveals an important functional linkage between NCX activity and the development of the Ca2+ paradox, mediated through SR Ca2+ loading in cardiac myocytes (Figure 10).
Recently, an increase in diastolic SR Ca2+ leak and resultant SR Ca2+ depletion have been implicated in the initiation of triggered electrical activity and depressed contractile function in cardiac disorders including heart failure (Shannon and Lew, 2009). The present findings further suggest that reduced SR Ca2+ content could be a trigger for the development of Ca2+ paradox-mediated cardiac injury. It is important to address the question as to whether the Ca2+ entry mechanism through TRPC channels actually mediates the Ca2+ overload in the myocardium under pathophysiological conditions such as ischaemia, where SR Ca2+ content is expected to be reduced (Carmeliet, 1999).
In conclusion, the present experiments identify a novel Ca2+ entry pathway (TRPC channels) for the development of the Ca2+ paradox and reveal an important functional linkage between NCX activity and the development of the Ca2+ paradox, mediated through SR Ca2+ loading in cardiac myocytes (Figure 10).
Acknowledgments
This study was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science.
Glossary
Abbreviations
- 2-APB
2-aminoethoxydiphenyl borate
- CAP
control antigen peptide
- CIB
cell isolation buffer
- DNP
2,4-dinitrophenol
- ER/SR
endoplasmic/sarcoplasmic reticulum
- IP3
inositol-1,4,5-trisphosphate
- NCX
Na+/Ca2+ exchange
- RyR2
cardiac ryanodine receptor
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+-ATPase
- SOCE
store-operated Ca2+ entry
- SR
sarcoplasmic reticulum
- STIM
stromal interaction molecule
- τ
time constant
- TRPC
transient receptor potential canonical
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Effect of pretreatment with normal IgG, anti-TRPC4 and anti-TRPC5 antibodies on the Ca2+ paradox. Ventricular myocytes were pretreated with normal IgG, anti-TRPC4 and anti-TRPC5 antibodies (1:200 dilution) for 15 min. There were no significant differences in the effects of IgG and these antibodies, compared with the control (N = 2–3 in each group).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Ahmmed GU, Mehta D, Vogel S, Holinstat M, Paria BC, Tiruppathi C, et al. Protein kinase Cα phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J Biol Chem. 2004;279:20941–20949. doi: 10.1074/jbc.M313975200. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez J, Coulombe A, Cazorla O, Ugur M, Rauzier JM, Magyar J, et al. ATP/UTP activate cation-permeable channels with TRPC3/7 properties in rat cardiomyocytes. Am J Physiol. 2008;295:H21–H28. doi: 10.1152/ajpheart.00135.2008. [DOI] [PubMed] [Google Scholar]
- Beech DJ. TRPC1: store-operated channel and more. Pflügers Arch. 2005;451:53–60. doi: 10.1007/s00424-005-1441-3. [DOI] [PubMed] [Google Scholar]
- Bénitah JP, Gomez AM, Bailly P, Da Ponte JP, Berson G, Delgado C, et al. Heterogeneity of the early outward current in ventricular cells isolated from normal and hypertrophied rat hearts. J Physiol. 1993;469:111–138. doi: 10.1113/jphysiol.1993.sp019807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bootman MD, Collins TJ, Mackenzie L, Roderick HL, Berridge MJ, Peppiatt CM. 2-Aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 2002;16:1145–1150. doi: 10.1096/fj.02-0037rev. [DOI] [PubMed] [Google Scholar]
- Busselen P. Effects of sodium on the calcium paradox in rat hearts. Pflügers Arch. 1987;408:458–464. doi: 10.1007/BF00585069. [DOI] [PubMed] [Google Scholar]
- Callewaert G, Cleemann L, Morad M. Caffeine-induced Ca2+ release activates Ca2+ extrusion via Na+-Ca2+ exchanger in cardiac myocytes. Am J Physiol. 1989;257:C147–C152. doi: 10.1152/ajpcell.1989.257.1.C147. [DOI] [PubMed] [Google Scholar]
- Carmeliet E. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol Rev. 1999;79:917–1017. doi: 10.1152/physrev.1999.79.3.917. [DOI] [PubMed] [Google Scholar]
- Chapman RA, Tunstall J. The calcium paradox of the heart. Prog Biophys Mol Biol. 1987;50:67–96. doi: 10.1016/0079-6107(87)90004-6. [DOI] [PubMed] [Google Scholar]
- Chatamra KR, Chapman RA. The effects of sodium-calcium exchange inhibitors on protein loss associated with the calcium paradox of the isolated Langendorff perfused guinea-pig heart. Exp Physiol. 1996;81:203–210. doi: 10.1113/expphysiol.1996.sp003925. [DOI] [PubMed] [Google Scholar]
- DeHaven WI, Jones BF, Petranka JG, Smyth JT, Tomita T, Bird GS, et al. TRPC channels function independently of STIM1 and Orai1. J Physiol. 2009;587:2275–2298. doi: 10.1113/jphysiol.2009.170431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz ME, Trafford AW, O'Neill SC, Eisner DA. Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J Physiol. 1997;501:3–16. doi: 10.1111/j.1469-7793.1997.003bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoso P, Mill JG, O'Neill SC, Eisner DA. Fluorescence measurements of cytoplasmic and mitochondrial sodium concentration in rat ventricular myocytes. J Physiol. 1992;448:493–509. doi: 10.1113/jphysiol.1992.sp019053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta AK, Sabirov RZ, Uramoto H, Okada Y. Role of ATP-conductive anion channel in ATP release from neonatal rat cardiomyocytes in ischaemic or hypoxic conditions. J Physiol. 2004;559:799–812. doi: 10.1113/jphysiol.2004.069245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Calculator programs for computing the composition of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. J Physiol (Paris) 1979;75:463–505. [PubMed] [Google Scholar]
- Fauconnier J, Lanner JT, Sultan A, Zhang SJ, Katz A, Bruton JD, et al. Insulin potentiates TRPC3-mediated cation currents in normal but not in insulin-resistant mouse cardiomyocytes. Cardiovasc Res. 2007;73:376–385. doi: 10.1016/j.cardiores.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, et al. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- Flemming R, Xu SZ, Beech DJ. Pharmacological profile of store-operated channels in cerebral arteriolar smooth muscle cells. Br J Pharmacol. 2003;139:955–965. doi: 10.1038/sj.bjp.0705327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Györke S, Lukyanenko V, Györke I. Dual effects of tetracaine on spontaneous calcium release in rat ventricular myocytes. J Physiol. 1997;500:297–309. doi: 10.1113/jphysiol.1997.sp022021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaszovich CR, Zitt C, Jungling E, Luckhoff A. Inhibition of TRP3 channels by lanthanides. Block from the cytosolic side of the plasma membrane. J Biol Chem. 2000;275:37423–37428. doi: 10.1074/jbc.M007010200. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved path-clamp techniques for high-resolution current recording from cells an cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hobai IA, Maack C, O'Rourke B. Partial inhibition of sodium/calcium exchange restores cellular calcium handling in canine heart failure. Circ Res. 2004;95:292–299. doi: 10.1161/01.RES.0000136817.28691.2d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang GN, Zeng W, Kim JY, Yuan JP, Han L, Muallem S, et al. STIM1 carboxyl-terminus activates native SOC, ICRAC and TRPC1 channels. Nat Cell Biol. 2006a;8:1003–1010. doi: 10.1038/ncb1454. [DOI] [PubMed] [Google Scholar]
- Huang J, van Breemen C, Kuo KH, Hove-Madsen L, Tibbits GF. Store-operated Ca2+ entry modulates sarcoplasmic reticulum Ca2+ loading in neonatal rabbit cardiac ventricular myocytes. Am J Physiol. 2006b;290:C1572–C1582. doi: 10.1152/ajpcell.00226.2005. [DOI] [PubMed] [Google Scholar]
- Imoto Y, Ehara T, Goto M. Calcium channel currents in isolated guinea-pig ventricular cells superfused with Ca-free EGTA solution. Jpn J Physiol. 1985;35:917–932. doi: 10.2170/jjphysiol.35.917. [DOI] [PubMed] [Google Scholar]
- Inoue R, Okada T, Onoue H, Hara Y, Shimizu S, Naitoh S, et al. The transient receptor potential protein homologue TRP6 is the essential component of vascular α1-adrenoceptor-activated Ca2+-permeable cation channel. Circ Res. 2001;88:325–332. doi: 10.1161/01.res.88.3.325. [DOI] [PubMed] [Google Scholar]
- Jansen MA, Van Echteld CJA, Ruigrok TJC. Na+/Ca2+ exchange during Ca2+ repletion is not a prerequisite for the Ca2+ paradox in isolated rat hearts. Pflügers Arch. 1998;436:515–520. doi: 10.1007/s004240050666. [DOI] [PubMed] [Google Scholar]
- Ju YK, Chu Y, Chaulet H, Lai D, Gervasio OL, Graham RM, et al. Store-operated Ca2+ influx and expression of TRPC genes in mouse sinoatrial node. Circ Res. 2007;100:1605–1614. doi: 10.1161/CIRCRESAHA.107.152181. [DOI] [PubMed] [Google Scholar]
- Jung S, Mühle A, Schaefer M, Strotmann R, Schultz G, Plant TD. Lanthanides potentiate TRPC5 currents by an action at extracellular sites close to the pore mouth. J Biol Chem. 2003;278:3562–3571. doi: 10.1074/jbc.M211484200. [DOI] [PubMed] [Google Scholar]
- Kaplan P, Hendrikx M, Mattheussen M, Mubagwa K, Flameng W. Effect of ischemia and reperfusion on sarcoplasmic reticulum calcium uptake. Circ Res. 1992;71:1123–1130. doi: 10.1161/01.res.71.5.1123. [DOI] [PubMed] [Google Scholar]
- Kimura J, Watano T, Kawahara M, Sakai E, Yatabe J. Direction-independent block of bi-directional Na+/Ca2+ exchange current by KB-R7943 in guinea-pig cardiac myocytes. Br J Pharmacol. 1999;128:969–974. doi: 10.1038/sj.bjp.0702869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, et al. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Cheng KT, Bandyopadhyay BC, Pani B, Dietrich A, Paria BC, et al. Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1(−/−) mice. Proc Natl Acad Sci USA. 2007;104:17542–17547. doi: 10.1073/pnas.0701254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López JJ, Salido GM, Pariente JA, Rosado JA. Interaction of STIM1 with endogenously expressed human canonical TRP1 upon depletion of intracellular Ca2+ stores. J Biol Chem. 2006;281:28254–28264. doi: 10.1074/jbc.M604272200. [DOI] [PubMed] [Google Scholar]
- Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev. 2007;87:165–217. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- North RA, Barnard EA. Nucleotide receptors. Curr Opin Neurobiol. 1997;7:346–357. doi: 10.1016/s0959-4388(97)80062-1. [DOI] [PubMed] [Google Scholar]
- Ohba T, Watanabe H, Murakami M, Takahashi Y, Iino K, Kuromitsu S, et al. Upregulation of TRPC1 in the development of cardiac hypertrophy. J Mol Cell Cardiol. 2007;42:498–507. doi: 10.1016/j.yjmcc.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Ozdemir S, Bito V, Holemans P, Vinet L, Mercadier JJ, Varro A, et al. Pharmacological inhibition of Na/Ca exchange results in increased cellular Ca2+ load attributable to the predominance of forward mode block. Circ Res. 2008;102:1398–1405. doi: 10.1161/CIRCRESAHA.108.173922. [DOI] [PubMed] [Google Scholar]
- Parekh AB. Cell biology: cracking the calcium entry code. Nature. 2006;441:163–165. doi: 10.1038/441163a. [DOI] [PubMed] [Google Scholar]
- Piper HM. The calcium paradox revisited: an artefact of great heuristic value. Cardiovasc Res. 2000;45:123–127. doi: 10.1016/s0008-6363(99)00304-1. [DOI] [PubMed] [Google Scholar]
- Poteser M, Graziani A, Rosker C, Eder P, Derler I, Kahr H, et al. TRPC3 and TRPC4 associate to form a redox-sensitive cation channel. Evidence for expression of native TRPC3-TRPC4 heteromeric channels in endothelial cells. J Biol Chem. 2006;281:13588–13595. doi: 10.1074/jbc.M512205200. [DOI] [PubMed] [Google Scholar]
- Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol Rev. 1998;50:413–492. [PubMed] [Google Scholar]
- Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado JA, Brownlow SL, Sage SO. Endogenously expressed Trp1 is involved in store-mediated Ca2+ entry by conformational coupling in human platelets. J Biol Chem. 2002;277:42157–42163. doi: 10.1074/jbc.M207320200. [DOI] [PubMed] [Google Scholar]
- Schaefer M, Plant TD, Obukhov AG, Hofmann T, Gudermann T, Schultz G. Receptor-mediated regulation of the nonselective cation channels TRPC4 and TRPC5. J Biol Chem. 2000;275:17517–17526. doi: 10.1074/jbc.275.23.17517. [DOI] [PubMed] [Google Scholar]
- Seki S, MacLeod KT. Effects of anoxia on intracellular Ca2+ and contraction in isolated guinea pig cardiac myocytes. Am J Physiol. 1995;268:H1045–H1052. doi: 10.1152/ajpheart.1995.268.3.H1045. [DOI] [PubMed] [Google Scholar]
- Seth M, Zhang ZS, Mao L, Graham V, Burch J, Stiber J, et al. TRPC1 channels are critical for hypertrophic signaling in the heart. Circ Res. 2009;105:1023–1030. doi: 10.1161/CIRCRESAHA.109.206581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon TR, Lew WY. Diastolic release of calcium from the sarcoplasmic reticulum: a potential target for treating triggered arrhythmias and heart failure. J Am Coll Cardiol. 2009;53:2006–2008. doi: 10.1016/j.jacc.2009.02.032. [DOI] [PubMed] [Google Scholar]
- Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91:594–600. doi: 10.1161/01.res.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- Shioya T. A simple technique for isolating healthy heart cells from mouse models. J Physiol Sci. 2007;57:327–335. doi: 10.2170/physiolsci.RP010107. [DOI] [PubMed] [Google Scholar]
- Strübing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron. 2001;29:645–655. doi: 10.1016/s0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- Tsien RY, Rink TJ. Neutral carrier ion-selective microelectrodes for measurement of intracellular free calcium. Biochim Biophys Acta. 1980;599:623–638. doi: 10.1016/0005-2736(80)90205-9. [DOI] [PubMed] [Google Scholar]
- Van Echteld CJ, Van Emous JG, Jansen MA, Schreur JH, Ruigrok TJ. Manipulation of intracellular sodium by extracellular divalent cations: a 23Na and 31P NMR study on intact rat hearts. J Mol Cell Cardiol. 1998;30:119–126. doi: 10.1006/jmcc.1997.0578. [DOI] [PubMed] [Google Scholar]
- Vassort G, Alvarez J. Transient receptor potential: a large family of new channels of which several are involved in cardiac arrhythmia. Can J Physiol Pharmacol. 2009;87:100–107. doi: 10.1139/Y08-112. [DOI] [PubMed] [Google Scholar]
- Vazquez G, Wedel BJ, Aziz O, Trebak M, Putney JW., Jr The mammalian TRPC cation channels. Biochim Biophys Acta. 2004;1742:21–36. doi: 10.1016/j.bbamcr.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Wihlborg AK, Balogh J, Wang L, Borna C, Dou Y, Joshi BV, et al. Positive inotropic effects by uridine triphosphate (UTP) and uridine diphosphate (UDP) via P2Y2 and P2Y6 receptors on cardiomyocytes and release of UTP in man during myocardial infarction. Circ Res. 2006;98:970–976. doi: 10.1161/01.RES.0000217402.73402.cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams IA, Allen DG. Intracellular calcium handling in ventricular myocytes from mdx mice. Am J Physiol. 2007;292:H846–H855. doi: 10.1152/ajpheart.00688.2006. [DOI] [PubMed] [Google Scholar]
- Xu SZ, Beech DJ. TrpC1 is a membrane-spanning subunit of store-operated Ca2+ channels in native vascular smooth muscle cells. Circ Res. 2001;88:84–87. doi: 10.1161/01.res.88.1.84. [DOI] [PubMed] [Google Scholar]
- Zhou H, Iwasaki H, Nakamura T, Nakamura K, Maruyama T, Hamano S, et al. 2-Aminoethyl diphenylborinate analogues: selective inhibition for store-operated Ca2+ entry. Biochem Biophys Res Commun. 2007;352:277–282. doi: 10.1016/j.bbrc.2006.10.174. [DOI] [PubMed] [Google Scholar]
- Zimmerman ANE, Hülsmann WC. Paradoxical influence of calcium ions on the permeability of the cell membranes of the isolated rat heart. Nature. 1966;211:646–647. doi: 10.1038/211646a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.