

Abstract
BACKGROUND AND PURPOSE
Recently, we identified etodolac as a possible ligand for the human intestinal proton-couple peptide transporter (hPEPT1). This raised the possibility that other non-steroidal anti-inflammatory drugs, and especially ibuprofen, could also interact with hPEPT1. Here, we have assessed the interactions of ibuprofen with hPEPT1.
EXPERIMENTAL APPROACH
The uptake of [14C]Gly-Sar, [3H]Ibuprofen and other radio-labelled compounds were investigated in Madin–Darby canine kidney cells (MDCK)/hPEPT1, MDCK/Mock, LLC-PK1 or Caco-2 cells. The transepithelial transport of ibuprofen and hPEPT1 substrates was investigated in Caco-2 cell monolayers.
KEY RESULTS
Ibuprofen concentration dependently inhibited hPEPT1-mediated uptake of Gly-Sar in MDCK/hPEPT1 cells (Kiapp= 0.4 mM) but uptake of ibuprofen in Caco-2 cells and MDCK/hPEPT1 cells was not inhibited by hPEPT1 substrates. The maximum uptake rate for Gly-Sar uptake was reduced from 522 pmol·min−1·cm−2 to 181 pmol·min−1·cm−2 and 78 pmol·min−1·cm−2 in the presence of 0.5 mM and 1 mM ibuprofen, respectively. The interaction between ibuprofen and hPEPT1 was thus non-competitive. In LLC-PK1 cells, ibuprofen (1 mM) did not influence the transporter-mediated uptake of glycine or α-methyl-D-glycopyranoside. In Caco-2 cell monolayers the absorptive transport of δ-aminolevulinic acid was reduced by 23% and 48% by ibuprofen (1 and 10 mM), respectively. Likewise the transport of Gly-Sar was reduced by 23% in the presence of ibuprofen (1 mM).
CONCLUSIONS AND IMPLICATIONS
Ibuprofen is a non-competitive inhibitor of hPEPT1. As ibuprofen reduced the transepithelial transport of δ-aminolevulinic acid, drug–drug interactions between ibuprofen and hPEPT1 drug substrates at their site of absorption are possible if administered together.
Keywords: Ibuprofen, hPEPT1 (SLC15A1), drug–drug interaction, intestinal absorption
Introduction
Ibuprofen is a widely used drug belonging to the structurally diverse, non-steroidal anti-inflammatory drug (NSAID) class, which also includes drugs such as naproxen, acetylsalicylic acid and etodolac. Recently, we identified etodolac as a possible ligand for the human proton-coupled peptide transporter (hPEPT1) (Larsen et al., 2009), and this led us to speculate that ibuprofen might also interact with this transporter. hPEPT1 is expressed in the apical membrane of the small intestinal enterocytes (Liang et al., 1995; Ogihara et al., 1996), where it facilitates the absorption of a number of drug substances such as β-lactam antibiotics, anti-viral pro-drugs and δ-aminolevulinic acid (ALA), a drug used in photodynamic cancer therapy (Addison et al., 1975; Nakashima et al., 1984; Balimane et al., 1998; Doring et al., 1998; de Vrueh et al., 1998; Sugawara et al., 2000). The recommended oral dose of ibuprofen is 400 mg, three times daily, resulting in an estimated initial intestinal luminal concentration of up to 8 mM (400 mg dose dissolved in 250 mL). Ibuprofen is absorbed rapidly and almost completely from the intestine (Martin et al., 1990; Cheng et al., 1994). In excised rat jejunum, mounted in an Ussing chamber, a net lumen-to-serosal transepithelial flux of ibuprofen was found. This net flux was caused by the intestinal acidic microclimate and was thus due to differential partitioning, caused by the pH gradient (Legen et al., 2003). Removing the luminal mucus abolished polarized transport in rat jejunum and, using artificial membranes, it was concluded that ibuprofen is transported passively (Legen et al., 2003). Ibuprofen is a substrate for the transporter OAT3 (Khamdang et al., 2002; Takeda et al., 2002b) and an inhibitor of OAT2, OAT4 and SMCT1 (Khamdang et al., 2002; Takeda et al., 2002b; Itagaki et al., 2006), and there are conflicting reports as to whether it is a substrate for OAT1 (Mulato et al., 2000; Khamdang et al., 2002). Ibuprofen has also been reported to inhibit the transport of substrates for a number of other transporters such as MRP2, MRP4, monocarboxylate transporter 1 (MCT1), oatp1a1, oatp1a3 and oatp1a4 (Tamai et al., 1995; Masuda et al., 1997; Mulato et al., 2000; Cha et al., 2001; Khamdang et al., 2002; Shitara et al., 2002; El-Sheikh et al., 2007; Cui and Morris, 2009). However, of these transporters only MRP2, MRP4, SMCT1 and MCT1 are expressed in the intestine.
The aim of the present study was to investigate whether ibuprofen interacted with hPEPT1 and if this could have implications for the transport of ibuprofen or hPEPT1 substrates across the intestinal epithelium. We found that ibuprofen interacted with hPEPT1 as a non-competitive inhibitor of hPEPT1-mediated transport. Ibuprofen was transported passively across Caco-2 cells, but significantly inhibited the intestinal transepithelial transport of hPEPT1 substrates such as Gly-Sar and the drug substance ALA, indicating that drug–drug interactions between hPEPT1 substrates and ibuprofen could occur in the small intestine.
Methods
Cell culture
Madin–Darby canine kidney cells (MDCK) stably transfected with the hPEPT1 cDNA (MDCK/hPEPT1) or the empty vector pcDNA3.1/V5&His (MDCK/Mock) were kindly donated by Bristol-Meyers Squibb Company (New Brunswick, NJ, USA) (Herrera-Ruiz et al., 2004). MDCK and Caco-2 cells were seeded in culture flasks in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, penicillin (100 U·mL−1), streptomycin (100 µg·mL−1), 1% L-glutamine and 1% non-essential amino acids and passaged once (Caco-2) or twice (MDCK) a week. Caco-2 cells (passage 26–36) were seeded onto either Transwell™ tissue culture (Corning Inc., Life Sciences, Lowell, MA, USA) treated polycarbonate membranes (pore size: 0.4 µm, growth area: 1.13 cm2) at a density of 105 cells per filter or in 24-well plates (1.9 cm2) at a density of 1.7 × 105 cells per well. Cells seeded onto polycarbonate membranes were used between day 25 and 28 and cells seeded into 24-well plates were used 6 days after seeding. MDCK/hPEPT1 cells (passage 17–31) and MDCK/Mock cells (passage 17–18) were seeded at a density of 1 × 105 cells per well in 24-well plates or 4 × 104 cells per well in 96-well plates and used 3 days post-seeding. LLC-PK1 cells were seeded in culture flasks and passaged in 1:1 DMEM : Nutrient mixture F-12 supplemented with 10% fetal bovine serum, penicillin (100 U·mL−1) and streptomycin (100 µg·mL−1). The LLC-PK1 cells were used in passage 110–112 and cells were seeded on to Transwell™ tissue culture treated polycarbonate filters at a density of 105 cells per filter and used 10 days post-seeding.
All cell lines were grown in an atmosphere of 5% CO2, 95% O2 and 37°C and the culture medium was changed every second or third day.
In vitro uptake studies in MDCK/hPEPT1 and MDCK/Mock cells
MDCK/hPEPT1 and MDCK/Mock cells were pre-incubated with Hanks' balanced salt solution (HBSS), pH 7.4 [HBSS supplemented with 10 mM HEPES and 0.05% bovine serum albumin (BSA)] for 15 min. After aspirating the HBSS, the experiment was initiated by addition of 400 µL HBSS, pH 6.0 [HBSS supplemented with 10 mM 2-(N-morpholino)ethanesulfonic acid (MES) and 0.05% BSA] containing 18 µM [14C]Gly-Sar (1 µCi·mL−1) or 0.1 µM [3H]ibuprofen (0.5 µCi·mL−1) and other compounds as indicated. During incubation the cells were continuously rotated at 37°C on an orbital shaker (Heidolph Unimax 2010, Kelheim, Germany). After 5 min, the solutions were removed and the cells were washed three times with ice-cold HBSS buffer. The cells were detached by adding 400 µL 0.1% Triton-X in H2O and incubating at 37°C for at least 30 min. The cell homogenate was transferred to a scintillation vial and 2 mL scintillation fluid was added. The radioactivity was counted by liquid scintillation spectrometry in a Packard TriCarb 2100TR liquid scintillation counter (Meriden, CT, USA).
In vitro uptake studies of ibuprofen in Caco-2 cells
Uptake experiments in Caco-2 cells were essentially performed as described in the previous section. The Caco-2 cells were pre-incubated in HBSS buffer pH 6.0 on the apical side and HBSS buffer pH 7.4 on the basolateral side. After the experiment the Caco-2 cells including polycarbonate filters were detached from the filter support and transferred to scintillation vials.
In vitro uptake studies of α-methyl-D-glycopyranoside and glycine in LLC-PK1 cells
Uptake studies in LLC-PK1 cells were performed essentially as described for Caco-2 cells. Exceptions were that uptake experiments with α-methyl-D-glycopyranoside were performed in HBSS buffer pH 7.4 without glucose [CaCl2 (1.26 mM), MgCl2 (0.49 mM), MgSO4 (0.41 mM), KCl (5.33 mM), KH2PO4 (0.44 mM), NaHCO3 (4.17 mM), NaCl (137.9 mM), Na2HPO4 (0.34 mM), HEPES (10 mM), BSA (0.05%)] and the uptake experiments were performed with HBSS buffer pH 7.4 on both apical and basolateral side.
Transepithelial transport studies in Caco-2 cell monolayers
The integrity of the Caco-2 cell monolayer was assessed by transepithelial electric resistance measurements at room temperature before initiating the experiments. The Caco-2 cells were pre-incubated in HBSS buffer pH 6.0 on the apical side and HBSS buffer pH 7.4 on the basolateral side. In the transport studies with [3H]ibuprofen, the cells were pre-incubated with experimental buffers 15 min before the start of the experiment. Apparent permeabilities [Papp (cm·s−1)] of the radiolabelled compounds across the Caco-2 cells were studied in both the absorptive [apical to basolateral (A–B)] and exsorptive (basolateral to apical) directions. Experiments were initiated by adding medium with radiolabelled compound with or without ibuprofen. When present, ibuprofen was added in both receiver and donor compartment. Samples from the receiver compartment were taken at various time points with or without 1 mM ibuprofen: 10, 20, 30, 40 and 60 min; and with or without 10 mM ibuprofen: 15, 30, 60, 90 and 120 min. After each sampling, the receiver compartment was replenished with the same volume of fresh medium. The radioactivity in samples was analysed by liquid scintillation spectrometry after adding 2 mL of scintillation fluid.
Fluorescence-based studies of hPEPT1 translocation
Translocation via hPEPT1 is electrogenic and changes in membrane potential may thus be used as a surrogate marker for substrate transport. The recently characterized and validated MDCK/hPEPT1 FLIPR® membrane potential assay (Molecular Devices, Berkshire, UK) (Faria et al., 2004; Frolund et al., 2010; Omkvist et al., 2010) was used to measure substrate transport via hPEPT1. Gly-Sar and ibuprofen were applied to the MDCK/hPEPT1 FLIPR® membrane potential assay in absence or presence of the recently identified high affinity hPEPT1 inhibitor, Met-Pro-Pro (Omkvist et al., 2010). Growth media were aspirated and replaced with 50 µl of FLIPR® membrane potential assay kit dye stock solution reconstituted in HBSS buffer pH 6.0. The cells were incubated for 30 min at 37°C to ensure dye equilibrium across the cell membrane. The assays were initiated by addition of 50 µl test solution in HBSS buffer pH 6.0 with FLIPR® membrane potential assay kit dye. Sample-induced changes in fluorescence (indicative of changes in membrane potential) were measured on a NOVOstar platereader (BMG LabTech, Offenburg, Germany) at 37°C for 72 s. The probe was excited at 544 nm and emission light was collected at 590 nm.
Evaluation of ibuprofen toxicity
Cellular toxicity of the investigated compounds was examined using a 3-(4, 5-dimethylthiazol-2–y1)2, 5-diphenyl tetrazolium bromide (MTT) assay (Mosmann, 1983). MDCK/hPEPT1 cells were grown for 3 days on the bottom of 96-well plates. The growth medium was aspirated, 100 µL test compound in HBSS (0.05–20 mM) or sodium lauryl sulphate in HBSS (0.05–10 mM) (positive control) or HBSS (negative control) were added and the cells were incubated at 37°C on an orbital shaker. After 5 min the solutions were removed and 100 µL 2.4 mM MTT was added and the cells were incubated in the dark at 37°C for 90 min. To dissolve the formed formazan salt 100 µL of a solution of 10 mM HCl, 50% isobutanol and 0.4 mM sodium lauryl sulfate was added and the cells were incubated for at least 3 h. Absorbance was measured at 590 nm on an elisa plate reader (Bie & Berntsen, Herlev, Denmark).
Data treatment
Affinity determinations
Cellular uptake of Gly-Sar in the presence of increasing concentrations of ibuprofen was fitted to a four-parameter logistic equation (Equation 1) to obtain an IC50 value:
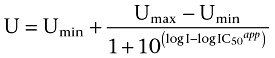 |
(1) |
U is % inhibition of the Gly-Sar flux at inhibitor concentration I, Umax is the initial flux value of Gly-Sar [(I) = 0 mM]∼100%, Umin is the lowest measured flux value of Gly-Sar at maximum inhibitor concentration and I is the concentration of inhibitor (mM).
According to Cheng and Prusoff (1973), IC50 equals Ki for non-competitive inhibitors. Results are given as the fitted Kiapp value ± standard error based on results from seven concentrations in three individual cell monolayers.
Fluorescence-based studies of hPEPT1 translocation
The fluorescence signal was normalized according to the initial fluorescence signal, to account for unequal loading of the monolayers or seeding of cells. The area under curve (AUC) of the normalized fluorescence signal was subtracted from the AUC caused by addition of a sample with HBSS, pH 6.0. The AUC values were then expressed as a percentage of the AUC caused by the addition of 20 mM Gly-Sar (later referred to as response), as described previously (Omkvist et al., 2010).
Determination of interaction type between ibuprofen and hPEPT1
The uptake rate of Gly-Sar in the presence or absence of 0.5 and 1 mM ibuprofen was fitted to the Eadie–Hofstee equation (Equation 2):
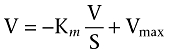 |
(2) |
V is the uptake rate of Gly-Sar (pmol·min−1·cm−2), Vmax is the maximum uptake rate (pmol·min−1·cm−2), Km is the Michaelis constant (mM) and S is the Gly-Sar concentration (mM).
Transepithelial transport across Caco-2 cell monolayers
The transepithelial transport of compounds across Caco-2 cell monolayers is expressed as the apparent permeability (Papp, cm·s−1) and was calculated using Equation 3:
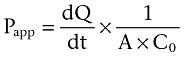 |
(3) |
dQ/dt is the mass of compound (Q, pmol) accumulated in the receiver compartment over time (t, minute), as a function of the area (A, cm2) and the initial concentration of compound in the donor compartment at steady-state conditions. Steady state was assumed when no more than 10% of the initial amount of compound was transported to the receiver compartment. When more than 10% was transported, non-steady-state calculations were used (Equation 4).
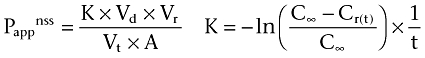 |
(4) |
Pappnss is the apparent permeability estimated at non-steady state (cm·s−1), Vd is the volume of the donor compartment (mL), Vr is the volume of the receiver compartment (mL), Vt is total volume in both compartments (mL), A is the area of the cell monolayers (cm2), C∞ is concentration in compartments at equilibrium (M), Cr(t) is the time-dependent concentration in receiver compartment (M) and t is the time (minute).
Statistical analysis
Statistical analysis was performed using either an unpaired Student's t-test or a one-way analysis of variance (anova) followed by Dunnett's post-test.
Statistical analysis and data fitting were performed using GraphPad Prism (version. 4.01, GraphPad Software Inc., San Diego, CA, USA).
Materials
DMEM, penicillin, streptomycin, L-glutamine, non-essential amino acids, HBSS and geneticin were from Invitrogen (Taastrup, Denmark). Fetal bovine serum was purchased from Biotech line (Slangerup, Denmark). The human colon adenocarcinoma cell line (Caco-2) and the proximal tubule porcine kidney cell line LLC-PK1 were obtained from American Type Culture Collection (Manassas, VA, USA). Gly-Pro, Gly-Sar, Triton X-100, HEPES, MES, BSA 2-(2-aminobenzoyl)benzoic acid, p-aminohippuric acid (PAH), oestrone-3-sulfate (E3S), MgSO4, MgCl2, CaCl2, 1:1 DMEM : Nutrient mixture F-12 and MTT were from Sigma-Aldrich (Brøndby, Denmark). [3H]mannitol (11.7 Ci·mmol−1), [14C]α-methyl-D-glycopyranoside (310 mCi·mmol−1), [14C]glycine (87 mCi·mmol−1) and Ultima Gold scintillation fluid were from PerkinElmer (Boston, MA, USA). [3H]ibuprofen (5 Ci·mmol−1), [3H]etoposide (20 Ci·mmol−1) and [14C]δ-aminolevulinic acid (ALA) (55 mCi·mmol−1) were from American Radiolabelled Chemicals Inc. (St. Louis, MO, USA). [3H]L-carnosine (10 Ci·mmol−1) was purchased from Moravek Biochemicals, Inc. (Brea, CA, USA). [14C]mannitol (56.5 mCi·mmol−1) was from Larodan (Malmö, Sweden). [3H]metoprolol (29.7 Ci·mmol−1) was purchased from Vitrax (Placentia, CA, USA). [14C]Gly-Sar (56 mC·mmol−1) was from GE-Healthcare (Freiburg, Germany). Sodium lauryl sulphate, KCl, KH2PO4, NaHCO3, Na2HPO4 and isobutanol were from Merck (Darmstad, Germany). MP probe included in the FLIPR® membrane potential assay kit was purchased from Molecular Devices (Berkshire, UK). NaCl was from VWR (Herlev, Denmark). Paracetamol, naproxen and piroxicam were obtained from Chr. Olesen Pharmaceuticals A/S (Gentofte, Denmark), whereas acetylsalicylic acid was from Fagron (Copenhagen, Denmark). Drug and molecular target nomenclature follows Alexander et al. (2009).
Results
Ibuprofen inhibits Gly-Sar uptake in MDCK/hPEPT1 cells
The ability of paracetamol and five structurally diverse NSAIDs (acetylsalicylic acid, etodolac, ibuprofen, naproxen and piroxicam) to inhibit hPEPT1-mediated [14C]Gly-Sar uptake was investigated in MDCK/hPEPT1 cells (Figure 1). Ibuprofen, piroxicam, etodolac and Gly-Pro, a high-affinity natural substrate for hPEPT1, inhibited the uptake of Gly-Sar significantly, whereas acetylsalicylic acid, paracetamol and naproxen did not interfere with Gly-Sar uptake. None of the tested compounds were toxic in a MTT assay at the concentration examined (0.5 mM) (data not shown). Ibuprofen was selected for further investigation and in MDCK/hPEPT1 cells [14C]Gly-Sar uptake was significantly inhibited to approximately 6% of the control uptake in the presence of 10 mM ibuprofen and 20 mM Gly-Pro (Figure 2A). The uptake of Gly-Sar in Caco-2 cells grown in 24-well plates was also investigated in absence or presence of 1 mM ibuprofen and 20 mM Gly-Pro (Figure 2B). Ibuprofen (1 mM) and Gly-Pro (20 mM) inhibited the uptake of Gly-Sar to 46% and 12% of the control uptake, respectively. The ability of ibuprofen to inhibit the uptake of [14C]Gly-Sar in MDCK/hPEPT1 cells was further investigated. As illustrated in Figure 2C, ibuprofen inhibits Gly-Sar uptake in a concentration-dependent manner with an estimated Kiapp of 0.4 mM (pKiapp= 0.4 ± 0.1 mM) (Figure 2C). The two stereoisomers of ibuprofen were investigated separately for their ability to inhibit Gly-Sar uptake, and they were found to be equally potent (data not shown). Ibuprofen is thus a high affinity ligand of hPEPT1-mediated transport. To verify that the effect of ibuprofen is not due to unspecific cellular toxicity, a MTT toxicity assay was performed. Ibuprofen concentrations below 10 mM did not affect MDCK/hPEPT1 cell viability, whereas cell viability was significantly affected by the presence of ibuprofen in concentrations above 10 mM (Figure 2D).
Figure 1.
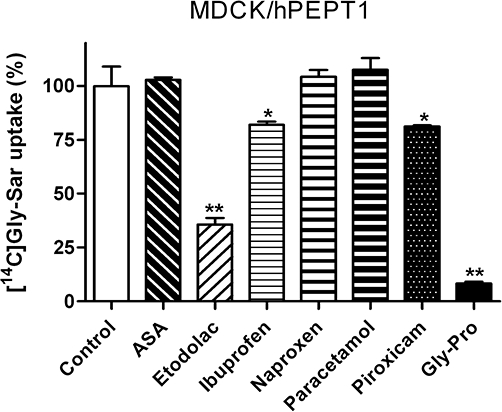
Uptake of 18 µM [14C]Gly-Sar in Madin–Darby canine kidney cells (MDCK)/human proton-coupled peptide transporter (hPEPT1) cell monolayers in presence or absence of 0.5 mM acetylsalicylic acid (ASA), etodolac, ibuprofen, naproxen, paracetamol, piroxicam or 20 mM Gly-Pro. The uptake was measured for 5 min with an extracellular pH of 6.0. Data represent mean ± SEM of six cell monolayers. *P < 0.05 and **P < 0.01, analysed by one-way anova followed by Dunnet's post-test (the control column used as control).
Figure 2.
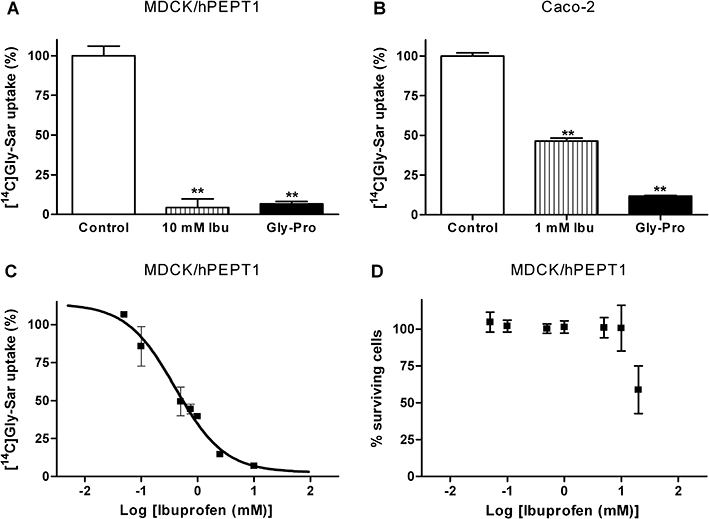
(A) Uptake of 18 µM [14C]Gly-Sar in the presence or absence of 20 mM Gly-Pro or 10 mM racaemic ibuprofen (Ibu) in Madin–Darby canine kidney cells (MDCK)/human proton-coupled peptide transporter (hPEPT1) cells. (B) Uptake of 18 µM [14C]Gly-Sar in the presence or absence of 20 mM Gly-Pro or 1 mM racaemic Ibu in Caco-2 cells grown in 24-well plates. (C) Uptake (%) of 18 µM [14C]Gly-Sar uptake as a function of increasing ibuprofen concentrations (0–10 mM). The solid line represents the fit of the experimental data to Equation 1 and Kiapp was determined to be 0.4 mM (pKiapp= 0.4 ± 0.1 mM). (D) The viability of MDCK/hPEPT1 cells in the presence of ibuprofen (0.05–20 mM) assessed via a 3-(4, 5-dimethylthiazol-2-y1)2, 5-diphenyl tetrazolium bromide test. Uptake was measured for 5 min using a 2-[N-morpholino]ethanesulfonic acid buffer with a pH of 6.0. Results in A + C are mean ± SD and in B + D are mean ± SEM of three individual cell monolayers in two to three passages. **P < 0.01, analysed by one-way anova followed by Dunnett's post-test.
Ibuprofen is not a substrate for hPEPT1
We then investigated if ibuprofen was a substrate for hPEPT1 using two different approaches; one using [3H]ibuprofen to measure direct cellular uptake and another using a fluorescence-based hPEPT1 translocation assay. Apical uptake of [3H]ibuprofen into MDCK/hPEPT1 cells was not inhibited by 20 mM Gly-Pro, whereas the uptake was significantly reduced in the presence of 20 mM ampicillin, 2-(2-aminobenzoyl)benzoic acid (ABBA) or ALA (Figure 3A). A similar pattern was observed in MDCK/Mock cells, where 20 mM of Gly-Pro did not inhibit the uptake of [3H]ibuprofen, whereas 20 mM of ABBA significantly reduced the uptake of ibuprofen (Figure 3B). The magnitude of cellular [3H]ibuprofen uptake was furthermore similar in MDCK/hPEPT1 and MDCK/Mock cells (Figure 3A,B). The [3H]ibuprofen uptake was investigated in MDCK/hPEPT1 cells using three concentrations: 0.0001, 0.1 and 0.5 mM ibuprofen. Here 0.1 mM reduced the uptake of [3H]ibuprofen by 39%, whereas 0.5 mM reduced it by 50% (Figure 3C). The uptake of [3H]ibuprofen into MDCK/hPEPT1 cells was further measured in the absence or presence of 1 mM PAH or 30 µM E3S (Figure 3D). The ibuprofen uptake was not affected by PAH, whereas the uptake was reduced approximately 50% by E3S. Collectively, these results point to the fact that ibuprofen is not a substrate for hPEPT1, but at the same time indicates that ibuprofen is a substrate for another transporter in MDCK cells.
Figure 3.
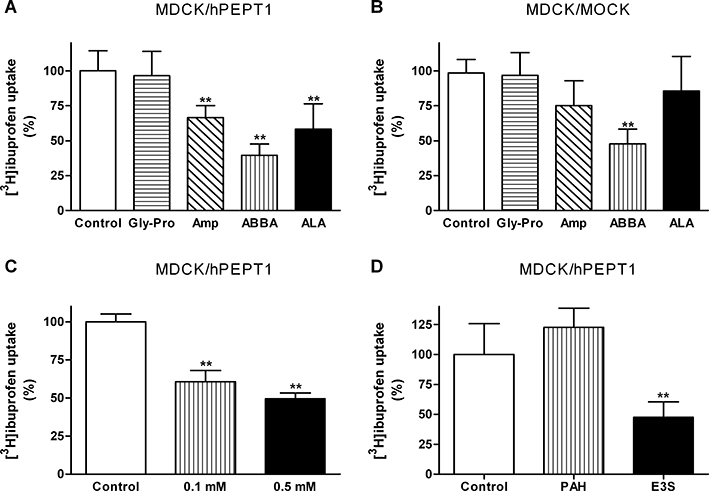
Uptake of ibuprofen in Madin–Darby canine kidney cells (MDCK)/human proton-coupled peptide transporter (hPEPT1) or MDCK/Mock cells. Uptake was measured for 5 min with a buffer pH of 6.0. (A,B) Uptake of [3H]ibuprofen (0.25 µCi·well−1, 0.1 µM) in MDCK/hPEPT1 (A) or MDCK/Mock (B) cell monolayers in the absence or presence of 20 mM Gly-Pro, 20 mM ampicillin (Amp), 20 mM 2-(2-aminobenzoyl)benzoic acid (ABBA) or 20 mM δ-aminolevulinic acid (ALA). The results are mean ± SD of four to five individual cell monolayers. (C) Uptake of [3H]ibuprofen (0.25 µCi·well−1, 0.1 µM) in MDCK/hPEPT1 cells in the absence or presence of 0.1 mM or 0.5 mM ibuprofen. Results are mean ± SEM of 9–10 individual cell monolayers from three different cell passages. (D) Uptake of [3H]ibuprofen (0.25 µCi·well−1, 0.1 µM) in MDCK/hPEPT1 cells in the absence or presence of 1 mM p-aminohippuric acid (PAH) or 30 µM oestrone-3-sulfate (E3S). The results are mean ± SD of four individual cell monolayers. **P < 0.01, analysed by one-way anova followed by Dunnett's post-test.
The apical uptake of [3H]ibuprofen in the intestinal Caco-2 cell line was investigated in the presence or absence of Gly-Pro and ampicillin. The apical uptake rate of [3H]ibuprofen was 10 ± 1 fmol·min−1·cm−2, and the uptake was not inhibited by neither 20 mM Gly-Pro (8 ± 3 fmol·min−1·cm−2) nor 20 mM ampicillin (9 ± 2 fmol·min−1·cm−2) (data not shown).
To provide further evidence that ibuprofen was an inhibitor of hPEPT1-mediated substrate transport, a membrane potential sensitive fluorescence based translocation assay was used. Met-Pro-Pro completely blocked the fluorescence response elicited by 5 mM Gly-Sar (Figure 4). Addition of 5 mM ibuprofen to the assay caused an increase in the fluorescence signal but no suppression of the signal was observed in presence of the hPEPT1 inhibitor Met-Pro-Pro (Figure 4). Ibuprofen caused a depolarization of the membrane potential by a mechanism that appeared to be unrelated to hPEPT1-mediated substrate translocation. Thus, ibuprofen inhibited translocation via hPEPT1 without being transported. Ibuprofen was therefore identified as a novel inhibitor of hPEPT1.
Figure 4.
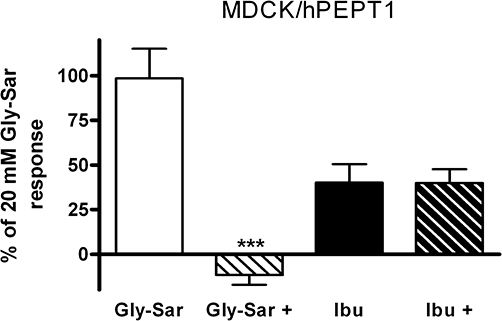
The percentage change in fluorescence, relative to the response elicited by 20 mM Gly-Sar, measured in the Madin–Darby canine kidney cells (MDCK)/human proton-coupled peptide transporter (hPEPT1) FLIPR® membrane potential translocation assay after addition of ibuprofen (Ibu) (5 mM) and Gly-Sar (5 mM) in the absence or presence of 0.25 mM Met-Pro-Pro (+). The cells were cultured for 3 days and change in fluorescence was measured for 72 s with an apical pH of 6.0. The results are mean ± SEM. of six cell monolayers. Statistical analysis was performed using an unpaired Student's t-test comparing the response in presence or absence of Met-Pro-Pro, ***P < 0.001.
Ibuprofen is a non-competitive inhibitor of hPEPT1
The mechanism of ibuprofen inhibition of hPEPT1 substrate transport was investigated. The concentration-dependent uptake of Gly-Sar was measured in the absence or presence of 0.5 mM or 1 mM ibuprofen in MDCK/hPEPT1 cells. In Figure 5 the results are displayed in an Eadie–Hofstee plot. The apparent Vmax values were 522 pmol·min−1·cm−2[95% confidence interval (CI) 453–618, pmol·min−1·cm−2], 181 pmol·min−1·cm−2 (95% CI 142–253 pmol·min−1·cm−2) and 78 pmol·min−1·cm−2 (95% CI 60–118 pmol·min−1·cm−2) for uptake of Gly-Sar, Gly-Sar with 0.5 mM ibuprofen and Gly-Sar with 1 mM ibuprofen, respectively. The Km values were 1.9 mM (95% CI 1.6–2.3 mM), 1.4 mM (95% CI 1.1–2.2 mM) and 1.0 mM (95% CI 0.7–1.6 mM) for uptake of Gly-Sar, Gly-Sar with 0.5 mM ibuprofen and Gly-Sar with 1 mM ibuprofen, respectively. The slopes of the Eadie–Hofstee plot were not significantly different from each other, whereas the intercepts were. The Vmax values for Gly-Sar uptake are thus significantly different for the concentrations of ibuprofen tested here, which suggested that ibuprofen inhibited substrate transport via hPEPT1 in a non-competitive manner.
Figure 5.
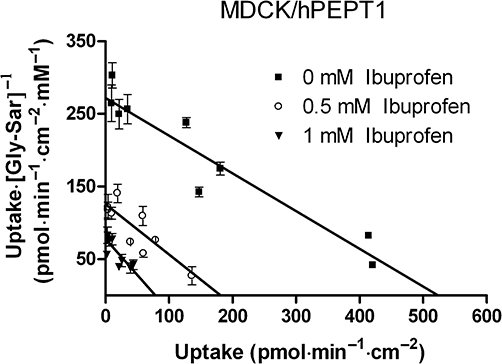
Concentration-dependent uptake of Gly-Sar (36 µM [14C]Gly-Sar) in the absence or presence of 0.5 mM or 1 mM ibuprofen. The uptake was measured for 5 min with a buffer pH of 6.0. Results are mean ± SD of three individual cell monolayers. Data are displayed in an Eadie–Hofstee plot. The solid lines represent the linear regression of the results. MDCK, Madin–Darby canine kidney cells; hPEPT1, human proton-coupled peptide transporter.
Ibuprofen does not inhibit α-methyl-D-glycopyranoside and glycine uptake in LLC-PK1 cells
To investigate if the ibuprofen inhibition of substrate transported via hPEPT1 was specific, the uptake of the SGLT1 substrate α-methyl-D-glycopyranoside into LLC-PK1 cells was investigated in the presence or absence of 1 mM ibuprofen and 1 mM of the SGLT1 inhibitor phlorizin (Figure 6A). Ibuprofen did not reduce the uptake of α-methyl-D-glycopyranoside at the concentration investigated, whereas 1 mM phlorizin completely abolished the uptake (Figure 6A). In addition, the uptake of [14C]glycine into LLC-PK1 cells was measured in presence or absence of 1 mM ibuprofen and 20 mM of unlabelled glycine (Figure 6B). Unlabelled glycine reduced the uptake to approximately 34% of the control while ibuprofen did not appear to inhibit the uptake (Figure 6B).
Figure 6.
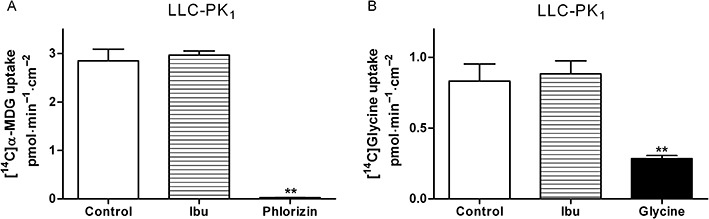
(A) Apical uptake of [14C]α-methyl-D-glycopyranoside (α-MDG) (3 µM) in LLC-PK1 cell monolayers in the absence or presence of 1 mM ibuprofen (Ibu) or 1 mM phlorizin. (B) Apical uptake of [14C]glycine (11 µM) in LLC-PK1 cell monolayers in the absence or presence of 1 mM Ibu or 20 mM unlabelled glycine. Uptake was measured for 5 min with an apical and basolateral pH of 7.4. Data represent mean ± SEM of two individual cell monolayers in three passages. **P < 0.01, analysed by one-way anova followed by Dunnett's post-test.
Uptake and transepithelial transport of ibuprofen across Caco-2 cell monolayers
The transepithelial transport of 0.1 µM ibuprofen was investigated in Caco-2 cell monolayers, grown for 27 days, in buffers of varying pH (Table 1). Without a pH gradient across the monolayer the absorptive and exsorptive permeability of ibuprofen was similar. When a pH gradient was present, the permeability of ibuprofen was approximately 30 times larger in the direction of increasing pH. The cellular concentration of ibuprofen was estimated to be 40 nM (cellular compartment 2 µL), whereas the initial concentration in donor compartment was 100 nM, hence no cellular accumulation of ibuprofen was observed. However, the uptake of [3H]ibuprofen into Caco-2 cells grown for 6 days in 24-well plates was decreased to 50% of the control value in the presence of 1 mM unlabelled ibuprofen (Figure 7).
Table 1.
Apparent permeability coefficients of the transepithelial transport of ibuprofen across Caco-2 cell monolayers
| pH | Apparent permeability (·10−5 cm·s−1) | Ratio | ||
|---|---|---|---|---|
| Apical | Basolateral | Absorptive Apical → basolateral | Exsorptive Basolateral → apical | Pabs/Pexs |
| 6.0 | 6.0 | 3.0 ± 0.1 | 2.4 ± 0.2 | 1.2 |
| 6.0 | 7.4 | 9.3 ± 0.7 | 0.3 ± 0.1 | 28 |
| 7.4 | 6.0 | 0.5 ± 0.1 | 9.8 ± 0.9 | 0.05 |
| 7.4 | 7.4 | 2.5 ± 0.1 | 2.3 ± 0.3 | 1.1 |
Values represent mean ± SEM of four to five individual cell monolayers in three individual Caco-2 cell passages.
Figure 7.
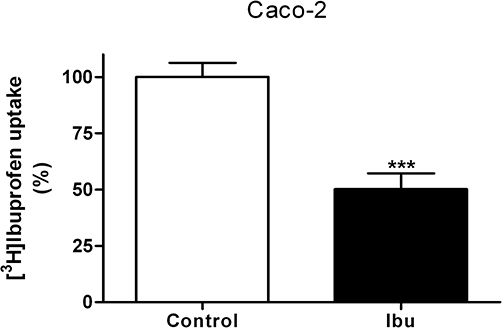
Uptake of 0.1 µM [3H]ibuprofen in Caco-2 cells in 24-well plates in the presence or absence of 1 mM racaemic ibuprofen. Uptake was measured for 5 min using a 2-[N-morpholino]ethanesulfonic acid buffer with a pH of 6.0. Results mean ± SEM of 3 individual cell monolayers in three passages. Statistical analysis was performed using an unpaired Student's t-test, ***P < 0.001.
Transepithelial transport of hPEPT1 substrates across Caco-2 cell monolayers in the presence of Ibuprofen
The possibility that ibuprofen could affect the transepithelial transport of hPEPT1 substrates was then investigated. The transepithelial transport of hPEPT1 substrates ALA, Gly-Sar and L-carnosine was measured in the presence or absence of ibuprofen. The A–B permeability of ALA and Gly-Sar was significantly larger than the basolateral to apical transport (Figure 8). The A–B transport of Gly-Sar and ALA was significantly inhibited by 1 mM ibuprofen (P < 0.05), whereas no change in A–B permeability was observed for L-carnosine (Figure 8). In the presence of 10 mM ibuprofen an increase in the A–B flux of all tested compounds were observed after approximately 40 min (data not shown). A time-dependent effect of ibuprofen on the barrier properties of the Caco-2 cell monolayers was observed and therefore the A–B Papp in the presence of 10 mM ibuprofen was calculated from data obtained within the first 30 min of the transport study. The presence of 10 mM ibuprofen inhibited the A–B transport of ALA and Gly-Sar to the same level as the B–A transport in absence of ibuprofen (Figure 8), thus abolishing the hPEPT1 mediated polarized transport. 10 mM ibuprofen did not affect A–B permeability of L-carnosine. Ibuprofen thus inhibits transepithelial transport of some hPEPT1 substrates. To verify that ibuprofen did not have additional effects on the barrier properties of Caco-2 cells the A–B transport of metoprolol (transcellular marker), mannitol (paracellular marker) and etoposide (marker for efflux transport) in the presence or absence of 1 and 10 mM ibuprofen were examined. In addition, the basolateral to apical (B–A) transport of etoposide was also investigated in the absence or presence of 1 mM ibuprofen. The A–B permeability of mannitol was 3.2 × 10−6± 0.4 × 10−6 cm·s−1 and the permeability was not significantly different in the presence of 1 or 10 mM ibuprofen, respectively. Metropolol had an A–B permeability of 4.6 × 10−6± 1.3 × 10−6 cm·s−1 and the permeability was not influenced by the presence of 1 mM ibuprofen. However, a significant (P < 0.01) increase in the absorptive permeability of metoprolol was seen in the presence of 10 mM ibuprofen. The A–B and B–A permeability of etoposide was 2.86 × 10−5± 0.3 × 10−5 cm·s−1 and 5.0 × 10−5± 0.4 × 10−5, respectively, and these permeabilities were not affected by the presence of ibuprofen.
Figure 8.
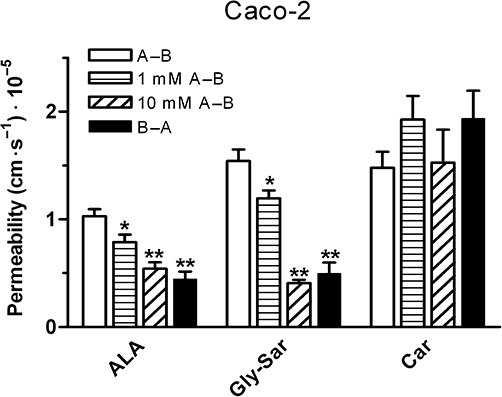
Apical to basolateral transport (A–B) of [14C]δ-aminolevulinic acid (ALA) (0.5 µCi·mL−1, 9 µM), [14C]Gly-Sar (0.5 µCi·mL−1, 9 µM) and [3H]L-carnosine (Car) (0.5 µCi·mL−1, 50 nM) in the absence or presence of 1 and 10 mM ibuprofen and basolateral to apical transport (B–A) of ALA, Gly-Sar and Car in Caco-2 cells. The apical pH was 6.0 and basolateral pH was 7.4. Apparent permeability coefficients (Papp) were calculated from flux data. To avoid possible toxic effects of ibuprofen at high concentrations, only data points from 15 and 30 min was used to calculate flux values in the presence of 10 mM ibuprofen. Data represent mean ± SEM of five to nine cell monolayers from three individual passages. *P < 0.05 and **P < 0.01, analysed by one-way anova followed by Dunnett's post-test (the A–B Papp used as control).
Discussion
Ibuprofen is administered orally in doses of 200–400 mg every 6 h when used as an over-the-counter medication (Davies, 1998). When the indication is rheumatoid arthritis the maximal dose is up to 3200 mg a day, that is 800 mg doses four times daily (Davies, 1998). Assuming that the ibuprofen dose is taken with 250 mL fluid, initial intestinal concentrations would be from 3.9 mM (200 mg dose) to 15.5 mM (800 mg dose) using a molecular weight of ibuprofen of 206.3 g·mol−1. We have determined the Ki-value for the non-competitive interaction of ibuprofen with hPEPT1, to be 0.4 mM in vitro. The intestinal concentrations of ibuprofen are thus likely to be much higher than the Ki-value of ibuprofen for hPEPT1. This indicates that, even at the lowest doses of ibuprofen, hPEPT1 will be fully saturated at the ibuprofen concentrations likely to be present in the intestine. Accordingly, ibuprofen was shown to reduce the absorptive transport of δ-aminolevulinic acid and Gly-Sar in Caco-2 cell monolayers. Therefore our in vitro data suggest that drug–drug interactions may occur, but obviously in vivo data are necessary to establish whether such drug–drug interactions are present in clinical settings. We therefore hypothesize that ibuprofen could reduce the absorption of hPEPT1 substrates when dosed together, and drug–drug interaction may occur in the small intestine at the level of intestinal absorption.
Ibuprofen is a non-competitive inhibitor of substrate transport via hPEPT1
The uptake of Gly-Sar was measured in the presence or absence of five NSAIDs and paracetamol. Ibuprofen inhibited Gly-Sar uptake via hPEPT1, and also piroxicam, which is a NSAID structurally unrelated to ibuprofen, likewise inhibited Gly-Sar uptake in MDCK/hPEPT1 cells. Etodolac, which we had previously identified as a possible hPEPT1 ligand (Larsen et al., 2009), was confirmed to interact with hPEPT1. Naproxen, which is structurally related to ibuprofen, did not inhibit Gly-Sar uptake nor did acetylsalicylic acid or paracetamol. This indicates that NSAIDs do not, in general, inhibit hPEPT1-mediated transport but specific structural features of individual NSAID drugs might interfere with hPEPT1-mediated transport. Ibuprofen was shown to have affinity for hPEPT1 with a Kiapp value of 0.4 mM, which is comparable to that of naturally occurring dipeptides and drug substances such as ceftibuten, L-cephalexin, L-valaciclovir and ALA (Nielsen et al., 2002a). We subsequently employed different strategies to investigate whether ibuprofen was a substrate or an inhibitor of hPEPT1. The uptake of [3H]ibuprofen was not inhibited by Gly-Pro in neither MDCK/hPEPT1 cells nor Caco-2 cells. In the MDCK/hPEPT1 FLIPR® membrane potential assay the hPEPT1 inhibitor Met-Pro-Pro did not affect the response elicited by ibuprofen. Collectively, the data all point to the conclusion that ibuprofen inhibits hPEPT1-mediated transport, without being transported itself. The type of inhibition of hPEPT1-mediated transport by ibuprofen was investigated, and based on Figure 5 we suggest that the interaction is of a non-competitive nature. To our knowledge the only other reported non-competitive inhibitors of hPEPT1 transport are quinapril, spirapril, nateglinide and glibenclamide (Sawada et al., 1999; Terada et al., 2000; Zhu et al., 2000; Knütter et al., 2008). The non-competitive inhibition of quinapril and glibenclamide may be caused by a direct interaction with hPEPT1 (Sawada et al., 1999; Zhu et al., 2000).
To assess whether the observed interaction between ibuprofen and hPEPT1 was a general effect of ibuprofen on membrane transporters, the uptake of α-methyl-D-glycopyranoside and glycine was measured in the presence or absence of ibuprofen using LLC-PK1 cell monalyers. Ibuprofen did not seem to interfere with either α-methyl-D-glycopyranoside or glycine uptake. Thus, ibuprofen does not appear to have a general effect on membrane transporters.
Renal MDCK cells stably transfected with hPEPT1 were used as an in vitro model to investigate ibuprofen interactions with hPEPT1. In MDCK/hPEPT1 cells the uptake of ibuprofen was concentration-dependent and inhibited by ALA, ampicillin, ABBA and E3S but not by PAH and Gly-Pro. This indicates that a non-hPEPT1 carrier-mediated mechanism is involved in the cellular uptake of ibuprofen in renal MDCK cells. Ibuprofen is reported to be a substrate for OAT3 and possibly OAT1 (Khamdang et al., 2002; Takeda et al., 2002a). However, PAH is also a substrate for both carriers excluding them as the carrier involved in ibuprofen transport in the MDCK/hPEPT1 cells (Cha et al., 2001; Groves et al., 2003). E3S is a substrate for both oatp1a1, oatp1a3 and oatp1a4 and PAH is not a ligand for oatp1a1 and oatp1a4 (the OATP/SLCO nomenclature defined by Hagenbuch and Meier, 2004, is used; Noe et al., 1997; Hagenbuch et al., 2000; van Montfoort et al., 2002; Hagenbuch and Meier, 2004). Oatp1a1, oatp1a4 and oatp1a5 have been identified at the mRNA level in rat kidney, whereas oatp1a1 and oatp1a4 have also been identified at the protein level (Hagenbuch and Meier, 2003).
Ampicillin significantly inhibited the uptake of ibuprofen in MDCK/hPEPT1 cells. The structures of penicillin G and ampicillin are very similar and only differ by the presence of an extra amine group in ampicillin. Penicillin G has been reported to inhibit the substrate uptake via oatp1a1 (Shitara et al., 2002). Whether any of the above-mentioned transporters are expressed in the MDCK cells is presently unknown, thus it remains purely speculative whether these carriers are mediating ibuprofen transport in the MDCK/hPEPT1 cells.
Uptake and transepithelial transport of ibuprofen in intestinal Caco-2 cell monolayers
The transepithelial transport of ibuprofen was strongly dependent on the pH values of the buffers used in the donor and receiver solution. We did not find any polarized transport of ibuprofen with similar pH values of 6.0/6.0 or 7.4/7.4 on the donor and receiver side. Even though the initial apical uptake of radiolabelled ibuprofen into Caco-2 cells was inhibited by unlabelled ibuprofen, no accumulation of ibuprofen in the Caco-2 cells could be observed at the end of the transport study. The predominant transcellular absorption mechanism for ibuprofen in Caco-2 cells thus appear to be passive diffusion driven by the pH gradient (pH partitioning) (Shore et al., 1957; Neuhoff et al., 2005). Comparable results were obtained by Legen et al. (2003) in rat jejunum and artificial membranes. In intestinal cells ibuprofen has been suggested to inhibit substrate transport via the proton-coupled MCT1 (Tamai et al., 1995). Therefore MCT1 might mediate the apical uptake of ibuprofen, but as no polarized transepithelial transport of ibuprofen was observed in the Caco-2 cell model, MCT1 does not seem to be a major determinant for transepithelial ibuprofen transport.
Interactions between ibuprofen and hPEPT1 substrates
The initial luminal concentration of ibuprofen was estimated to be between 3.9 mM and 15.5 mM. This corresponds to approximately 10–40 times the Kiapp of ibuprofen determined for hPEPT1. As ibuprofen is a non-competitive inhibitor of hPEPT1, the theoretical maximum transport rate would be reduced by at least 90% at the luminal ibuprofen concentrations likely to occur in vivo. Therefore, we investigated the effect on transepithelial transport of hPEPT1 substrates across Caco-2 cell monolayers using 1 or 10 mM ibuprofen. The relatively high permeability coefficients obtained in our Caco-2 cell transport experiments suggest a relatively high paracellular transport. We observed absorptive polarized transepithelial transport for ALA and Gly-Sar, whereas no polarized transport was observed for L-carnosine. For L-carnosine this pattern was expected, since even though L-carnosine is a substrate for hPEPT1, the transepithelial absorption mechanism for L-carnosine is not hPEPT1 mediated (Nielsen et al., 2002b). ALA was recently also identified as a substrate for the proton-coupled amino acid transporter, hPAT1 (Anderson et al., 2010; Frolund et al., 2010). In Caco-2 cell monolayers, Frolund et al. (2010) found that only the proton-coupled carriers hPEPT1 and hPAT1 contributed to intestinal absorption of ALA. However, at the concentration used here, 9 µM ALA and 9 µM Gly-Sar, only hPEPT1-mediated transport accounts for the total apical transport. In Caco-2 cell monolayers 1 mM of ibuprofen significantly inhibited the absorptive transport of ALA and Gly-Sar. This confirms that the transport of hPEPT1 substrates is affected by the presence of ibuprofen in concentrations likely to be present in the lumen, following oral administration of ibuprofen. At 1 mM, ibuprofen did not affect the A–B transport of the passively transported metoprolol and mannitol, which confirmed that ibuprofen did not cause a non-specific effect on Caco-2 cells. Etoposide is a substrate for at least three efflux transporters, that is P-gp (ABCB1), BCRP (ABCG2) and MRP2 (ABCC2), which are expressed in the apical membrane of intestinal cells (Cui et al., 1999; Guo et al., 2002; Yuan et al., 2009). Ibuprofen (1 mM) did not alter the A–B or B–A transport of etoposide and therefore did not appear to interact with the efflux transporters responsible for etoposide transport. In dose regimens including ibuprofen and a hPEPT1 substrate, inhibition of hPEPT1-mediated transport may be expected. The pharmacokinetic consequence of this may be an increased time to reach maximal plasma concentration and/or a reduced absorption fraction. In vivo studies are needed to clarify if the proposed interaction between ibuprofen and hPEPT1-substrates has clinical relevance in humans.
In conclusion, we have identified ibuprofen as a non-competitive inhibitor of hPEPT1 and drug–drug interactions at the level of intestinal drug absorption via hPEPT1 are anticipated in dose regimens consisting of ibuprofen and hPEPT1 substrates.
Acknowledgments
This project was funded by a grant from the Carlsberg Foundation. The excellent technical support of Bettina Dinitzen, Birgitte Eltong and Maria D. Læssøe Pedersen is highly appreciated. In addition, we are grateful to Steen Honoré Hansen for supplying us with the R and S isomers of ibuprofen.
Glossary
Abbreviations
- A–B
apical to basolateral
- ABBA
2-(2-aminobenzoyl)benzoic acid
- ALA
δ-aminolevulinic acid
- B–A
basolateral to apical
- BSA
bovine serum albumin
- Caco-2
Human colon adenocarcinoma cells
- E3S
oestrone-3-sulfate
- HBSS
Hanks' balanced salt solution
- MDCK
Madin–Darby canine kidney cells
- MES
2-[N-morpholino]ethanesulfonic acid
- MTT
3-(4, 5-dimethylthiazol-2-y1)2, 5-diphenyl tetrazolium bromide
- NSAID
Non-steroidal anti-inflammatory drug
- PAH
p-aminohippuric acid
Conflicts of interest
The authors state no conflict of interest.
Supporting Information
Teaching Materials; Figs 1–8 as PowerPoint slide.
References
- Addison JM, Burston D, Dalrymple JA, Matthews DM, Payne JW, Sleisenger MH, et al. A common mechanism for transport of di- and tri-peptides by hamster jejunum in vitro. Clin Sci Mol Med. 1975;49:313–322. doi: 10.1042/cs0490313. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, Jevons M, Thangaraju M, Edwards N, Conlon NJ, Woods S, et al. Transport of the photodynamic therapy agent 5-aminolevulinic acid by distinct H+-coupled nutrient carriers coexpressed in the small intestine. J Pharmacol Exp Ther. 2010;332:220–228. doi: 10.1124/jpet.109.159822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balimane PV, Tamai I, Guo A, Nakanishi T, Kitada H, Leibach FH, et al. Direct evidence for peptide transporter (PepT1)-mediated uptake of a nonpeptide prodrug, valacyclovir. Biochem Biophys Res Commun. 1998;250:246–251. doi: 10.1006/bbrc.1998.9298. [DOI] [PubMed] [Google Scholar]
- Cha SH, Sekine T, Fukushima J, Kanai Y, Kobayashi Y, Goya T, et al. Identification and characterization of human organic anion transporter 3 expressing predominantly in the kidney. Mol Pharmacol. 2001;59:1277–1286. doi: 10.1124/mol.59.5.1277. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Cheng H, Rogers JD, Demetriades JL, Holland SD, Seibold JR, Depuy E. Pharmacokinetics and bioinversion of ibuprofen enantiomers in humans. Pharm Res. 1994;11:824–830. doi: 10.1023/a:1018969506143. [DOI] [PubMed] [Google Scholar]
- Cui D, Morris ME. The drug of abuse gamma-hydroxybutyrate is a substrate for sodium-coupled monocarboxylate transporter (SMCT) 1 (SLC5A8): characterization of SMCT-mediated uptake and inhibition. Drug Metab Dispos. 2009;37:1404–1410. doi: 10.1124/dmd.109.027169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Konig J, Buchholz JK, Spring H, Leier I, Keppler D. Drug resistance and ATP-dependent conjugate transport mediated by the apical multidrug resistance protein, MRP2, permanently expressed in human and canine cells. Mol Pharmacol. 1999;55:929–937. [PubMed] [Google Scholar]
- Davies NM. Clinical pharmacokinetics of ibuprofen. The first 30 years. Clin Pharmacokinet. 1998;34:101–154. doi: 10.2165/00003088-199834020-00002. [DOI] [PubMed] [Google Scholar]
- Doring F, Walter J, Will J, Focking M, Boll M, Amasheh S, et al. Delta-aminolevulinic acid transport by intestinal and renal peptide transporters and its physiological and clinical implications. J Clin Invest. 1998;101:2761–2767. doi: 10.1172/JCI1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sheikh AAK, van den Heuvel JJMW, Koenderink JB, Russel FGM. Interaction of nonsteroidal anti-inflammatory drugs with multidrug resistance protein (MRP) 2/ABCC2- and MRP4/ABCC4-mediated methotrexate transport. J Pharmacol Exp Ther. 2007;320:229–235. doi: 10.1124/jpet.106.110379. [DOI] [PubMed] [Google Scholar]
- Faria TN, Timoszyk JK, Stouch TR, Vig BS, Landowski CP, Amidon GL, et al. A novel high-throughput PepT1 transporter assay differentiates between substrates and antagonists. Mol Pharm. 2004;1:67–76. doi: 10.1021/mp034001k. [DOI] [PubMed] [Google Scholar]
- Frolund S, Marquez OC, Larsen M, Brodin B, Nielsen CU. delta-Aminolevulinic acid is a substrate for the amino acid transporter SLC36A1 (hPAT1) Br J Pharmacol. 2010;159:1339–1353. doi: 10.1111/j.1476-5381.2009.00620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves CE, Munoz L, Bahn A, Burckhardt G, Wright SH. Interaction of cysteine conjugates with human and rabbit organic anion transporter 1. J Pharmacol Exp Ther. 2003;304:560–566. doi: 10.1124/jpet.102.043455. [DOI] [PubMed] [Google Scholar]
- Guo A, Marinaro W, Hu P, Sinko PJ. Delineating the contribution of secretory transporters in the efflux of etoposide using Madin-Darby canine kidney (MDCK) cells overexpressing P-glycoprotein (Pgp), multidrug resistance-associated protein (MRP1), and canalicular multispecific organic anion transporter (cMOAT) Drug Metab Dispos. 2002;30:457–463. doi: 10.1124/dmd.30.4.457. [DOI] [PubMed] [Google Scholar]
- Hagenbuch B, Meier PJ. The superfamily of organic anion transporting polypeptides. Biochim Biophys Acta. 2003;1609:1–18. doi: 10.1016/s0005-2736(02)00633-8. [DOI] [PubMed] [Google Scholar]
- Hagenbuch B, Meier PJ. Organic anion transporting polypeptides of the OATP/SLC21 family: phylogenetic classification as OATP/SLCO superfamily, new nomenclature and molecular/functional properties. Pflugers Arch. 2004;447:653–665. doi: 10.1007/s00424-003-1168-y. [DOI] [PubMed] [Google Scholar]
- Hagenbuch B, Adler ID, Schmid TE. Molecular cloning and functional characterization of the mouse organic-anion-transporting polypeptide 1 (Oatp1) and mapping of the gene to chromosome X. Biochem J. 2000;345(Pt 1):115–120. [PMC free article] [PubMed] [Google Scholar]
- Herrera-Ruiz D, Faria TN, Bhardwaj RK, Timoszyk J, Gudmundsson OS, Moench P, et al. A novel hPepT1 stably transfected cell line: establishing a correlation between expression and function. Mol Pharm. 2004;1:136–144. doi: 10.1021/mp034011l. [DOI] [PubMed] [Google Scholar]
- Itagaki S, Gopal E, Zhuang L, Fei YJ, Miyauchi S, Prasad PD, et al. Interaction of ibuprofen and other structurally related NSAIDs with the sodium-coupled monocarboxylate transporter SMCT1 (SLC5A8) Pharm Res. 2006;23:1209–1216. doi: 10.1007/s11095-006-0023-1. [DOI] [PubMed] [Google Scholar]
- Khamdang S, Takeda M, Noshiro R, Narikawa S, Enomoto A, Anzai N, et al. Interactions of human organic anion transporters and human organic cation transporters with nonsteroidal anti-inflammatory drugs. J Pharmacol Exp Ther. 2002;303:534–539. doi: 10.1124/jpet.102.037580. [DOI] [PubMed] [Google Scholar]
- Knütter I, Wollesky C, Kottra G, Hahn M, Fischer W, Zebisch K, et al. Transport of angiotensin-converting enzyme inhibitors by H+/peptide transporters revisited. J Pharmacol Exp Ther. 2008;327:432–441. doi: 10.1124/jpet.108.143339. [DOI] [PubMed] [Google Scholar]
- Larsen SB, Omkvist DH, Brodin B, Nielsen CU, Steffansen B, Olsen L, et al. Discovery of ligands for the human intestinal Di-/Tripeptide transporter (hPEPT1) using a QSAR-assisted virtual screening strategy. ChemMedChem. 2009;4:1439–1445. doi: 10.1002/cmdc.200900145. [DOI] [PubMed] [Google Scholar]
- Legen I, Zakelj S, Kristl A. Polarised transport of monocarboxylic acid type drugs across rat jejunum in vitro: the effect of mucolysis and ATP-depletion. Int J Pharm. 2003;256:161–166. doi: 10.1016/s0378-5173(03)00073-5. [DOI] [PubMed] [Google Scholar]
- Liang R, Fei YJ, Prasad PD, Ramamoorthy S, Han H, Yang-Feng TL, et al. Human intestinal H+/peptide cotransporter. Cloning, functional expression, and chromosomal localization. J Biol Chem. 1995;270:6456–6463. doi: 10.1074/jbc.270.12.6456. [DOI] [PubMed] [Google Scholar]
- Martin W, Koselowske G, Toberich H, Kerkmann T, Mangold B, Augustin J. Pharmacokinetics and absolute bioavailability of ibuprofen after oral administration of ibuprofen lysine in man. Biopharm Drug Dispos. 1990;2:265–278. doi: 10.1002/bdd.2510110311. [DOI] [PubMed] [Google Scholar]
- Masuda S, Saito H, Inui KI. Interactions of nonsteroidal anti-inflammatory drugs with rat renal organic anion transporter, OAT-K1. J Pharmacol Exp Ther. 1997;283:1039–1042. [PubMed] [Google Scholar]
- van Montfoort JE, Schmid TE, Adler ID, Meier PJ, Hagenbuch B. Functional characterization of the mouse organic-anion-transporting polypeptide 2. Biochim Biophys Acta. 2002;1564:183–188. doi: 10.1016/s0005-2736(02)00445-5. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Mulato AS, Ho ES, Cihlar T. Nonsteroidal anti-inflammatory drugs efficiently reduce the transport and cytotoxicity of adefovir mediated by the human renal organic anion transporter 1. J Pharmacol Exp Ther. 2000;295:10–15. [PubMed] [Google Scholar]
- Nakashima E, Tsuji A, Mizuo H, Yamana T. Kinetics and mechanism of in vitro uptake of amino-beta-lactam antibiotics by rat small intestine and relation to the intact-peptide transport system. Biochem Pharmacol. 1984;33:3345–3352. doi: 10.1016/0006-2952(84)90104-7. [DOI] [PubMed] [Google Scholar]
- Neuhoff S, Ungell AL, Zamora I, Artursson P. pH-Dependent passive and active transport of acidic drugs across Caco-2 cell monolayers. Eur J Pharm Sci. 2005;25:211–220. doi: 10.1016/j.ejps.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Nielsen CU, Brodin B, Jorgensen FS, Frokjaer S, Steffansen B. Human peptide transporters: therapeutic applications. Expert Opin Ther Pat. 2002a;12:1329–1350. [Google Scholar]
- Nielsen CU, Supuran CT, Scozzafava A, Frokjaer S, Steffansen B, Brodin B. Transport characteristics of L-carnosine and the anticancer derivative 4-toluenesulfonylureido-carnosine in a human epithelial cell line. Pharm Res. 2002b;19:1337–1344. doi: 10.1023/a:1020306926419. [DOI] [PubMed] [Google Scholar]
- Noe B, Hagenbuch B, Stieger B, Meier PJ. Isolation of a multispecific organic anion and cardiac glycoside transporter from rat brain. Proc Natl Acad Sci USA. 1997;94:10346–10350. doi: 10.1073/pnas.94.19.10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogihara H, Saito H, Shin BC, Terado T, Takenoshita S, Nagamachi Y, et al. Immuno-localization of H+/peptide cotransporter in rat digestive tract. Biochem Biophys Res Commun. 1996;220:848–852. doi: 10.1006/bbrc.1996.0493. [DOI] [PubMed] [Google Scholar]
- Omkvist DH, Larsen SB, Nielsen CU, Steffansen B, Olsen L, Jorgensen FS, et al. A quantitative structure-activity relationship for translocation of tripeptides via the human proton-coupled peptide transporter, hPEPT1 (SLC15A1) AAPS J. 2010;12:385–396. doi: 10.1208/s12248-010-9195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawada K, Terada T, Saito H, Hashimoto Y, Inui KI. Effects of glibenclamide on glycylsarcosine transport by the rat peptide transporters PEPT1 and PEPT2. Br J Pharmacol. 1999;128:1159–1164. doi: 10.1038/sj.bjp.0702895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shitara Y, Sugiyama D, Kusuhara H, Kato Y, Abe T, Meier PJ, et al. Comparative inhibitory effects of different compounds on rat oatpl (slc21a1)- and Oatp2 (Slc21a5)-mediated transport. Pharm Res. 2002;19:147–153. doi: 10.1023/a:1014264614637. [DOI] [PubMed] [Google Scholar]
- Shore PA, Brodie BB, Hogben CA. The gastric secretion of drugs: a pH partition hypothesis. J Pharmacol Exp Ther. 1957;119:361–369. [PubMed] [Google Scholar]
- Sugawara M, Huang W, Fei YJ, Leibach FH, Ganapathy V, Ganapathy ME. Transport of valganciclovir, a ganciclovir prodrug, via peptide transporters PEPT1 and PEPT2. J Pharm Sci. 2000;89:781–789. doi: 10.1002/(SICI)1520-6017(200006)89:6<781::AID-JPS10>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Takeda M, Babu E, Narikawa S, Endou H. Interaction of human organic anion transporters with various cephalosporin antibiotics. Eur J Pharmacol. 2002a;438:137–142. doi: 10.1016/s0014-2999(02)01306-7. [DOI] [PubMed] [Google Scholar]
- Takeda M, Khamdang S, Narikawa S, Kimura H, Hosoyamada M, Cha SH, et al. Characterization of methotrexate transport and its drug interactions with human organic anion transporters. J Pharmacol Exp Ther. 2002b;302:666–671. doi: 10.1124/jpet.102.034330. [DOI] [PubMed] [Google Scholar]
- Tamai I, Takanaga H, Maeda H, Sai Y, Ogihara T, Higashida H, et al. Participation of a proton-cotransporter, MCT1, in the intestinal transport of monocarboxylic acids. Biochem Biophys Res Commun. 1995;214:482–489. doi: 10.1006/bbrc.1995.2312. [DOI] [PubMed] [Google Scholar]
- Terada T, Sawada K, Saito H, Hashimoto Y, Inui KI. Inhibitory effect of novel oral hypoglycemic agent nateglinide (AY4166) on peptide transporters PEPT1 and PEPT2. Eur J Pharmacol. 2000;392:11–17. doi: 10.1016/s0014-2999(00)00119-9. [DOI] [PubMed] [Google Scholar]
- de Vrueh RL, Smith PL, Lee CP. Transport of L-valine-acyclovir via the oligopeptide transporter in the human intestinal cell line, Caco-2. J Pharmacol Exp Ther. 1998;286:1166–1170. [PubMed] [Google Scholar]
- Yuan J, Lv H, Peng B, Wang C, Yu Y, He Z. Role of BCRP as a biomarker for predicting resistance to 5-fluorouracil in breast cancer. Cancer Chemother Pharmacol. 2009;63:1103–1110. doi: 10.1007/s00280-008-0838-z. [DOI] [PubMed] [Google Scholar]
- Zhu T, Chen XZ, Steel A, Hediger MA, Smith DE. Differential recognition of ACE inhibitors in Xenopus laevis oocytes expressing rat PEPT1 and PEPT2. Pharm Res. 2000;17:526–532. doi: 10.1023/a:1007556630189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.