

Abstract
C—H stretching bands, νCH, in the infrared spectrum of single crystals of nominally high purity, of laboratory-grown MgO, and of natural upper mantle olivine, provide an “organic” signature that closely resembles the symmetrical and asymmetrical C—H stretching modes of aliphatic —CH2 units. The νCH bands indicate that H2O and CO2, dissolved in the matrix of these minerals, converted to form H2 and chemically reduced C, which in turn formed C—H entities, probably through segregation into defects such as dislocations. Heating causes the C—H bonds to pyrolyze and the νCH bands to disappear, but annealing at 70°C causes them to reappear within a few days or weeks. Modeling dislocations in MgO suggests that the segregation of C can lead to Cx chains, x = 4, with the terminal C atoms anchored to the MgO matrix by bonding to two O−. Allowing H2 to react with such Cx chains leads to [O2C(CH2)2CO2] or similar precipitates. It is suggested that such Cx—Hy—Oz entities represent protomolecules from which derive the short-chain carboxylic and dicarboxylic and the medium-chain fatty acids that have been solvent-extracted from crushed MgO and olivine single crystals, respectively. Thus, it appears that the hard, dense matrix of igneous minerals represents a medium in which protomolecular units can be assembled. During weathering of rocks, the protomolecular units turn into complex organic molecules. These processes may have provided stereochemically constrained organics to the early Earth that were crucial to the emergence of life.
Of all natural environments where chemical reactions occur that produce organic molecules, the dense hard matrix of igneous minerals may appear as the most unlikely place. Yet, our earlier research has shown that a suite of medium- to long-chain fatty acids, C6—C12, can be identified among the organics extracted from crushed olivine single crystals from the CO/CO2/H2O-laden high temperature, high pressure environment of the upper mantle (1). Freshly crushed olivine single crystals, when heated in vacuum, were found to release a range of organic molecules, including aromatic compounds (2). Solvent extraction of crushed MgO crystals, grown in the laboratory at 1 bar from a CO/CO2/H2O-saturated MgO melt (3), produced short-chain carboxylic and dicarboxylic acids up to C4 (4).
When minerals grow either in the laboratory or in nature, their environments are always “contaminated” and often saturated with CO2 and H2O. The presence of CO2 and H2O introduces the low-z elements carbon and hydrogen as “impurities” into the mineral matrix. As will be shown in this report, solute C and H2 participate in reactions that lead to the precipitation of protomolecular Cx entities and formation of C—H bonds inside the hard, dense mineral matrix. These solid state reactions are different from the reactions that lead to the synthesis of lipids under hydrothermal conditions by Fischer-Tropsch-type reactions (5) or to the reduction of CO2 during serpentinization of olivine and the production of organics with the help of catalysts such as magnetite (6) or to any other abiogenic reaction that has been considered for the early Earth (7–9).
Dissolution of H2O and CO2 in Mineral Matrices
H2O becomes incorporated into the matrix of minerals that crystallize in H2O-laden environments, even of minerals that are nominally anhydrous. The basic reaction controlling the uptake of “impurity” H2O can be described as a proton transfer from H2O onto an O2− of the host oxide/silicate matrix:
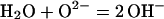 |
1 |
A large body of literature exists about OH− in nominally anhydrous minerals from various geological settings (10, 11). The most widely used method of analysis is IR spectroscopy. If transparent crystals are available, IR can detect very small amounts of OH− by way of their O—H stretching bands, νOH, in the range of 3000–3700 cm−1. Because the νOH bands lie at relatively high wavenumbers and are decoupled from the lower frequency lattice modes, they are generally sharp and unambiguously identifiable.
The amounts of OH− in olivine (Mg,Fe)2SiO4, the dominant upper mantle mineral, range from 10–1000 H/106 Si (ppm) (11). Similar but generally low OH− concentrations have been found in other petrologically important minerals (10). Under the assumption that Eq. 1 completely describes the dissolution reaction of H2O, the OH− concentrations determined by IR have been used to calculate the total amount of chemically bound “water.”
CO2 is the dominant gas in many volcanoes
and the dominant gas/fluid component in the magmas that feed them.
The important role that CO2 plays in the
petrogenesis of igneous rocks has also long been recognized. At high
pressures, it can lower the melting points of mineral assemblies, i.e.,
the temperatures at which partial melts form, by hundreds of degrees
(12). Whereas CO2 is known to dissolve in
quenched high-pressure silicate melts, maybe in form of carbonate
anions, CO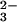 (13, 14), some researchers maintain that
C or CO2 will not enter as “impurity” into
the solid matrix of minerals (15, 16). The formation of a carbonate
anion can be described as the merging of a CO2
molecule with a lattice O2− without changing the
oxidation state of the carbon:
(13, 14), some researchers maintain that
C or CO2 will not enter as “impurity” into
the solid matrix of minerals (15, 16). The formation of a carbonate
anion can be described as the merging of a CO2
molecule with a lattice O2− without changing the
oxidation state of the carbon:
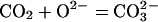 |
2 |
At closer inspection, dissolution of H2O and CO2 in solid matrix turns out to be more complicated than Eqs. 1 and 2 might suggest. This finding is exemplified by Fig. 1, which displays the IR spectrum of a nominally high purity MgO single crystal,¶ grown from an MgO melt equilibrated with an atmosphere of CO/CO2 plus H2O and N2 at 1 bar pressure (3). In the center, a group of νOH bands is seen that consists of (i) a relatively strong band or group of bands around 3300 cm−1, (ii) a weaker band at 3560 cm−1, and (iii) a very broad band extending from below 3000 cm−1 to 3700 cm−1. Because MgO crystallizes in the simple, face-centered cubic rocksalt structure, the νOH bands i–iii have been assigned to single OH− adjacent to an Mg2+ vacancy site, to OH− pairs adjacent to an Mg2+ vacancy site, and to interstitial OH−, i.e., H+ associated with O2− at regular O2− sites, respectively (17).
Figure 1.

Infrared spectrum of a nominally high purity MgO crystal, grown from a CO/CO2/H2O-laden melt, in the 2800–4200 cm−1 range covering the H—H, O—H, and C—H stretching frequencies.
Fig. 1 also shows a weak but distinct band on the left, at 4152 cm−1 (enlarged in the Inset). It has the characteristic signature of a νHH band arising from the H—H stretching mode of lattice-bound H2 molecules similar to the νHH band of H2 in noble gas matrices (18). Because the νHH band is intrinsically weak, the fact that it can be observed in the MgO crystal under study suggests a high concentration of H2 molecules.
This νHH band and the νCH bands on the right of Fig. 1, between 2800–3000 cm−1, jointly point at the complexity of the solid state dissolution of H2O and CO2. The νCH bands suggest that some form of C—H entities exist in the MgO crystal, providing an “organic” signature. These C—H entities do not come from surface contamination as has been suggested (16, 19, 20) but are associated with C in the bulk that remains detectable even on heating in ultrahigh vacuum up to 700–900°C (21–23).
The main focus of this study will be to address the nature this “organic” signature and how the presence of C—H entities is linked to the dissolution mechanism of H2O and CO2 in mineral matrices.
Experimental Procedures
We chose MgO crystals for the basic study because MgO crystallizes in the simple, face-centered cubic rocksalt-type (NaCl) structure, consisting of a close packing of O2− anions with the Mg2+ cations occupying all available octahedral sites. A further advantage of MgO is that large single crystals can be grown from the melt in high purity grades. The structure of olivine (Mg,Fe)2SiO4, although orthorhombic, is similarly dense, deriving from a hexagonal close packing of O2− anions with Mg2+ and Fe2+ cations in two differently distorted octahedral sites and Si4+ in tetrahedral sites. Both MgO and olivine tend not to develop internal cleavage planes as some other minerals do, in particular, pyroxenes.
The MgO crystals used in this study were grown at 1 bar pressure from a CO/CO2/H2O-saturated melt (3). Nominally, i.e., with respect to metal impurities, these MgO crystals were of 99.9% purity, colorless, with some turbidity because of micrometer- and sub-micrometer-sized cavities that decorate a dense network of subgrain boundaries and dislocations. They were available in form of large cubes, 20–30 mm in size, reflecting the perfect cleavage of MgO along (100). The olivine single crystals used in this study came from Afghanistan. They were recovered from peridotite nodules brought up by volcanic eruptions (24). The crystals were 20–30 mm in size, irregular in shape, olive-green and partly turbid because of a decorated network of subgrain boundaries and dislocations. A selected large olivine crystal was cut with a low-speed diamond saw to a rectangular shape of about 20 × 10 × 6 mm. This olivine crystal and a similarly sized MgO crystal were cut into several identical pieces, about 5 × 10 × 6 mm, so that the study to be described below could be done with pieces of the same single crystals. The cut surfaces were left “as is”, i.e., without further grinding or polishing.
The single crystal pieces were cleaned with organic solvents. They were mounted in Al blocks that fit into the sample holder of the Nicolet Nexus 670 Fourier transform (FT)-IR spectrometer. The MgO and olivine crystals were heated for 12 h to 400°C and 45 min to 300°C in a stream of high purity N2 gas, respectively. Previously it had been shown by gas chromatographic techniques (1, 4) that, after drying, no solvents are retained, even on finely crushed single crystal powders. By measuring the νCH intensities from a thick MgO crystal and then cutting the same crystal into several slices, it had been shown earlier (17) that the νCH band strength correlates with the length of the optical path through the bulk, not with the number of surfaces, indicating that the signal came from C—H entities in the bulk. All IR spectra (before and after heating) were recorded at 30°C, acquiring data during 20 scans over the range 400–4000 cm−1.
The heat treatment pyrolyzed their C—H entities in the MgO and olivine crystals, causing their νCH bands to disappear or nearly disappear as determined from the IR spectra recorded immediately after cooling to room temperature. For the next 32 days, sets of these MgO and olivine crystals (wrapped in Al foil) were stored in air at 70°C, whereas one control set was stored at 24°C. A run started at 45°C was lost because of a malfunction of the temperature controller. The IR spectra of each sample were recorded, first in daily intervals, later in weekly intervals. In the case of MgO, the background in the νCH region was fitted linearly between 2785 cm−1 and 3025 cm−1. In the case of olivine, a best-fit polynomial was used to compensate for the more steeply sloping background.
Dislocations in MgO were modeled by using the crystalmaker 4.0 program by David C. Palmer (Crystalmaker Software, Bicester, U.K.) modified in such a way as to allow the introduction of defects into the perfect structure. No lattice relaxation around the dislocation cores was taken into account.
Results
Fig. 2 shows the νCH bands in the “as received” MgO and olivine crystals, i.e., before heating. In both cases, the bands are similar in number and with respect to their position and relative intensities. The two strongest νCH bands lie at 2926 and 2855 cm−1 in MgO and at 2922 and 2852 cm−1 in olivine. Minor bands occur at 2955 and 2870 and a shoulder at 2895 cm−1. MgO exhibits an additional weak band at 3008 cm−1, which the spectrum of olivine does not show. In MgO, all νCH bands are slightly broader than in olivine.
Figure 2.

C—H stretching bands, νCH, in the synthetic MgO and an upper mantle derived olivine single crystal recorded before heating. The strongest νCH bands appear to arise from symmetrical and asymmetrical C—H stretching modes of —CH2-entities with minor νCH bands probably because of —CH3. The νCH bands of MgO are broader than those of olivine.
The different νCH bands may arise from C—H entities in different local environments or from Cx entities in which some C atoms are bonded to two or more H, thereby giving rise to a set of symmetrical and asymmetrical C—H stretching modes. Indeed, the two strongest bands at 2926 and 2855 cm−1 in MgO and at 2922 and 2852 cm−1 in olivine agree with the symmetrical and asymmetrical C—H stretching modes of —CH2 units in aliphatic hydrocarbon chains such as in polyethylene (25). The weak band at 2955 cm−1 and a companion at 2870 cm−1 agree with the symmetrical and asymmetrical C—H stretching modes of —CH3 units.
The weakness of the νCH signature does not necessarily mean that the amount of solute Cx entities is small. The strength of the νCH bands solely depends on the number of C—H bonds formed, not on the number of C atoms in the Cx entities. The total C concentration in the laboratory-grown MgO single crystals is probably of the order of 50–100 ppm (21, 26). Similar total C concentrations have been reported for upper mantle-derived olivine crystals (27, 28), and at least 5 ppm C2—C6 hydrocarbons.
Heating the MgO crystal to 400°C and the olivine crystal to 350°C caused their νCH bands to nearly completely disappear, because of in situ pyrolysis of the C—H bonds. Fig. 3 shows how the νCH bands reappear in the MgO crystal during annealing at 70°C. The band positions are slightly shifted. At the end of 32 days at 70°C the integral intensity of the νCH bands reached about 10% of the initial intensity. Annealing at 90°C and 24°C caused the νCH bands to reappear faster and slower, respectively. The new νCH bands lie at nearly the same positions as the ones observed before heating. This result suggests that, whereas heating pyrolyzed the C—H bonds, it left the Cx entities intact to which the H atoms had bonded.
Figure 3.

After heating for 14 h to 400°C, the νCH bands in MgO vanish nearly completely but reappear during annealing at 70°C over a period of 32 days.
Fig. 4 plots the intensity of the νCH bands in MgO as a function of annealing time at 70°C (solid circles), at 24°C, and 45°C (open circles and solid diamonds), and 90°C after the temperature controller malfunctioned, raising the temperature of the 45°C run to 90°C (open diamonds). The solid and broken lines represent parabolic fits to the data. Obviously, the pyrolysis of the C—H bonds had caused H to disperse in the MgO matrix adjacent to the sites of the Cx entities. We do not know, however, whether hydrogen remains as H after pyrolysis or forms H2. On annealing, H or H2 diffuse back to the Cx chains, forming C—H bonds. If this process is controlled by 1-dimensional diffusion, the νCH intensity should increase linearly with the square root of time. Indeed, the parabolic fit to the 70°C data describes rather well the overall increase in the integral intensity of the νCH bands. During the first 32 days at 70°C the νCH bands regain about 10% of their original intensity. Assuming the same diffusion rate, 50% of the original intensity would be reached after 4500 days or 12.5 yr.
Figure 4.

Integrated intensity of the νCH bands in the MgO crystal annealed at 70°C and other temperatures, plotted as a function of time. The symbols represent measured data, the lines parabolic fits.
The νCH bands of olivine disappear or nearly disappear on heating to 300°C for 45 min. They reappear on annealing, shifted by about 7 cm−1 to lower wavenumbers, with a similar time constant as in MgO or slightly faster. After 32 days at 70°C the νCH bands of olivine had regained about 15% of their original intensity.
Discussion
The IR observations presented here can be summarized as follows: (i) the νCH bands in the 2800–3050 cm−1 window arise from C—H entities in the crystal matrix; (ii) nearly identical νCH bands are seen in the IR spectrum of laboratory-grown MgO and natural olivine crystals from the H2O/CO2-laden high pressure environment of the upper mantle; (iii) the complexity of the νCH bands suggests polyatomic Cx entities with —CH2— and —CH3 units; (iv) the νCH bands disappear on heating because of the pyrolysis of the C—H bonds; and (v) the νCH bands reappear relatively rapidly, within a few days and weeks, on annealing at moderate temperatures between room temperature and 70°C.
The presence of the νCH bands, their disappearance on heating, and their reappearance on annealing jointly point at a sequence of physical and chemical processes that occur in solid matrix. The νCH bands are consistent with Cx entities containing —CH2— and —CH3. These Cx entities may represent “protomolecules” of those carboxylic, dicarboxylic, and fatty acids that have been extracted from MgO and olivine crystals (1, 4). To understand how such protomolecules form, we review earlier work on the dissolution mechanism of H2O and CO2 that has laid the foundation for the study presented here.
Redox Conversion of Solute H2O and CO2.
Classically, MgO crystallizing in the presence of
H2O and CO2 can be treated
as a three-component system with Mg(OH)2,
MgCO3, and Mg-hydroxy-carbonates as distinct
compounds (29). This result suggests that, even if consideration is
given to the possibility that H2O and
CO2 may enter into solid solution, the only
oxyanions will be OH− and
CO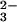 as in Eqs. 1 and 2.
However, the presence of H2 molecules and of C—H entities
suggests that the uptake of H2O and
CO2 into solid solution leads to reactions that
change the oxidation state of some of the solutes, producing
H2 and reduced C.
as in Eqs. 1 and 2.
However, the presence of H2 molecules and of C—H entities
suggests that the uptake of H2O and
CO2 into solid solution leads to reactions that
change the oxidation state of some of the solutes, producing
H2 and reduced C.
An early observation (30), made during the study of the degassing of finely divided MgO heavily doped with OH−, provided the first hint toward a truly unusual reaction. In accordance with Eq. 1, MgO containing OH− should release nothing but H2O. However, the finely divided MgO was found to release substantial amounts of molecular H2, about 5,000 H2/106 O (ppm). Such a large number of H2 could not be accounted for by the very small number of transition metal impurities Men+, <5 ppm, that could have oxidized to Me(n+1)+ by reducing H2O according to: 2 Men+ + H2O = 2 Me(n+1)+ + O2− + H2.
A further hint of what was happening came from the observation that the
MgO began to emit O atoms above 600°C (30). This result suggested
peroxy anions, O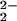 , decomposing according to the
reaction O
, decomposing according to the
reaction O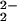 = 1/2 O2 +
O2− with O atoms being the primary product of
disproportionation (29). This result led to the proposition that the
formation of H2 molecules in the MgO matrix was
coupled to the formation of peroxy anions by way of a hitherto unknown
redox conversion involving OH− pairs:
= 1/2 O2 +
O2− with O atoms being the primary product of
disproportionation (29). This result led to the proposition that the
formation of H2 molecules in the MgO matrix was
coupled to the formation of peroxy anions by way of a hitherto unknown
redox conversion involving OH− pairs:
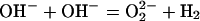 |
3 |
The validity of this reaction was independently confirmed by a group at Oxford University (31) measuring the oxidizing properties of MgO prepared in a similar manner.
Eq 3 is remarkable in as much as it implies a redox conversion in which the oxygens of OH− act as electron donors transferring electron density to the protons, thereby reducing them to H2. Because a peroxy anion represents an excess O atom, this effectively describes a “water splitting” reaction, H2O = H2 + O. Although well documented (30, 31), this redox conversion has so far not been considered in the geosciences as an entry to better understand the interactions between H2O and minerals.
Details of this reaction were further elaborated through an IR
study of MgO crystals (17), which provided evidence that the conversion
takes place among pairs of OH− at specific
defect sites, where Mg2+ vacancies are chargewise
compensated by two OH−. Around 500°C the
OH− pairs convert to O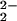 plus
H2. Because of this conversion, the
νOH band at 3560 cm−1 in
Fig. 1, assigned to OH− pairs at
Mg2+ vacancy sites (17), is relatively weak.
Because a majority of the OH− pairs in the MgO
matrix is affected, this result leads to such a large number of
H2 molecules that the νHH
band at 4150 cm−1 becomes observable, as
demonstrated by Fig. 1.
plus
H2. Because of this conversion, the
νOH band at 3560 cm−1 in
Fig. 1, assigned to OH− pairs at
Mg2+ vacancy sites (17), is relatively weak.
Because a majority of the OH− pairs in the MgO
matrix is affected, this result leads to such a large number of
H2 molecules that the νHH
band at 4150 cm−1 becomes observable, as
demonstrated by Fig. 1.
In the same IR study (17), evidence was obtained that solute
CO2 in the MgO matrix exists in a chemically
reduced form, probably as formate anions, CO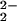 , with
C occupying Mg2+ vacancy sites, and in an even
more reduced form as CO− anions with C on
interstitial sites. This finding led to the proposition that
dissolution of CO2 in solid matrix is accompanied
by a redox conversion similar to Eq. 3 in as much as
O2− acts as electron donor to reduce the
C-bearing solutes:
, with
C occupying Mg2+ vacancy sites, and in an even
more reduced form as CO− anions with C on
interstitial sites. This finding led to the proposition that
dissolution of CO2 in solid matrix is accompanied
by a redox conversion similar to Eq. 3 in as much as
O2− acts as electron donor to reduce the
C-bearing solutes:
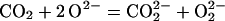 |
4 |
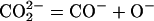 |
5 |
These reactions are not limited to MgO, but apparently also occur in olivine (Mg,Fe)2SiO4 (23), even though, containing about 10% Fe2+, upper mantle olivine may appear to be unlikely to contain oxygen oxidized to the peroxy stage (32). The νCH bands in olivine and their similarity to the νCH bands in MgO suggest that redox conversions whereby O2− acts as electron donor may be common when H2O and CO2 dissolve in mineral matrices.
Segregation.
Dissolution of H2O and CO2 in mineral matrices takes place during crystallization when the gas/fluid components partition between the melt and the growing crystals. The amount of H2O and CO2 taken up into solid solution depends on the temperature (T) and partial pressures of the gas/fluid components (12). However, the equilibrium concentrations of the solute H2O and CO2 species decreases with decreasing T. This sets up a thermodynamic driving force to segregate the solutes to the surface or to any other sink that might be available inside the bulk (33, 34). The denser the crystal structure, the larger is the driving force. As long as the diffusion of the major cationic and anionic lattice constituents remains activated, i.e., at high T, segregation will simply lead to degassing of H2O and CO2. At lower T, as diffusion of cations and anions freezes, only those solutes can respond to the thermodynamic driving force that remain diffusively mobile. If some solutes that derive from H2O and CO2 retain diffusive mobility, they will segregate to the surface as well as to dislocations and other defects.
H2 molecules are diffusively highly mobile in fused silica and quartz, which have relatively open structures (35). Although no diffusion coefficients for H2 in MgO and olivine have been reported, given their small size and high polarizability, H2 molecules are expected to retain diffusive mobility even in such dense structures down to relatively low temperatures. The case of C diffusion requires additional comments. By studying the temperature-time dependent behavior of solute C in MgO and olivine by x-ray photoelectron spectroscopy, 12C(d,p)13C depth profiling (22, 23), and secondary ion mass spectrometry (21) evidence was obtained that the diffusion of C involves the CO− complex postulated in Eq. 4, i.e., a C atom bonding to one O− (21). When C occupies an interstitial site and bonds to O−, the short C—O− bond (≈1.2 Å) will create a local volume contraction that lowers the activation energy barrier for the C atom to execute a diffusional jump to the next interstitial site. By bonding to a succession of O− and executing a succession of interstitial jumps, solute C would thus be able to diffuse even through a densely packed O2− matrix. Such a mechanism involves only transport of C atoms, because an O− represents nothing but an electronic charge, i.e., a defect electron, moving from O2− to O2− in an otherwise stationary O2− matrix.
Experimentally, during heating of MgO and olivine crystals, surface segregation of C sets in around 200°C (21–23), implying that, during cooling under geological conditions, solute C can be expected to segregate down to relatively low temperatures. The most widely available segregation sites inside crystals, however, are dislocations.
Dislocations can be classified into screw and edge dislocations (36). Fig. 5A depicts an ao/2[100] screw dislocation and Fig. 5B the projection of two edge dislocations marking a subgrain boundary in MgO. The arrows in Fig. 5B point at the rows of Mg2+ cations along the edge dislocations that are under high compressive stress and therefore energetically unfavorable. To reduce the stress, two possibilities exist: either remove this one highly stressed row of Mg2+ or remove in addition a row of O2− next to it. In the first case, the core of the dislocation becomes negatively charged. In the second case, charge neutrality is maintained but at the expense of creating a larger void.
Figure 5.

(A) An ao/2[100] screw dislocation in MgO showing how the (100) plane is displaced by ao/2 after one turn. The large dark spheres indicate O2− anions, the smaller light spheres Mg2+ cations. (B) Projection of two idealized edge dislocations generated as part of a subgrain boundary in MgO by the insertion of two half-planes of Mg2+ and O2−. The arrows point at rows of Mg2+ cations that are under high compressive stress and therefore energetically unfavorable (after Harding et al., ref. 38).
To model dislocations in compound crystals and avoid complications from net charges, it is common practice to remove an equal number of cations and anion from the dislocation core (37, 38). We have chosen to remove only Mg2+ cations. This decision was justified by the fact that dislocations in compound crystals are generally charged because, as they form and sweep through the structure, they collect cation vacancies (33, 34). Therefore, as their local stoichiometry deviates from the overall stoichiometry of the crystal, they become negatively charged. The CO− complex postulated in Eq. 4 is positively charged. Carrying a negative charge, dislocations will attract CO− through long-range Coulomb interaction, facilitating the formation of polyatomic Cx entities through segregation (21).
Modeling Dislocations.
As part of the work described here, we modeled screw and edge dislocations in MgO and their interaction with CO− by decomposing the process into three steps: first, we remove one row of Mg2+ cations; second, we convert the two adjacent rows of O2− to O−, thus providing full charge compensation for the Mg2+ vacancies; and third, we allow C to segregate into the dislocation cores and to bond to O−.
When we add C atoms one by one to the dislocation cores, allowing them to also bond to each other, we build short Cx chains with the terminal C atoms bonded to two O− of the MgO matrix. Taking into account the lattice parameter of MgO, ao = 4.21 Å, the C—C bond length and bond angles, we can build aliphatic Cx chains with n = 4 that are strain-free, even without taking into account a possible relaxation of the MgO matrix around the dislocation cores (37, 38). In the case of the ao/2[100] screw dislocation, we find the best fit for C4 units in trans configuration. In the case of subgrain boundary dislocations, to fit into the dislocation core, the C4 units have to buckle into a cis configuration. For n > 4 the C—O− bonds at the terminal C positions become progressively more strained and go out of phase with respect to the surrounding MgO matrix.
Fig. 6A shows a cut through a stack of MgO planes containing an ao/2[100] screw dislocation. Its core is decorated with a C4 unit in trans configuration and its terminal C atoms bonding to two O− each, resulting in an [O2C—C—C—CO2] unit. Fig. 6B provides an oblique view of an edge dislocation with one row of Mg2+ cations removed and two rows of O− lining the core. In Fig. 6C, C atoms are segregated into the axis of the dislocation core forming [O2C—C—C—CO2] units in cis configuration.
Figure 6.

(A) Stack of MgO (100) planes with a vertical ao/2[100] screw dislocation in the center, viewed at right angle. The screw dislocation is decorated by one [O2C—C—C—CO2] unit in the energetically favorable trans configuration with the medium size light gray spheres representing O−. (B) Oblique view of an edge dislocation in MgO with one row Mg2+ removed and two rows of O2− changed to O− (lighter gray spheres). (C) Same edge dislocation with C4 units segregated into its core. Because of mismatch with the surrounding MgO matrix, the [O2C—C—C—CO2] units would have to adopt the energetically less favorable cis configuration.
Stereochemical Control.
Dislocations provide for a stereochemically constrained environment that forces the segregating C atoms into Cx chains. Depending on the specific conditions and/or the mineral structure, different chain length Cx entities might form. In other types of lattice defects, solute C atoms may segregate to form cyclic or branched Cx entities. We are here confronted with the possibility that the mineral matrices in which Cx entities precipitate influence or even control the shape and size of these Cx segregates. Hence, when these minerals weather, they will release a set of stereochemically preselected organic molecules. Such a mechanism is expected to produce a smaller number of different compounds than reactions in the gas phase, liquid phase, or by surface catalysis.
On the basis of the simulations presented here, it appears that linear Cx entities with n = 4 in trans configuration might be energetically favored in screw dislocations in MgO. With their terminal C atoms bonding stress-free to two O− each, they would anchor the [O2C—C—C—CO2] entity to the MgO structure. Adding H2 leads to —CH2— and to entities that may be described as [O2C(CH2)2CO2] or, more generally, as Cx—Hy—Oz entities. The —CH2— therein would give rise to two νCH bands arising from the symmetrical and asymmetrical C—H stretching mode similar to these modes in the IR spectrum of polyethylene (25). The two strongest νCH bands at 2926 and 2855 cm−1 in the IR spectrum of MgO (see Fig. 2) would then have to be assigned to —CH2— sections of linear aliphatic chains of the general formula Cx—Hy—Oz, formed through segregation of C and H2 into dislocations.
Solvent extraction experiments of crushed MgO single crystals produced a suite of carboxylic and dicarboxylic acids from C2 to C4 with succinic acid, HOOC(CH2)2COOH, being a major component (4), pointing at dislocation-bound [O2C(CH2)2CO2] entities as possible protomolecules. Extraction experiments with crushed olivine crystals yielded longer chain-length fatty acids, C6 to C12 (1), suggesting that longer-chain Cx—Hy—Oz entities had formed in the olivine matrix. The difference in chain length may reflect differences between the structures of MgO and olivine. Alternatively, because the olivine crystals had cooled at a much slower rate in a volcanic pipe (24), there was more time for the segregation of solute C into the dislocations, thus producing longer chain Cx—Hy—Oz entities. These longer Cx—Hy—Oz entities may be the reason why, as seen in Fig. 2, the νCH bands from the olivine crystal are narrower than the νCH bands from the MgO crystal, even though MgO has a simpler structure and would thus be expected to provide a more uniform local lattice environment.
Prebiotic Evolution.
Many pathways are known by which organic matter can be synthesized through gas phase, liquid phase, and gas-solid and liquid-solid reactions. How much could have been produced under early Earth conditions depends on whether or not the atmosphere at that time was reducing. Table 1 gives estimated production rates (8) for two cases: (i) a highly reducing atmosphere rich in methane, hydrogen, and ammonia, and (ii) an intermediate atmosphere, still reducing, with an H2/CO2 ratio of 1:10. More likely, however (39), the early atmosphere was non-reducing. In this case almost no organics would have been produced in the atmosphere by the processes indicated.
Table 1.
Major source of prebiotic organics for two candidate early atmospheres (after 8)
| Source | Highly reducing atmosphere, g⋅yr−1 | Intermediate atmosphere, g⋅yr−1 |
|---|---|---|
| UV photolysis | 1 × 1015 | 3 × 1011 |
| Electric discharges | 3 × 1012 | 3 × 1010 |
| Shocks from large impacts | 2 × 1013 | 4 × 105 |
| Shocks from meteors | 4 × 1012 | 8 × 104 |
| Totals | ≈1 × 1015 | ≈5 × 1011 |
Interplanetary dust particles (IDPs) in the size range 0.6–60 μm and containing up to 10% carbon provide an exogenous source for organics. Because they are slowly decelerated on entry in the Earth's atmosphere, their organics survive the entry. Today, the Earth captures between 3 × 109 and 1 × 1010 g⋅yr−1 IDPs, contributing between 3 × 108 and 1 × 109 g⋅yr−1 organics (40, 41). The rate of IDP capture after the period of heavy bombardment was probably much higher (8), maybe 2–3 orders of magnitude higher, thus delivering about 1011–1012 g⋅yr−1 of organics.
The work presented here points at a source of organics that is different from all other sources discussed so far in the literature. If Cx—Hy—Oz protomolecules form in mineral matrices, they will turn into organic molecules during weathering (1, 4). The organic molecules thus liberated may be difficult to synthesize by any of the other reaction pathways given in Table 1. Note that these reaction pathways invariably involve large departures from thermodynamic equilibrium, mostly in form of very high temperatures for a very short period. By contrast, the assembly of Cx—Hy—Oz protomolecules inside the minerals and their release during weathering are processes that occur under much more benign conditions, at moderate or even ambient temperatures, and not too far from equilibrium.
We can estimate how much organics could have been supplied to the Earth through the weathering cycle. On the early continents many of the rocks exposed at the surface were probably peridotites, rich in olivine and other minerals that weather rapidly under the effect of CO2-saturated meteoric water. Assume that the volume of rock recycled was of the order of 3 km3/yr (1016 g/yr), the same as today (42), and that their solute C content was of the order of 100 ppm as in olivine (27, 28). If 1/10 of this solute C or 10 ppm were in form of Cx—Hy—Oz protomolecules, the weathering cycle would have produced organics at a rate of about 1011 g/yr. The production would have been unaffected by the atmospheric composition and independent of any delivery of meteors and comets to the early Earth (43). Furthermore, the production rate would have been sustained over long time or even increased with the growth of the continents. If we include subsurface weathering such as serpentinization of peridotites and leaching of rocks by hydrothermal fluids, the production rate of organics would be even higher.
Conclusions
The assembly of protomolecules by way of solid state processes shows that “organic” chemistry can take place in the dense, hard matrix of igneous minerals. This finding opens new aspects to the study of stereochemically constrained complex organic molecules, synthesized under prebiotic conditions. Such processes may have provided large amounts of biochemically relevant organics to the early Earth.
Acknowledgments
We thank Bishun Khare for the opportunity to use the Nicolet Nexus 670 FT-IR spectrometer. J.S. thanks the NASA Astrobiology Academy for the opportunity to spend the Summer 2000 at the NASA Ames Research Center. This work was supported by the Exobiology Program of the National Aeronautics and Space Administration under RTop 344-38-22-15. A.S. participated through Grant PHY-9605147 from the National Science Foundation as part of the REU (Research Experience for Undergraduates) Program at the Department of Physics, San Jose State University.
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Nominal high purity refers to metal cation impurities only, which are routinely measured. It does not take into account impurities that may arise from the dissolution of gases in solid matrix.
References
- 1.Freund F, Gupta A, Kumar D. Origins Life Evol Biosphere. 1999;29:489–509. doi: 10.1023/a:1006698710710. [DOI] [PubMed] [Google Scholar]
- 2.Freund F, Ho R. In: Circumstellar Habitable Zones. Doyle L R, editor. Menlo Park, CA: Travis House; 1996. pp. pp.71–98. [Google Scholar]
- 3.Butler C T, Sturm B J, Quincy R B. J Cryst Growth. 1971;8:197–204. [Google Scholar]
- 4.Gupta A, Freund F. Lunar & Planetary Science Conference. Vol. 29. Houston, TX: Lunar & Planetary Institute; 1998. [Google Scholar]
- 5.McCollom T M, Ritter G, Simoneit B R T. Origins Life Evol Biosphere. 1999;29:153–166. doi: 10.1023/a:1006592502746. [DOI] [PubMed] [Google Scholar]
- 6.Bernd M E, Allen D E, Seyfied W E. Geology. 1996;24:351–354. [Google Scholar]
- 7.Hennet R J C, Holm N G, Engel M H. Naturwissenschaften. 1992;79:361–365. doi: 10.1007/BF01140180. [DOI] [PubMed] [Google Scholar]
- 8.Chyba C F, Sagan C. Nature (London) 1992;355:125–132. doi: 10.1038/355125a0. [DOI] [PubMed] [Google Scholar]
- 9.Chang S. In: The Chemistry of Life's Origins. Greenberg J M, editor. Dordrecht, The Netherlands: Kluwer; 1993. pp. 259–299. [Google Scholar]
- 10.Rossman G R. Phys Chem Miner. 1996;23:299–304. [Google Scholar]
- 11.Bell D R, Rossman G R. Science. 1992;255:1391–1397. doi: 10.1126/science.255.5050.1391. [DOI] [PubMed] [Google Scholar]
- 12.Burnham W C. In: The Evolution of Igneous Rocks: Fifthieth Anniversary Perspectives. Yoder H S, editor. Princton, NJ: Princeton Univ. Press; 1979. pp. 439–482. [Google Scholar]
- 13.Spera F J, Bergman S C. Contrib Miner Petrol. 1980;74:55–66. [Google Scholar]
- 14.Fine G, Stolper E. Contrib Miner Petrol. 1985;91:105–121. [Google Scholar]
- 15.Mathez E A. Geochim Cosmochim Acta. 1987;51:2339–2347. [Google Scholar]
- 16.Tingle T N, Mathez E A, Hochella M F. Geochim Cosmochim Acta. 1991;55:1345–1352. doi: 10.1016/0016-7037(93)90537-7. [DOI] [PubMed] [Google Scholar]
- 17.Freund F, Wengeler H. J Phys Chem Solids. 1982;43:129–145. [Google Scholar]
- 18.De Remigi J, Welsh H L. Can J Phys. 1970;48:1622–1627. [Google Scholar]
- 19.Tsong I S T, Knipping U, Loxton C M, Magee C W, Arnold G W. Phys Chem Miner. 1985;12:261–270. [Google Scholar]
- 20.Mathez E A, Blacic J D, Berry J, Maggiore C, Hollander M. J Geophys Res. 1987;92:3500–3506. [Google Scholar]
- 21.Freund F. Phys Chem Miner. 1986;13:262–276. [Google Scholar]
- 22.Kathrein H, Gonska H, Freund F. Appl Phys. 1982;30:33–41. [Google Scholar]
- 23.Oberheuser G, Kathrein H, Demortier G, Gonska H, Freund F. Geochim Cosmochim Acta. 1983;47:1117–1129. [Google Scholar]
- 24.Frey F A, Prinz M. Earth Planet Sci Lett. 1978;38:129–176. [Google Scholar]
- 25.Spectra S S. Infrared Spectra Atlas of Monomers and Polymers. New York: Wiley; 1980. [Google Scholar]
- 26.Freund F. Phys Chem Miner. 1986;13:280. [Google Scholar]
- 27.Minaev V M, Shilobreeva S N, Kadik A A. J Radioanal Nucl Chem. 1995;189:147–155. [Google Scholar]
- 28.Kadik A A, Shilobreeva S S, Minaev V M, Kovalenko V I. Lunar and Planetary Science XXVII. Vol. 27. Houston, TX: Lunar and Planetary Institute; 1996. pp. 631–632. [Google Scholar]
- 29.Wells A F. Structural Inorganic Chemistry. Oxford: Clarendon; 1984. [Google Scholar]
- 30.Martens R, Gentsch H, Freund F. J Catalysis. 1976;44:366–372. [Google Scholar]
- 31.Praliaud H, Coluccia S, Deane A M, Tench A J. Chem Phys Lett. 1979;66:44–47. [Google Scholar]
- 32.Freund F, Oberheuser G. J Geophys Res. 1986;91:745–761. [Google Scholar]
- 33.Nowotny J. In: Surfaces and Interfaces of Ceramic Materials. Dufour L C, Monty C, Petot-Ervas G, editors. Dordrecht, The Netherlands: Kluwer; 1989. pp. 205–239. [Google Scholar]
- 34.Pennycock S J, Chisholm M F, Yan Y, Duscher G, Pantelides S T. Phys B. 1999;274:453–457. [Google Scholar]
- 35.Shelby J E. J Appl Phys. 1977;48:3387–3394. [Google Scholar]
- 36.Philibert J. In: Basic Properties of Binary Oxides. Dominiguez-Rodrigues A, Castaing J, Marques R, editors. Sevilla, Spain: Univ. Sevilla; 1983. pp. 279–296. [Google Scholar]
- 37.Harris D J, Khan M A, Parker S C. Phys Chem Miner. 1999;27:133–137. [Google Scholar]
- 38.Harding J H, Harris D J, Parker S C. Phys Rev B. 1999;60:2740–2746. [Google Scholar]
- 39.Kasting J F. Science. 1993;259:920–926. doi: 10.1126/science.11536547. [DOI] [PubMed] [Google Scholar]
- 40.Anders E. Nature(London) 1989;342:255–257. doi: 10.1038/342255a0. [DOI] [PubMed] [Google Scholar]
- 41.Love S G, Brownlee D E. Science. 1993;262:550–553. doi: 10.1126/science.262.5133.550. [DOI] [PubMed] [Google Scholar]
- 42.Koster von Groos A F. J Geophys Res. 1988;93:8952–8958. [Google Scholar]
- 43.Chyba C F, Thomas P J, Brookshaw L, Sagan C. Science. 1990;249:366–373. doi: 10.1126/science.11538074. [DOI] [PubMed] [Google Scholar]