

Abstract
Investigations of K+-occlusion by the phosphoenzyme of Na+,K+-ATPase from shark rectal gland and pig kidney by stopped-flow fluorimetry reveal major differences in the kinetics of the two enzymes. In the case of the pig enzyme, a single K+-occlusion step could be resolved with a rate constant of 342 (±26) s−1. However, in the case of the shark enzyme, two consecutive K+-occlusions were detected with rate constants of 391 (±19) s−1 and 48 (±2) s−1 at 24°C and pH 7.4. A conformational change of the phosphoenzyme associated with K+-occlusion is, thus, the major rate-determining step of the shark enzyme under saturating concentrations of all substrates, whereas for the pig enzyme the major rate-determining step under the same conditions is the E2 → E1 transition and its associated K+ deocclusion and release to the cytoplasm. The differences in rate constants of the K+ occlusion reactions of the two enzymes are paralleled by compensating changes to the rate constant for the E2 → E1 transition, which explains why the differences in the enzymes' kinetic behaviors have not previously been identified.
Introduction
The Na+,K+-ATPase is a crucial enzyme of animal physiology. It is responsible for maintaining Na+ and K+ electrochemical potential gradients across the plasma membrane of all animal cells, which are essential for the maintenance of cell volume and for a variety of physiological processes, e.g., nerve, muscle, and kidney function.
Crystal structures of the Na+,K+-ATPase from both pig (1) and shark (2,3) have recently been reported. These structures contain MgF42−, as an analog of phosphate, and Rb+ or K+ in the cation-binding sites. They are thought to represent enzyme in the E2[K2]·Pi state, an enzyme state intermediate between the nonoccluded E2PK2 state and the occluded E2[K2] state, i.e., after hydrolysis of the enzyme-phosphate bond but before the dissociation of Pi from the enzyme. An occluded state (signified here by square brackets) is defined as one in which the bound ions are enclosed within the protein with no direct access to either the cytoplasm or the extracellular medium.
Here we report kinetic measurements using the electric-field-sensitive fluorescence probe RH421 on partial reactions of the Na+,K+-ATPase purified from shark rectal gland and the outer medulla of pig kidney, i.e., preparations from the same animals for which the crystal structures have been determined. In previous studies it has been found (4) that the Na+,K+-ATPase of these two preparations have similar turnover numbers of ∼200 s−1 at 37°C (or 50–70 s−1 at 24°C) at saturating substrate concentrations. However, this does not necessarily mean that the kinetics of the partial reactions of the two enzymes are the same. In fact, it has recently been found (5) that there are significant differences in the kinetics of the E2 → E1 conformational transition of the two enzymes. Whereas the pig enzyme displays a maximum rate constant for this reaction of ∼40 s−1 at saturating ATP concentrations at 24°C and pH 7.4, for the shark enzyme the value was found to be 182 (±6) s−1 under the same conditions. Thus, the E2 → E1 transition is a major rate-determining step of the pig enzyme, but its contribution to rate-limitation must be much less in the case of the shark enzyme. Another difference found (5) was that the kinetics of pig enzyme suggested it behaved as an (αβ)2 dimer of the enzyme's α- and β-subunits at low ATP concentrations, whereas the shark enzyme's kinetic behavior was consistent with an (αβ) monomer at all ATP concentrations.
Because of the significant differences in the kinetics of the E2 → E1 transition of the pig and shark enzyme, there must be other differences within the enzyme cycle which counteract these to lead to the similar turnover numbers of both enzymes. Previous kinetic studies (6,7) have shown no significant difference in the kinetics of enzyme phosphorylation and conversion to the E2P state, i.e., the sequence of reactions
In this article we, therefore, concentrate on subsequent steps of the cycle, i.e., binding of K+ to the E2P state, K+ occlusion within the protein, and dephosphorylation of the enzyme to the state E2[K2]. Preliminary measurements on these reactions indicated (8) that differences in the kinetics between the two enzymes do in fact exist.
Another reason to study K+ binding to the E2P state is that apparent discrepancies are present in the literature regarding the affinity of K+ to this state. From kinetic studies on enzyme from pig kidney, Kane et al. (9) reported an apparent dissociation constant of 1.3 (±0.2) mM (pH 7.4, 24°C, [NaCl] = 130 mM). A similar value of 1.9 mM was reported by Gropp et al. (10) for enzyme from shark rectal gland from kinetic measurements conducted under different conditions (pH 6.2, 24°C, [NaCl] = 130 mM or 40 mM). However, based on steady-state titrations of rabbit kidney enzyme, a 10-fold higher apparent affinity has been reported by Bühler and Apell (11), who found a half-saturating K+ concentration of 0.192 mM (pH 7.2, 20°C, [NaCl] = 21 mM).
Materials and Methods
Enzyme and reagents
Na+,K+-ATPase-containing membrane fragments from shark rectal gland and pig kidney were purified as described by Skou and Esmann (12) and Klodos et al. (13), respectively. The specific ATPase activities at 37°C and pH 7.4 were measured according to Ottolenghi (14). The activities of the preparation used were 1765 and 1365 μmol ATP hydrolyzed h−1 (mg of protein)−1 at saturating substrate concentrations for shark and pig enzyme, respectively. The protein concentrations were 6.8 mg mL−1 for shark and 4.7 mg mL−1 for pig enzyme. The concentrations were determined according to the Peterson modification (15) of the Lowry method (16) using bovine serum albumin as a standard.
N-(4-Sulfobutyl)-4-(4-(p-(dipentylamino)phenyl)butadienyl)-pyridinium salt (RH421) was obtained from Molecular Probes (Eugene, OR) and was used without further purification. RH421 was added to Na+,K+-ATPase-containing membrane fragments from an ethanolic stock solution. The dye spontaneously partitions into the membrane fragments.
The origins of the various reagents used were: imidazole (≥99%; Sigma, Castle Hill, Australia), NaCl (suprapure; Merck, Kilsyth, Australia), KCl (analytical grade; Merck), choline chloride (99%; Sigma), MgCl2·6H2O (analytical grade; Merck), EDTA (99%; Sigma), ATP disodium·3H2O (special quality; Roche, Castle Hill, Australia), NaOH (analytical grade; Merck), and HCl (0.1 N Titrisol solution; Merck).
Stopped-flow spectrofluorimetry
Stopped-flow experiments were carried out using an SF-61 stopped-flow spectrofluorimeter from Hi-Tech Scientific (Salisbury, UK). In one type of experiment (sequential mixing), measurements of the kinetics of K+ binding, occlusion, and K+-induced dephosphorylation of the enzymes were carried out by first phosphorylating the enzyme with ATP and then stimulating dephosphorylation by subsequently mixing the phosphorylated enzyme with KCl. This was performed using a modification of a method described previously (9). Briefly, Na+,K+-ATPase-containing membrane fragments (60 μg/mL of enzyme) from either shark rectal gland or pig kidney, labeled with RH421 (800 nM), were premixed in one of the drive syringes with 0.5 M Na2ATP to a final ATP concentration of 4 mM.
After phosphorylation of the enzyme had reached a steady state (this occurs within 1 s), the enzyme was mixed with an equal volume of a KCl solution from the other drive syringe. To avoid any change in ionic strength or Na+ concentration on mixing, the KCl solutions contained 40 mM NaCl and choline chloride of varying concentrations such that the total concentration of KCl, NaCl, plus choline chloride was constant at 130 mM. This ionic strength was chosen to agree with physiological conditions. The enzyme suspension and the KCl solutions were prepared in the same buffer containing, in addition to choline chloride, 30 mM imidazole, 40 mM NaCl, 5 mM MgCl2, and 1 mM EDTA. In the case of the enzyme suspension, 90 mM of choline chloride was added to the buffer so that the total concentration of NaCl and choline chloride was 130 mM, i.e., identical to the sum of the KCl, NaCl, and choline chloride concentrations of the KCl solutions. The concentration of 40 mM NaCl was chosen to ensure the maximal rate of enzyme phosphorylation and hence the maximum conversion of enzyme into the E2P state (6,7). The pH of the buffer was adjusted to 7.4 by the addition of HCl.
To improve the signal/noise ratio, typically between 9 and 21 traces were averaged before the observed rate constants, kobs, or the amplitudes, ΔF/F0, were evaluated. The observed rate constants and the magnitudes of the amplitudes were determined from nonlinear least-squares fits of either one or a sum of two exponential functions to the averaged experimental trace. The choice between a single- or a double-exponential fit was made based on the presence or absence of any observed deviation from random fluctuation in residual plots and the values of the χ2 parameter.
In a second type of experiment (simultaneous mixing), Na+,K+-ATPase-containing membrane fragments labeled with RH421 in one of the drive syringes were mixed simultaneously with both Na2ATP (4 mM) and KCl (varying concentrations) in the other drive syringe. Equal volumes of the enzyme-containing suspension and the ATP/KCl solution were mixed from both drive syringes in all cases. Before mixing, the enzyme was incubated in a buffer containing 40 mM NaCl and 90 mM choline chloride, so that the enzyme can be considered to be initially in the state E1Na3. To avoid any changes in ionic strength on mixing, the ATP/KCl solutions contained varying concentrations of choline chloride such that the sum of the KCl and choline chloride concentrations was always 90 mM. The enzyme suspension and the ATP/KCl solutions were prepared in the same buffer containing, in addition to choline chloride, 30 mM imidazole, 40 mM NaCl, 5 mM MgCl2, and 1 mM EDTA. The pH of the buffer was adjusted to 7.4 by the addition of HCl.
Results
Kinetics of sequential K+-mixing experiments
Typical stopped-flow kinetic traces obtained with both the pig kidney and shark rectal gland enzymes are shown in Fig. 1. Both enzymes display a drop in fluorescence of the probe RH421 on mixing of phosphorylated enzyme with KCl. A significant difference between the two enzymes is that, in the case of the pig kidney enzyme, the fluorescence drop is monophasic, whereas for the shark rectal gland it is biphasic. To adequately fit the shark kinetic data, it was found that, at all K+ concentrations above 0.20 mM (after mixing), the fitting function had to include two exponentials.
Figure 1.
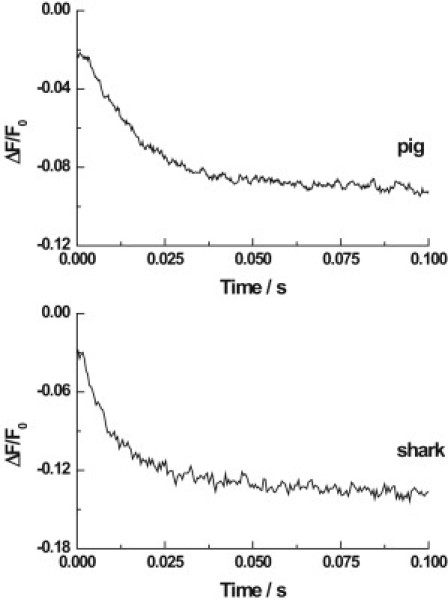
Sequential mixing stopped-flow fluorescence transients of Na+,K+-ATPase from pig kidney and shark rectal gland noncovalently labeled with RH421. Na+,K+-ATPase (60 μg/mL) was premixed with a small volume of 0.5 M Na2ATP to a final concentration of 4 mM ATP. A few seconds later, the enzyme was mixed with an equal volume of a KCl solution (0.6 mM after mixing). The final Na+,K+-ATPase concentration was 30 μg/mL, and the RH421 concentration was 400 nM. Both the enzyme suspensions and the KCl solutions were in a buffer containing 30 mM imidazole, 40 mM NaCl, 5 mM MgCl2, and 1 mM EDTA (pH 7.4, 24°C). The total ionic strength was maintained at a constant value by the addition of choline chloride to the enzyme suspension and the KCl solutions ([choline chloride] + [KCl] = 90 mM). The fluorescence of membrane-bound RH421 was measured at an excitation wavelength of 577 nm at emission wavelengths of ≥665 nm (RG665 glass cutoff filter). The calculated observed rate constants were 67 (±2) s−1 for the pig enzyme and 120 (±11) s−1 (82% of the total amplitude) and 16 (±3) s−1 (18%) for the shark enzyme.
The biphasic nature of the shark kinetic data shown in Fig. 1 may not be obvious. The necessity of a double-exponential fit is, however, quite clear when one inspects the residuals of both single- and double-exponential fits of the data (see Fig. 2). There it can be seen that only the double-exponential fit results in a random fluctuation of the experimental data points about the fitted curve. To test the appropriateness of the double-exponential fit of the shark enzyme data further, we carried out statistical F-tests, as described previously (5). For the shark kinetic curve shown in Fig. 1, which is an average of 18 individual traces, each consisting of 1024 data points, the F value for a double-exponential fit was found to be 316. This value is significantly greater than the critical value of 18 for a 1% probability of the null hypothesis that the double-exponential fit is no better than the single-exponential fit for these data. F tests were also carried out on all of the other averaged shark enzyme stopped-flow transients measured over the K+ concentration 0.2–20 mM after mixing. Representative transients are shown in Fig. 3. For each K+ concentration in this range, the F value was in the range 148–5892, i.e., all above the critical value of 18. Based on this quantitative assessment, we conclude that the biphasic behavior of the shark rectal gland enzyme is statistically significant for all K+ concentrations from 0.2 to 20 mM.
Figure 2.
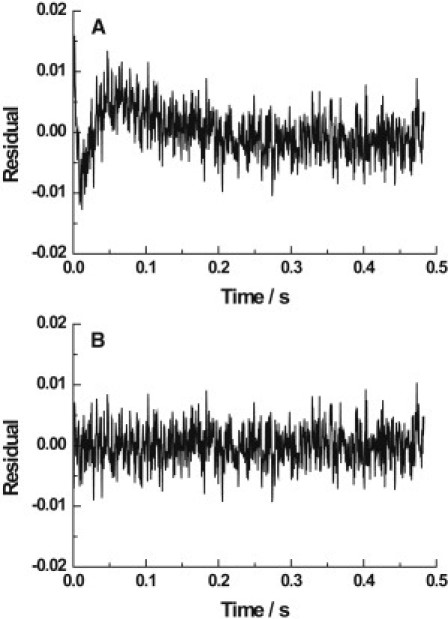
Residual plots of the best (A) single- and (B) double-exponential fits to the stopped-flow fluorescence transient (see Fig. 1, lower panel) obtained for shark rectal gland Na+,K+-ATPase under the conditions described in Fig. 1. The χ2 values were 1.52 for the single-exponential fit and 0.939 for the double-exponential fit.
Figure 3.
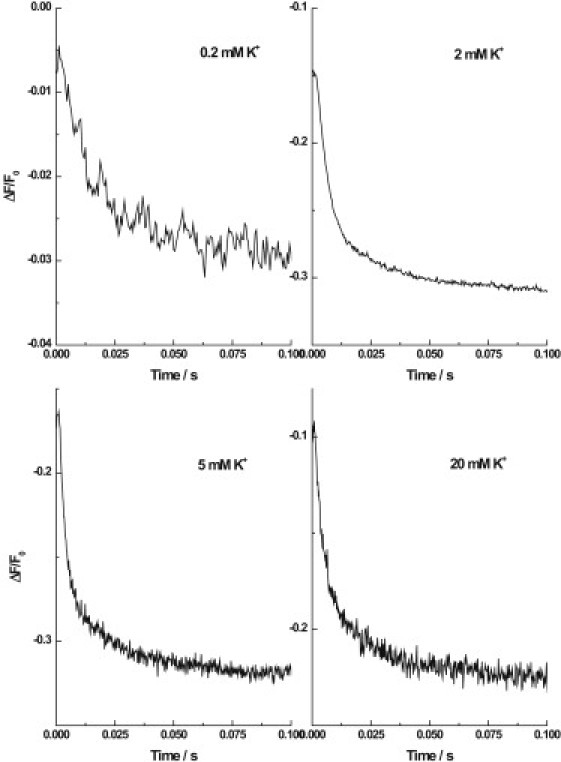
Sequential mixing stopped-flow fluorescence transients of Na+,K+-ATPase from shark rectal gland noncovalently labeled with RH421. All experimental conditions were as described in Fig. 1. The number of averaged transients and the F values for double-exponential fits to the data were: 0.2 mM K+ (17 transients, F = 148), 2 mM K+ (19 transients, F = 3167), 5 mM K+ (14 transients, F = 1416), and 20 mM K+ (15 transients, F = 404). The K+ concentrations are all after mixing.
It is generally agreed that the electric-field-sensitive probe RH421 used in this study detects changes in electric field strength within the membrane-spanning domain of the protein (17–19). It seems unlikely that the probe responds to changes in the phosphorylation state of the enzyme, because experiments including K+ and phosphate to allow the enzyme to operate in the K+,K+-exchange mode (as indicated by radioactive K+ data), which is still associated with enzyme phosphorylation and dephosphorylation but does not involve net charge transport, showed no fluorescence changes after phosphate addition (H.-J. Apell, unpublished results).
Electrophysiological studies (20–22) on the physiological Na+,K+-exchange mode of the enzyme, however, have shown that in this mode of function, K+ binding from the extracellular solution to sites within the protein, before enzyme dephosphorylation, is electrogenic, i.e., it causes a change in electric field strength within the protein. Furthermore, it has been found (23) that the fluorescence change of RH421 is linearly correlated to the amount of Rb+ (a K+ congener) occluded by the enzyme. Therefore, K+ occlusion within the protein is the most likely cause for the drop in fluorescence of the probe. This does not necessarily mean, however, that the protein conformational change responsible for K+ occlusion itself directly causes the fluorescence change. The conformational change could allow the K+ ions to penetrate further into the protein interior, and it may be the electric field-strength change associated with this increased K+ penetration that the dye is responding to. If one accepts K+-occlusion within the protein interior as the cause of the RH421 fluorescence change, the question, however, still remains as to why the drop in fluorescence caused by K+ occlusion by the native enzyme should occur in two steps for the shark enzyme, but only in a single step for the pig enzyme.
In principle, there are two possible causes for a biphasic fluorescence decrease: two consecutive reactions or two independent parallel reactions. In both cases, the two reactions must each cause a fluorescence decrease of the probe. Multiphasic decays in the amount of phosphoenzyme on mixing with K+ have previously been observed using enzyme isolated from electric eel and from shark (24–27). Hobbs et al. (26) considered the possibility that the two fastest phases were associated with dephosphorylation of enzyme distributed between the E1P and E2P states. However, after finding that the proportions of these phases were independent of the lag time between the initiation of phosphorylation and that of dephosphorylation, they concluded that the E1P → E2P transition is very rapid and that neither could be due to conversion of E1P to E2P before dephosphorylation. Another relevant result is that, although K+-stimulated dephosphorylation of the eel enzyme is multiphasic, spontaneous dephosphorylation of the enzyme in the absence of K+ is monophasic (24,25). Therefore, multiphasicity in the phosphoenzyme decay seems to be induced by K+ interaction with the enzyme.
To gain further insight into the origin of the biphasic RH421 fluorescence decay of the shark enzyme and to determine K+ affinities of both enzymes, sequential and simultaneous mixing experiments have been conducted over a wide range of K+ concentrations.
K+ concentration dependence of the sequential K+-mixing kinetic experiments
The K+ concentration dependences of the kinetics of the observed rate constants, kobs, for the single phase drop in fluorescence observed for the pig kidney enzyme and for both phases observed for the shark rectal gland enzyme are shown in Figs. 4 and 5. In all cases it was found that kobs increases with increasing K+ concentration at low concentrations but saturates at a constant value at high K+ concentrations. This is in agreement with previous observations on the pig enzyme (9). This indicates that none of the kinetic phases being observed can be attributed alone to a second-order binding of K+ to the protein surface, because such a process would be expected to result in an indefinite linear increase in kobs with increasing K+ concentration. Binding of K+ to the protein surface would also not be expected by itself to induce a fluorescence change of RH421, because, as described above, to cause a change in electric field strength within the protein the ions must reach their binding sites within the protein matrix where they are occluded.
Figure 4.
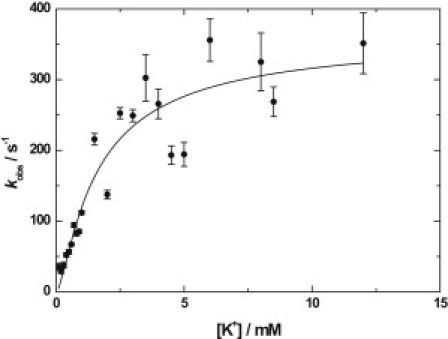
Dependence of the observed rate constant, kobs, of the RH421 fluorescence change on the concentration of K+ (after mixing) for sequential mixing stopped-flow experiments in which Na+,K+-ATPase from pig kidney was premixed with Na2ATP to a concentration of 4 mM for a few seconds to allow for phosphorylation and then subsequently mixed with KCl. All experimental conditions were as described in the caption to Fig. 1. (Solid line) Nonlinear least-squares fit of Eq. 1 to the data. The fit parameters were k = 342 (±26) s−1 and Kd = 0.67 (±0.14) mM.
Figure 5.

Dependence of the observed rate constant, kobs, of the fast phase (A) and the slow phase (B) of the RH421 fluorescence change on the concentration of K+ (after mixing) for sequential mixing stopped-flow experiments in which Na+,K+-ATPase from shark kidney was premixed with Na2ATP to a concentration of 4 mM for a few seconds to allow for phosphorylation and then subsequently mixed with KCl. All experimental conditions were as described in the caption to Fig. 1. (Solid lines) Nonlinear least-squares fits of either Eq. 2 (fast phase, A) or Eq. 1 (slow phase, B) to the data. The fit parameters derived for the fast phase (A) were kfast = 392 (±19) s−1 and Kd1 = 1.0 (±0.2) mM. For the slow phase (B), the fit parameters derived were kslow = 48 (±2) s−1 and Kd = 0.38 (±0.05) mM.
The saturation in kobs at high K+ concentrations implies a first-order reaction, but the fact that kobs increases at low K+ concentrations suggests that the first-order reaction is undergoing rate-limitation by a preceding second-order reaction at low K+ concentrations. Possible first-order reactions are:
-
1.
The restricted diffusion of K+ from the protein surface to the ion binding sites within the protein,
-
2.
Dephosphorylation of the protein stimulated by K+ occlusion, and
-
3.
A conformational change of the protein responsible for K+ occlusion.
Restricted diffusion of K+ to its binding sites is unlikely to be responsible for the observed kinetics, because one would expect this to occur on a much faster timescale. For example, diffusion of ions through channels occurs with rate constants of up to ∼108 s−1 (28), in comparison to the maximum kobs values measured here of only ∼400 s−1.
If the fluorescence change of RH421 is due to K+ occlusion (23), K+-stimulated dephosphorylation cannot be responsible for the kinetics of the fluorescence change observed for the pig enzyme or for the rapid phase of the shark enzyme because this would only occur after occlusion. Dephosphorylation could be a possible origin of the slow phase of the fluorescence change observed with the shark enzyme, but only if the fluorescence level of the dephosphorylated state, E2[K2], were lower than that of the E2P[K2] state. However, as discussed earlier, this is unlikely to be the case because of the absence of any observed RH421 response to the phosphorylation or dephosphorylation reactions in other experiments.
The most likely explanation for the first-order reaction responsible for the kobs values of 300–400 s−1 observed for both enzymes is that it is due to a protein conformational change which occludes K+ within the protein. Such a rate constant would be consistent with the values of rate constants observed for protein conformational changes responsible for the opening and closing of ion channels, which are in the range 100–1000 s−1 (28).
If the first-order process causing the saturation in kobs arises from K+-occlusion, the preceding second-order reaction must be due to the binding of K+ to its binding sites within the protein (still open to the extracellular medium). Hence, an estimate of the binding affinity of K+ can be determined from the K+ concentration dependence of kobs.
Considering first the monophasic pig kidney enzyme results, the following reaction scheme is sufficient to explain the observed kinetic behavior,
where Kd represents the microscopic dissociation constant for binding of K+ to the sites within the protein but still open to the extracellular medium, and k represents the apparent rate constant for an enzyme conformational change leading to the occlusion of the K+ ions within the phosphoenzyme. If the first step is in a rapid equilibrium on the timescale of the second reaction, the dependence of kobs on the concentration of K+ is given by (10)
| (1) |
Fitting Eq. 1 to the values of kobs experimentally determined for the pig enzyme (see Fig. 4) yields the values: k = 342 (±26) s−1 and Kd = 0.67 (±0.14) mM. The value of k is in quite good agreement with previous measurements on pig enzyme (9), which yielded saturating values of kobs in the range 366–420 s−1. The value of Kd is somewhat lower than the value previously reported of 1.33 (±0.22) mM for pig enzyme (9). A possible reason for this is that in this study a lower NaCl concentration of 40 mM was used in comparison to 130 mM previously (9) in an attempt to reduce competition between K+ and Na+ for the same binding sites on E2P. However, some variation in Kd between different preparations of the same enzyme could also be possible, particularly if the value of Kd depends on the surface charge density of the membrane fragments, which would be affected by the lipid composition.
The simple model described above for the pig enzyme is insufficient to explain the experimental behavior observed for the shark enzyme. Because of its biphasic fluorescence decay, an expanded kinetic model is required. We propose a two-step K+ occlusion mechanism:
Because in this model the first step involves the binding and occlusion of a single K+ ion, the relevant equation is
| (2) |
Fitting Eq. 2 to the data obtained for the fast phase of the shark enzyme yields the values kfast = 392 (±19) s−1 and Kd1 = 1.0 (±0.2) mM (see Fig. 5 A). Because the second step of the above mechanism requires the binding of two K+ ions, Eq. 1 was fitted to the slow phase data. This yields the values kslow = 48 (±2) s−1 and Kd = 0.38 (±0.05) mM (see Fig. 5 B). Kd in this case is an apparent value because it would incorporate Kd1, Kd2, and kfast of the above two-step mechanism. The value of kfast obtained for the shark is very similar to the value of k determined for the pig enzyme. This could indicate that the same reaction (probably K+ occlusion) is responsible for the observed kinetics in both enzymes.
The value of Kd is significantly lower than the value of 1.9 mM previously determined by Gropp et al. (10) from kinetic measurements on enzyme from the same source. However, the measurements of Gropp et al. (10) were performed at pH 6.2, whereas those reported here were at pH 7.4, i.e., at a >10-fold lower H+ concentration. There are indications from previous work (18,29) that H+ can compete with K+ for binding to the transport sites. This could possibly explain the differences in the Kd values.
The values of k and kfast for the pig and shark enzymes of 342 (±26) s−1 and 392 (±19) s−1, respectively, are consistent with the observed rate constant for the rapid phase of K+-stimulated dephosphorylation measured via quenched-flow for eel enzyme at 21°C, which was found (24) to be in the range of several hundred to >1000 s−1. The value of kslow determined here of 48 (±2) s−1 is similar to the observed rate constant of the slower phase of K+-stimulated phosphoenzyme decay of eel enzyme of ≤60 s−1 (25).
K+ concentration dependence of the fluorescence amplitudes of the sequential K+-mixing kinetic experiments
In the case of the pig enzyme and the fast phase of the shark enzyme, a significant proportion of the fluorescence change is lost in the dead-time of the stopped-flow instrument, particularly at high K+ concentrations. This precludes accurate measurement of relative fluorescence changes, ΔF/F0, for these phases. However, this is not the case for the slow phase observed for the shark enzyme. The K+-concentration dependence for this phase is shown in Fig. 6. There it can be seen that ΔF/F0 increases to a saturating level as the K+ concentration increases. This behavior indicates an increasing accumulation of enzyme in K+-occluded states as the K+ concentration increases, i.e., an increase in the rate constants for the formation of K+-occluded states relative to their breakdown.
Figure 6.
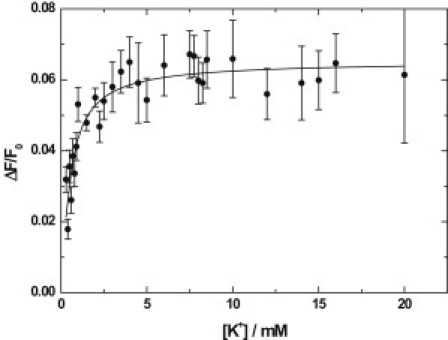
Amplitudes (ΔF/F0) of the fluorescence change associated with the slow phase of sequential mixing stopped-flow measurements on shark rectal gland Na+,K+-ATPase under the conditions described in the caption of Fig. 1 as a function of the K+ concentration (after mixing). ΔF is the fluorescence change associated with the slow phase, and F0 is the initial fluorescence level before mixing with KCl. The values of ΔF/F0 were determined from extrapolations to infinite time of double-exponential fits to the kinetic data. (Solid line) Nonlinear least-squares fit of Eq. 2 to the data. The fit parameters derived were (ΔF/F0)max = 0.065 (±0.002) and Kd = 0.22 (±0.03) mM.
Fitting of Eq. 1 to the fluorescence amplitude data (see Fig. 6) yields an apparent Kd value for K+ binding to the E2P state of 0.22 (±0.03) mM. This is almost half that found from the analysis of the K+ concentration dependence of kobs for the same kinetic phase. However, the comparison is not valid. In the case of the kinetic data, the value of Kd determined depends only on the binding of K+ to the E2P state and the occlusion of the first K+ ion, whereas, in the case of the amplitudes, it depends also on the occlusion of the second K+ ion (assuming this is the reaction responsible for the fluorescence change of the slow phase). Furthermore, because the fluorescence amplitudes reflect the relative rates of formation and disappearance of K+-occluded enzyme states as a function of K+, they could depend on several reaction steps of the enzymatic cycle. Saturation of the fluorescence amplitudes at a much lower K+ concentration than in the case of the kobs values has previously been found from fluorescence titrations using the pig enzyme (9).
For the reasons just described, the values of Kd determined here from the K+-concentration dependence of kobs also cannot be compared with the half-saturating K+ concentration of 0.192 mM determined by Bühler and Apell (11), which was determined from the amplitudes of fluorescence titrations. The value of Bühler and Apell (11) is, however, in relatively good agreement with the apparent Kd of 0.22 (±0.03) mM derived here from amplitude data.
Kinetics of simultaneous K+-mixing experiments
Experiments performed by the simultaneous mixing of the enzyme with both ATP and KCl would be expected to produce the same final steady state as in the case of sequential mixing experiments. The difference is that, in the case of simultaneous mixing, the fluorescence changes observed arise from the phosphorylation and deocclusion (and fast release) of Na+ from the E2P state as well as the subsequent occlusion of K+ and dephosphorylation, and the observed rate of the fluorescence changes depends on the rate constants of all reactions in the reaction sequence:
The steps from E1Na3 to E2P are known from previous measurements on both pig and shark enzymes to yield an increase in fluorescence (6,7), due to the deocclusion of Na+ from the enzyme. Here it has already been shown that occlusion of K+ by E2P, and hence, the steps from E2P to E2P[K2] and E2[K2], lead to a decrease in fluorescence. Therefore, one would expect these opposing fluorescence changes to partially cancel each other out. However, independent measurements using RH421 to follow the transition E2[K2] → E1Na3 have shown (5,6,30,31) that there is a decrease in fluorescence associated with this reaction. Because the sequence of reactions followed by the simultaneous mixing experiments described here results in a conversion of E1Na3 to E2[K2], i.e., the reverse of the E2[K2] → E1Na3 transition, one would expect that the fluorescence change should also be the reverse, i.e., the fluorescence should increase. That is, in fact, what is experimentally observed (see Fig. 7). However, in the case of the shark enzyme it can clearly be seen that there is not simply a gradual increase in fluorescence, but there is an overshoot before the fluorescence decays back down to its final level. This indicates that one of the reactions leading to the fluorescence decrease—i.e., occlusion of the first or second K+ ion by the phosphoenzyme,
—must be slower than those producing the fluorescence increase, i.e., E1Na3 → E2P.
Figure 7.
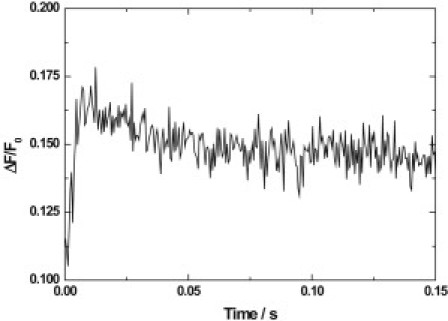
Simultaneous mixing stopped-flow fluorescence transient of Na+,K+-ATPase membrane fragments from shark rectal gland, noncovalently labeled with RH421. Na+,K+-ATPase (60 μg/mL) was rapidly mixed with an equal volume of a solution containing 4.0 mM Na2ATP and 20 mM KCl (i.e., 10 mM after mixing). Each solution was in a buffer containing 30 mM imidazole, 40 mM NaCl, 5 mM MgCl2 and 1 mM EDTA; pH 7.4, 24°C. The total ionic strength was maintained at a constant value by the addition of 90 mM choline chloride to the enzyme suspension and 70 mM choline chloride to the ATP/KCl solution. The wavelengths used were as described in the caption to Fig. 1. The calculated observed rate constants were 391 (±100) s−1 for the rapid phase, associated with an 18% increase in fluorescence, and 42 (±11) s−1 for the slower phase, associated with a 3% decrease in fluorescence.
Similar overshoots have been observed with the pig kidney enzyme (9), but only at nonsaturating K+ concentrations. At saturating K+ concentrations of 5 mM (after mixing) or above, the overshoot disappeared (see Fig. 5 of (9)), indicating that under those conditions K+ occlusion is faster than enzyme phosphorylation by ATP, the rate-determining step leading from E1(Na+)3 to E2P, which has a rate constant of ∼200 s−1 (6) under the conditions used here. However, the situation is very different for the shark enzyme. Here the overshoot is still present at the higher K+ concentration of 10 mM after mixing, which, as one can see from Fig. 5, is close to 100% saturating. Therefore, the observation of the overshoot for the shark enzyme indicates that one of the K+ occlusion steps,
must be slow, i.e., k significantly <200 s−1.
From the sequential mixing experiments on the shark enzyme, it has already been shown that, at saturating K+ concentrations, the enzyme undergoes two first-order reactions after K+ binding to E2P with rate constants of 391 (±19) s−1 and 48 (±2) s−1. Only the second of these is slower than enzyme phosphorylation and, therefore, could be considered as a cause for the overshoot. Nonlinear least-squares fitting of the simultaneous-mixing stopped-flow curve shown in Fig. 7 yields an observed rate constant of 42 (±11) s−1 for the fluorescence decay into a steady state after the overshoot. This agrees very well with the slow phase of the sequential mixing experiments, strongly suggesting that we are observing the same process in both cases, and that the slow phase of the sequential mixing experiments is truly due to a reaction on the pathway leading to E2[K2].
Based on the facts that occlusion of K+ within the phosphorylated enzyme must precede dephosphorylation, that the saturating value of kobs for the fast phase of the shark enzyme (391 s−1) agrees with that observed in sequential mixing on the pig enzyme (342 s−1), and that the fluorescence drop on mixing with K+ has been attributed to K+-occlusion (24), it would seem logical to conclude that, for the shark enzyme, the rate constants of 391 (±19) s−1 and 48 (±2) s−1 refer to consecutive K+ occlusion steps by E2P, i.e.,
Discussion
The stopped-flow experiments described here, together with those which we recently published on the kinetics of the E2 → E1 transition (5), have revealed major differences in the kinetics of the K+-branch of the enzymatic cycle of Na+,K+-ATPase derived from pig kidney and shark rectal gland. In our previous publication (5) we showed that, whereas the E2 → E1 transition and its associated K+ deocclusion and release to the cytoplasmic medium is the rate-determining step at saturating substrate concentrations with a rate constant of ∼40 s−1 for the pig enzyme at 24°C, this is not the case for the shark enzyme, where the rate constant for this step is ∼180 s−1 under the same conditions. In contrast, it has been found here that one of the rate constants for K+ occlusion by the phosphorylated shark enzyme is much lower than that of the pig. Unfortunately, it is not possible to determine actual values of the rate constant for the two separate K+ occlusion steps by phosphorylated pig enzyme.
However, the fact that only a monophasic fluorescence decay is observed in sequential K+-mixing experiments (see Fig. 1, upper panel) implies that the second K+ occlusion step by E2P must be faster than the first, so that it follows virtually instantaneously on the timescale of the first occlusion step. The overall process of K+-occlusion was found here to occur with a rate constant of 365 s−1 for the pig kidney enzyme. Therefore, we conclude that the rate constant for occlusion of the second K+ ion by phosphorylated pig enzyme should be >365 s−1. The phosphorylated shark enzyme, on the other hand, displays a much lower rate constant for occlusion of the second K+ ion, with a value of 48 s−1. This makes K+ occlusion by the phosphoenzyme the rate-determining step of the shark enzyme in the presence of saturating K+ concentrations. For the pig enzyme, K+ occlusion by the phosphoenzyme and dephosphorylation are only rate-determining at low or zero K+ concentrations.
The lower rate constant of the E2 → E1 transition for the pig relative to the shark enzyme and the higher rate constant of K+ occlusion for the pig phosphoenzyme relative to that of the shark means that, in steady-state kinetic measurements under saturating conditions, these two effects cancel each other out to a large extent. This explains why the differences between the two enzymes have not previously been reported. The differences only become obvious when one carries out measurements on the isolated partial reactions, as has been done here.
As mentioned earlier, the kinetics of E2P decay of the shark enzyme has also been observed to be biphasic in quenched-flow measurements (27). To explain both biphasic RH421 fluorescence transients and biphasic dephosphorylation, we suggest the mechanism shown in Fig. 8. This mechanism is consistent with the experimental observation of biphasic behavior in the sequential mixing stopped-flow experiments all the way up to a K+ concentration of 20 mM (see Fig. 3), because at saturating K+ concentrations the enzyme would pass through both rate-limiting K+ occlusion steps.
Figure 8.
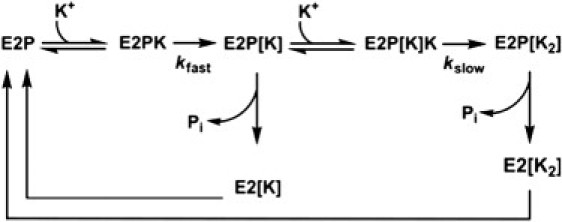
Reaction cycle of shark Na+,K+-ATPase incorporating rate-limiting consecutive K+ occlusions by the phosphoenzyme and parallel pathways of dephosphorylation with either one or two occluded K+ ions (occlusion indicated by square brackets).
Therefore, the observed experimental results imply that the dephosphorylation reactions themselves are very fast and are rate-limited by K+ occlusion. The mechanism shown in Fig. 8 is also consistent with biphasic quenched-flow data, because the species E2P[K] and E2P[K2] could undergo separate dephosphorylation reactions in parallel pathways, as has been suggested for eel enzyme (25). The return of the species E2[K] and E2[K2] to the E2P state in the mechanism is assumed to occur via the normal physiological route, i.e., conversion back to E1 and rephosphorylation by ATP (E2 → E1[Na3] → E1P[Na3] → E2P). Such a return pathway is required to prevent total accumulation of enzyme in K+-occluded states at all K+ concentrations, which would contradict the experimental amplitude results (see Fig. 6), indicating that the proportion of enzyme with occluded K+ increases with the K+ concentration. Although the mechanism shown in Fig. 8 would seem to predict qualitatively all of the experimental observations on the shark enzyme, a more detailed analysis is still required.
The differences in kinetic behavior of the shark and pig enzymes must relate to some structural differences, either in the protein itself or in its lipid environment. Both the shark and pig Na+,K+-ATPases are composed of α1 and β1 isoforms of the enzyme's major catalytic α-subunit and the smaller β-subunit, but they are associated with different regulatory FXYD proteins. The shark enzyme associates with a phospholemman-like FXYD10 (32), which contains a C-terminal multisite phosphorylation domain for protein kinases, whereas the pig kidney enzyme contains the shorter FXYD2, which lacks a regulatory phosphorylation domain (33,34). Therefore, the different FXYD proteins of the two enzymes is a possible origin for the kinetic differences in their partial reactions.
Comparing the recently published structure of the Na+,K+-ATPase of enzyme derived from shark rectal gland at 2.4 Å (2) with the previously published one from pig kidney that was resolved to 3.5 Å (1) does not reveal any obvious differences in the K+-binding sites. However, the β-subunit has also been implicated in modulation of K+ binding (35). Unfortunately the extracellular domain of the β-subunit was not resolved in the pig Na+,K+-ATPase crystals (1), thus precluding structural comparisons.
Whether or how protein and membrane structural differences can explain the kinetic differences in the reactions of the K+-branch of the enzymatic cycle observed here is still unclear. Molecular dynamics simulations or reconstitution into liposomes of defined lipid composition may be able to shed some light on this question. Detailed comparisons of these two enzymes' structures and kinetics could, thus, yield valuable information on the molecular detail of the mechanisms of K+-stimulated dephosphorylation, the E2 → E1 transition, and cytoplasmic K+ release by the Na+,K+-ATPase.
Acknowledgments
The authors thank Assoc. Prof. Serdar Kuyucak for valuable discussions.
The authors thank Prof. Helge Rasmussen, Royal North Shore Hospital, Sydney, for financial assistance supporting enzyme transport. R.J.C. acknowledges, with gratitude, financial support from the Australian Research Council/National Health and Medical Research Council-funded Research Network “Fluorescence Applications in Biotechnology and Life Sciences”, No. RN0460002.
References
- 1.Morth J.P., Pedersen B.P., Nissen P. Crystal structure of the sodium-potassium pump. Nature. 2007;450:1043–1049. doi: 10.1038/nature06419. [DOI] [PubMed] [Google Scholar]
- 2.Shinoda T., Ogawa H., Toyoshima C. Crystal structure of the sodium-potassium pump at 2.4 A resolution. Nature. 2009;459:446–450. doi: 10.1038/nature07939. [DOI] [PubMed] [Google Scholar]
- 3.Ogawa H., Shinoda T., Toyoshima C. Crystal structure of the sodium-potassium pump (Na+,K+-ATPase) with bound potassium and ouabain. Proc. Natl. Acad. Sci. USA. 2009;106:13742–13747. doi: 10.1073/pnas.0907054106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lüpfert C., Grell E., Clarke R.J. Rate limitation of the Na+,K+-ATPase pump cycle. Biophys. J. 2001;81:2069–2081. doi: 10.1016/S0006-3495(01)75856-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khalid M., Cornelius F., Clarke R.J. Dual mechanisms of allosteric acceleration of the Na+,K+-ATPase by ATP. Biophys. J. 2010;98:2290–2298. doi: 10.1016/j.bpj.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kane D.J., Fendler K., Clarke R.J. Stopped-flow kinetic investigations of conformational changes of pig kidney Na+,K+-ATPase. Biochemistry. 1997;36:13406–13420. doi: 10.1021/bi970598w. [DOI] [PubMed] [Google Scholar]
- 7.Cornelius F. Rate determination in phosphorylation of shark rectal Na,K-ATPase by ATP: temperature sensitivity and effects of ADP. Biophys. J. 1999;77:934–942. doi: 10.1016/S0006-3495(99)76944-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khalid M., Fouassier G., Clarke R.J. Interaction of ATP with the phosphoenzyme of the Na+,K+-ATPase. Biochemistry. 2010;49:1248–1258. doi: 10.1021/bi9019548. [DOI] [PubMed] [Google Scholar]
- 9.Kane D.J., Grell E., Clarke R.J. Dephosphorylation kinetics of pig kidney Na+,K+-ATPase. Biochemistry. 1998;37:4581–4591. doi: 10.1021/bi972813e. [DOI] [PubMed] [Google Scholar]
- 10.Gropp T., Cornelius F., Fendler K. K+-dependence of electrogenic transport by the NaK-ATPase. Biochim. Biophys. Acta. 1998;1368:184–200. doi: 10.1016/s0005-2736(97)00162-4. [DOI] [PubMed] [Google Scholar]
- 11.Bühler R., Apell H.J. Sequential potassium binding at the extracellular side of the Na,K-pump. J. Membr. Biol. 1995;145:165–173. doi: 10.1007/BF00237374. [DOI] [PubMed] [Google Scholar]
- 12.Skou J.C., Esmann M. Preparation of membrane Na+,K+-ATPase from rectal glands of Squalus acanthias. Methods Enzymol. 1988;156:43–46. doi: 10.1016/0076-6879(88)56006-8. [DOI] [PubMed] [Google Scholar]
- 13.Klodos I., Esmann M., Post R.L. Large-scale preparation of sodium-potassium ATPase from kidney outer medulla. Kidney Int. 2002;62:2097–2100. doi: 10.1046/j.1523-1755.2002.00654.x. [DOI] [PubMed] [Google Scholar]
- 14.Ottolenghi P. The reversible delipidation of a solubilized sodium-plus-potassium ion-dependent adenosine triphosphatase from the salt gland of the spiny dogfish. Biochem. J. 1975;151:61–66. doi: 10.1042/bj1510061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peterson G.L. A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal. Biochem. 1977;83:346–356. doi: 10.1016/0003-2697(77)90043-4. [DOI] [PubMed] [Google Scholar]
- 16.Lowry O.H., Rosebrough N.J., Randall R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 17.Stürmer W., Bühler R., Läuger P. Charge translocation by the Na,K-pump: II. Ion binding and release at the extracellular face. J. Membr. Biol. 1991;121:163–176. doi: 10.1007/BF01870530. [DOI] [PubMed] [Google Scholar]
- 18.Apell H.-J., Roudna M., Trentham D.R. Kinetics of the phosphorylation of Na,K-ATPase by inorganic phosphate detected by a fluorescence method. Biochemistry. 1996;35:10922–10930. doi: 10.1021/bi960238t. [DOI] [PubMed] [Google Scholar]
- 19.Klodos I. Partial reactions in Na+/K+- and H+/K+-ATPase studied with voltage-sensitive fluorescent dyes. In: Bamberg E., Schoner W., editors. The Sodium Pump: Structure Mechanism, Hormonal Control and Its Role in Disease. Steinkopff; Darmstadt, Germany: 1994. pp. 517–528. [Google Scholar]
- 20.Bielen F.V., Glitsch H.G., Verdonck F. Dependence of Na+ pump current on external monovalent cations and membrane potential in rabbit cardiac Purkinje cells. J. Physiol. 1991;442:169–189. doi: 10.1113/jphysiol.1991.sp018788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rakowski R.F., Vasilets L.A., Schwarz W. A negative slope in the current-voltage relationship of the Na+/K+ pump in Xenopus oocytes produced by reduction of external [K+] J. Membr. Biol. 1991;121:177–187. doi: 10.1007/BF01870531. [DOI] [PubMed] [Google Scholar]
- 22.Peluffo R.D., Berlin J.R. Electrogenic K+ transport by the Na+-K+ pump in rat cardiac ventricular myocytes. J. Physiol. 1997;501:33–40. doi: 10.1111/j.1469-7793.1997.033bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schvartz P.G., Monti J.L.E., Rossi R.C. Kinetic characterization of intermediates during Na-ATPase and Na,K-ATPase activity using RH421 and Rb+ occlusion. J. Gen. Physiol. 2005;126:30a–31a. [Google Scholar]
- 24.Hobbs A.S., Albers R.W., Froehlich J.P. Potassium-induced changes in phosphorylation and dephosphorylation of (Na+ + K+)-ATPase observed in the transient state. J. Biol. Chem. 1980;255:3395–3402. [PubMed] [Google Scholar]
- 25.Froehlich J.P., Hobbs A.S., Albers R.W. Evidence for parallel pathways of phosphoenzyme formation in the mechanism of ATP hydrolysis by electrophorus Na,K-ATPase. Curr. Top. Membr. Transp. 1983;19:513–535. [Google Scholar]
- 26.Hobbs A.S., Albers R.W., Froehlich J.P. Quenched-flow determination of the E1P to E2P transition rate constant in electric organ Na,K-ATPase. In: Glynn I.M., Ellory C., editors. The Sodium Pump. The Company of Biologists; Cambridge, UK: 1985. pp. 355–361. [Google Scholar]
- 27.Cornelius F. Phosphorylation/dephosphorylation of reconstituted shark Na+,K+-ATPase: one phosphorylation site per αβ protomer. Biochim. Biophys. Acta. 1995;1235:197–204. doi: 10.1016/0005-2736(95)80005-z. [DOI] [PubMed] [Google Scholar]
- 28.Hille B. 2nd Ed. Sinauer; Sunderland, MA: 1992. Ionic Channels of Excitable Membranes. 476–478. [Google Scholar]
- 29.Apell H.J., Diller A. Do H+ ions obscure electrogenic Na+ and K+ binding in the E1 state of the Na,K-ATPase? FEBS Lett. 2002;532:198–202. doi: 10.1016/s0014-5793(02)03675-x. [DOI] [PubMed] [Google Scholar]
- 30.Humphrey P.A., Lüpfert C., Clarke R.J. Mechanism of the rate-determining step of the Na+,K+-ATPase pump cycle. Biochemistry. 2002;41:9496–9507. doi: 10.1021/bi025836o. [DOI] [PubMed] [Google Scholar]
- 31.Clarke R.J., Kane D.J., Bamberg E. Kinetics of Na+-dependent conformational changes of rabbit kidney Na+,K+-ATPase. Biophys. J. 1998;75:1340–1353. doi: 10.1016/S0006-3495(98)74052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahmmoud Y.A., Vorum H., Cornelius F. Identification of a phospholemman-like protein from shark rectal glands. Evidence for indirect regulation of Na,K-ATPase by protein kinase c via a novel member of the FXYD family. J. Biol. Chem. 2000;275:35969–35977. doi: 10.1074/jbc.M005168200. [DOI] [PubMed] [Google Scholar]
- 33.Cornelius F., Mahmmoud Y.A. Functional modulation of the sodium pump: the regulatory proteins “Fixit”. News Physiol. Sci. 2003;18:119–124. doi: 10.1152/nips.01434.2003. [DOI] [PubMed] [Google Scholar]
- 34.Garty H., Karlish S.J.D. Role of FXYD proteins in ion transport. Annu. Rev. Physiol. 2006;68:431–459. doi: 10.1146/annurev.physiol.68.040104.131852. [DOI] [PubMed] [Google Scholar]
- 35.Geering K. The functional role of β subunits in oligomeric P-type ATPases. J. Bioenerg. Biomembr. 2001;33:425–438. doi: 10.1023/a:1010623724749. [DOI] [PubMed] [Google Scholar]