

Abstract
This unit shows how to use the Bowtie package to align short sequencing reads, such as those output by second-generation sequencing instruments. It also includes protocols for building a genome index and calling consensus sequences from Bowtie alignments using SAMtools.
Keywords: Short reads, read alignment, alignment, read mapping, mapping, genome indexing, comparative genomics, software package
INTRODUCTION
The Bowtie (Langmead et al. 2009) package enables ultrafast and memory-efficient alignment of large sets of sequencing reads to a reference sequence, such as the human genome. The package contains tools for building indexes of reference genomes and for aligning short reads using the index as a guide. This is the first step of many comparative genomics workflows, including variant detection and digital gene expression. In what follows, the term read refers to a short DNA sequence, typically as output by a sequencing instrument. A read may be accompanied by a corresponding string of quality values, where each value estimates the probability that the corresponding base was miscalled by the instrument software. The term subject sequence refers to the true sequence of the sample(s) from which the reads were drawn. The term reference sequence or reference genome refers to the sequence to which the subject is to be compared. An example of a commonly used reference sequence is the public human genome assembly GRCh37.
To achieve both speed and memory efficiency, Bowtie aligns reads with the aid of an index of the reference genome. The Bowtie index is a refinement of the FM Index (Ferragina and Manzini 2000), which uses the Burrows-Wheeler Transform (Burrows and Wheeler 1994) to achieve both speed and space efficiency. The user must have built or otherwise obtained an appropriate index before reads can be aligned to the reference. Once an index is built, it can be queried any number of times. Indexes for commonly used reference genomes are also available for download from the Bowtie website at http://bowtie-bio.sf.net.
The Basic Protocol describes how to use the bowtie tool to align a collection of reads to a reference genome, assuming an index is available. The Indexing Protocol (Alternate Protocol 1) describes how to build an index for a user-specified reference genome with the bowtie-build tool. The Consensus and SNP Calling Protocol (Alternate Protocol 2) describes how to call consensus sequence, including SNPs, from Bowtie’s output using SAMtools (Li et al. 2009). The command line option protocol (Alternate Protocol 3) demonstrates a variety of commonly used alignment options. Finally, the Support Protocols describe how to obtain and install Bowtie software (Support Protocol 1), how to build Bowtie from source code (Support Protocol 2), and how to obtain pre-built indexes from the Bowtie website (Support Protocol 3). Each of these protocols runs in a variety of Unix or Unix-like environments, including Linux, Mac OS X, and Windows.
Bowtie is open source and can be used free of charge, subject to the terms of the Artistic License at http://www.opensource.org/licenses/artistic-license-1.0.php.
BASIC PROTOCOL 1
ALIGNING A SET OF SHORT READS TO A REFERENCE GENOME
This protocol uses the bowtie tool to take a collection of short reads and search for each read’s best alignment to a reference genome. In typical comparative genomics applications, an alignment is interpreted as a hypothesis for where the read originated in the genome. Once reads are aligned to the reference, the resulting set of alignments becomes a source of evidence for other features of interest, such as DNA sequence differences or gene expression measurements.
Bowtie has many command-line options that allow the user to tune its behavior according to the inputs and the desired result. This protocol describes a few commonly used options and gives some advice regarding how other options should be set. For a complete description of Bowtie’s command-line options, see the MANUAL file included in the Bowtie package.
The first example uses an E. coli index that comes with the Bowtie package. Alternate Protocol 1 describes how to build an index from a user-specified reference genome.
All Bowtie options are specified on the command line when invoking Bowtie. A full list of Bowtie options can be found in the MANUAL file included with the Bowtie package; these examples focus on the shorter list of important options shown in Table 11.7.1.
Table 11.7.1.
Descriptions of important command line options for the bowtie alignment tool. For the full list of options, see the MANUAL file included with the version of Bowtie being used.
| Option | Description |
|---|---|
| Alignment policy | |
| −v <int> | Only alignments with up to <int> mismatches in the entire alignment are considered “valid.” −v is mutually exclusive with −n. Default: −n mode is enabled and −v mode is disabled. |
| OR: | |
| −n <int> | Only alignments with up to <int> mismatches in the seed portion of the alignment are considered “valid.” −n is mutually exclusive with −v. Default: 2. |
| −l <int> | When −n is specified, −l sets the length of the seed portion of the alignment to <int>. Default: 28. |
| −e <int> | When −n is specified, −e sets the limit on the sum of quality scores at the mismatched positions. Default: 70. |
| Reporting policy | |
| −k <int> | If Bowtie finds multiple valid alignments for a read, only report the first <int> alignments found. Default: 1. |
| −m <int> | If Bowtie finds more than <int> valid alignments for a read, suppress all alignments for the read. Default: no limit. |
| −M <int> | If Bowtie finds more than <int> valid alignments for a read, suppress the alignments but report one of them at random. Default: no limit. |
| Other options | |
| −S | Output alignments in SAM format (Li et al, 2009). |
| −u <int> | Stop and exit after the first <int> reads have been processed. |
Necessary Resources
Hardware
A Unix, Linux, Mac OS X or Windows 32-bit workstation.
Software
Bowtie 0.12.5 package (see Support Protocols 1 and 2 for download and installation instructions).
Files
A set of one or more files containing the reads. Reads can be in the FASTA format (see APPENDIX 1B for information on FASTA), FASTQ format (Cock et al., 2009), or in a raw one-sequence-per-line format. The reads used in this example are a set of simulated reads included in the Bowtie package.
A set of index files containing the index of the reference genome. The index file format is unique to Bowtie, and FASTA formats are converted to this format using the bowtie-build tool (discussed in Alternate Protocol 1). The index files used for this example encode the whole genome of E. coli strain 536. These files are included in the Bowtie package.
Running Bowtie Interactively from the Command Line
-
1aChange to the directory containing Bowtie. For instance, if Bowtie is installed in a directory named /Users/joe/software/bowtie, type:
- cd /Users/joe/software/bowtie
- The example index files are located in the indexes subdirectory, and reads are located in the reads subdirectory.
-
2aRun Bowtie on the example E. coli data with the −u 10 command line option:
- ./bowtie −u 10 indexes/e_coli reads/e_coli_1000.fq
- When invoking Bowtie, the user specifies an index file “basename,” i.e. the prefix shared by all the index files (indexes/e_coli in this case), and the path(s) to the FASTQ read files to be aligned (reads/e_coli_1000.fq in this case). All other arguments to Bowtie are interpreted as options (see Table 11.7.1 for a list of a few important options).
- This command aligns all reads in the specified FASTQ file to the E. coli 536 reference genome, using the specified index as a guide. The −u 10 option causes Bowtie to exit after processing the first 10 reads from the file.
-
3a
Examine and interpret the output, also shown in Figure 11.7.1.
Note that in practice, users rarely need to examine the alignment output manually. Rather, users will typically redirect Bowtie’s output to a file or directly to another program for downstream processing. See Alternate Protocol 2 for an example.
The output consists of two sets of lines, those printed to the “standard out” file handle and those printed to the “standard error” file handle. Each line printed to “standard out” is a valid, reportable alignment found by Bowtie. A valid alignment is one that satisfies the alignment policy specified in the −n, −l, −e or −v options. A reportable alignment is one that is not suppressed by any other option, such as the −k, −m, or −M options. Lines printed to “standard error” contain summary information about the entire alignment run, e.g., the total number of reads that were processed and the percentage of total reads for which Bowtie found at least one alignment.
- The format for an alignment consists of 8 fields, separated by tabs. The fields, from left to right, are:
- Read name
- Reference strand aligned to (“+” denotes forward strand, “−” denotes reverse strand)
- Name of reference sequence aligned to
- Offset of the leftmost position on the forward reference strand covered by the alignment
- Read sequence aligned (or its reverse complement, if the read aligned to the reverse strand)
- Quality sequence aligned (or its reverse, if the read aligned to the reverse strand)
- See the MANUAL file included with the Bowtie package for information on this field
- A string representing the differences between the read and the corresponding characters of the reference genome. Each difference is described in a comma-separated field.
-
4aTo obtain alignment results in SAM format, a file format recognized by SAMTools and many other sequence analysis packages, add the −S option to the bowtie command-line:
- ./bowtie −S −u 10 indexes/e_coli reads/e_coli_1000.fq
- This will produce output similar to the earlier example, except that the alignments printed to standard output will be in SAM format (Fig. 11.7.2). More information about the SAM format is available in the MANUAL file included with the Bowtie package and on the SAMtools website at http://samtools.sourceforge.net/.
-
5aWhen aligning a large number of reads, the user will wish to capture the standard output to a file rather than to have it appear on the screen. The user can do this by redirecting standard output as shown here:
- ./bowtie −S indexes/e_coli reads/e_coli_1000.fq > alignments.sam
- This will align all reads contained in the E. coli FASTQ file and save them in SAM format to the file named alignments.sam.
Figure 11.7.1.
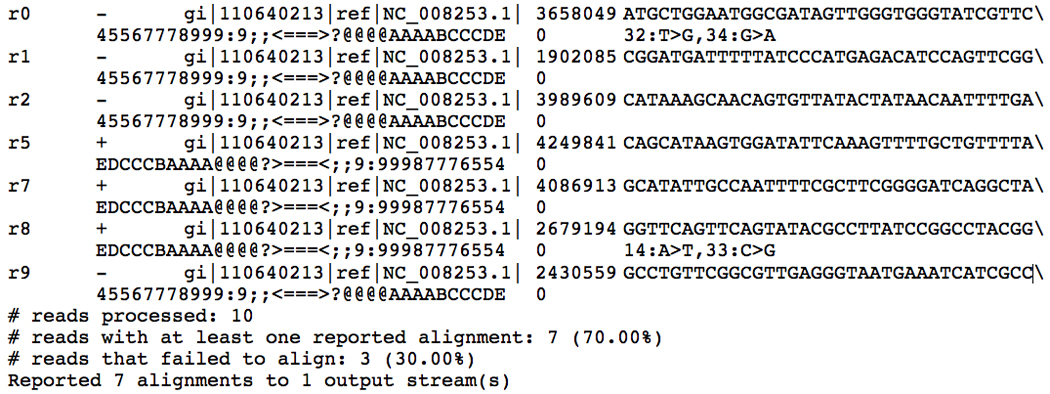
Sample Bowtie session. The last few lines are written to the “standard error” file handle and convey summary information about the run. The other lines are written to the “standard out” file handle and show valid, reportable alignments found by Bowtie. In this and subsequent figures, the backslash character is used to indicate that some long lines are wrapped.
Figure 11.7.2.
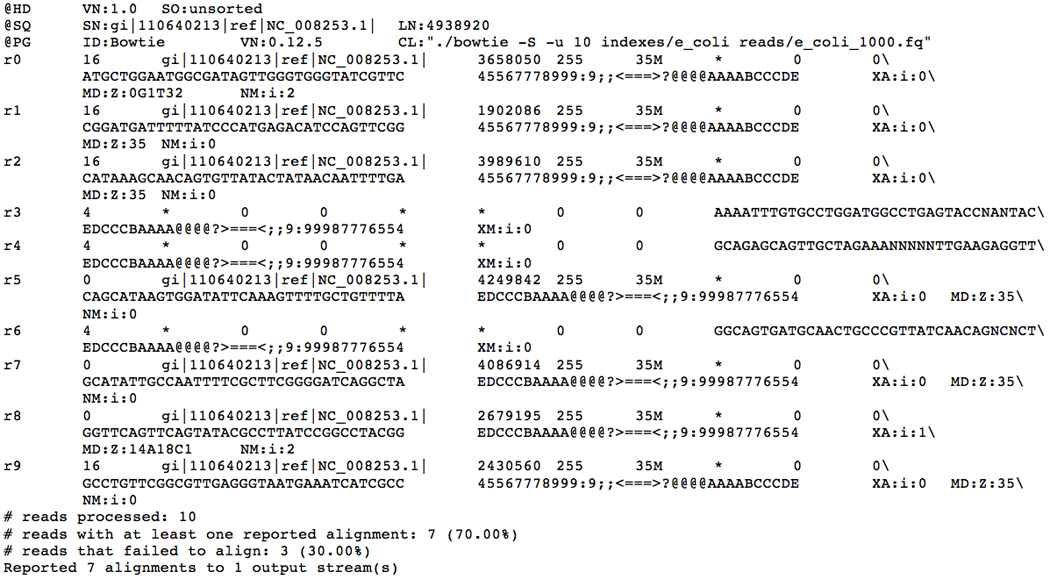
Sample Bowtie session when SAM output mode is enabled. The last few lines are written to the “standard error” file handle and convey summary information about the run and are not part of the SAM format. The first few lines beginning with “@” are the SAM header. The remaining lines are alignments.
ALTERNATE PROTOCOL 1
BUILDING AN INDEX FOR A SET OF REFERENCE SEQUENCES
This protocol uses the bowtie-build tool to take a collection of FASTA files for a reference genome and generate a collection of index files. Index files can then be used by bowtie to align reads to the reference genome. The same set of index files can be used across multiple runs of bowtie.
bowtie-build has command-line options that allow the user to tune its behavior, but this protocol will use the defaults. For a complete description of bowtie-build’s command-line options, see the MANUAL file included with the Bowtie package.
Necessary Resources
Hardware
A Unix, Linux, Mac OS X or Windows 32-bit workstation.
Software
Bowtie 0.12.5 package (see Support Protocols 1 and 2 for download and installation instructions).
Download the two compressed FASTA files corresponding to the human sex chromosomes (chrX.fa.gz and chrY.fa.gz) from the UCSC FTP site at http://hgdownload.cse.ucsc.edu/goldenPath/hg19/chromosomes/.
- In the same directory, use gunzip to decompress the compressed FASTA files that were just downloaded:
- gunzip *.gz
- Run bowtie-build to build an index of the two human sex chromosomes:
- bowtie-build chrX.fa,chrY.fa human_xy
- This builds an index consisting of the sequences in the chrX.fa and chrY.fa files and stores the results in a set of six files with prefix “human_xy”. If the bowtie-build executable is not in the search path, specify the full path to bowtie-build instead. This command typically takes about ten or fifteen minutes.
- In this example each FASTA file contains one sequence. FASTA files with multiple sequences (“multi-FASTA” files) are also permitted, in which case all sequences in the multi-FASTA file are included as separate sequences in the index.
- Confirm that the output files were created:
- ls −l human_xy*
- The output should be similar to Figure 11.7.3. Note the index consists of 6 distinct files with the same prefix (human_xy), and suffix (.ebwt).
- Inspect the index using bowtie-inspect:
- bowtie-inspect −s human_xy
- The output should be similar to Figure 11.7.4. Among other things, the output tells the user that the index is not for colorspace alignment (first line), and shows the names and lengths of the two reference sequences included in the index (last two lines). See the MANUAL file included with the Bowtie package for detailed description of bowtie-inspect’s output.
Figure 11.7.3.
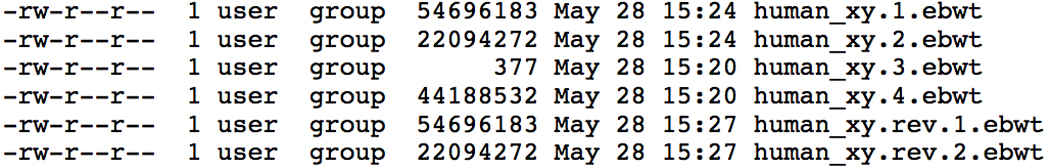
Listing of files output by bowtie-build after indexing the human sex chromosomes.
Figure 11.7.4.

Output of bowtie-inspect when inspecting the index consisting of the two human sex chromosomes.
ALTERNATE PROTOCOL 2
ALIGNMENT AND VARIATION DETECTION USING BOWTIE AND SAMTOOLS
A common task in comparative genomics is to take a collection of sequencing reads derived from genomic DNA and obtain a list of variants, or differences between the subject’s genome sequence and the reference genome sequence. This protocol outlines how to accomplish this using the E. coli index and simulated E. coli reads that comes with the Bowtie package, together with the SAMtools package.
Necessary Resources
Hardware
A Unix, Linux, Mac OS X or Windows 32-bit workstation.
Software
Bowtie 0.12.5 package (see Support Protocol 1 for download and installation instructions).
SAMtools 0.1.7 package (see instructions on the SAMtools website at http://samtools.sourceforge.net for how to download and install SAMtools)
Running Bowtie
- Change to the directory containing Bowtie. For instance, if Bowtie is installed in a directory named /Users/joe/software/bowtie, type:
- cd /Users/joe/software/bowtie
- Align the reads to the E. coli genome with Bowtie and save the SAM output to a temporary file:
- ./bowtie −S indexes/e_coli reads/e_coli_10000snp.fq > /tmp/e_coli.sam
- Convert the SAM output to BAM format:
- samtools view −bS −o /tmp/e_coli.bam /tmp/e_coli.sam
- BAM is the compressed, binary encoded counterpart to SAM.
- If the samtools executable is not in the search path, specify the full path to samtools instead.
- Convert the BAM file to a sorted BAM file:
- samtools sort /tmp/e_coli.bam /tmp/e_coli.sorted
- Sorted BAM is a useful format because alignments are both compressed, which is convenient for long-term storage, and sorted, which is convenient for variant discovery and other downstream analyses.
- If the samtools executable is not in the search path, specify the full path to samtools instead.
- Call variants from the sorted BAM file:
- samtools pileup −cvf genomes/NC_008253.fna /tmp/e_coli.sorted.bam
- The output should be similar to Figure 11.7.5, consisting of 10 SNPs. See the SAMtools website at http://samtools.sourceforge.net for details about SAMtools output and command line options.
Figure 11.7.5.
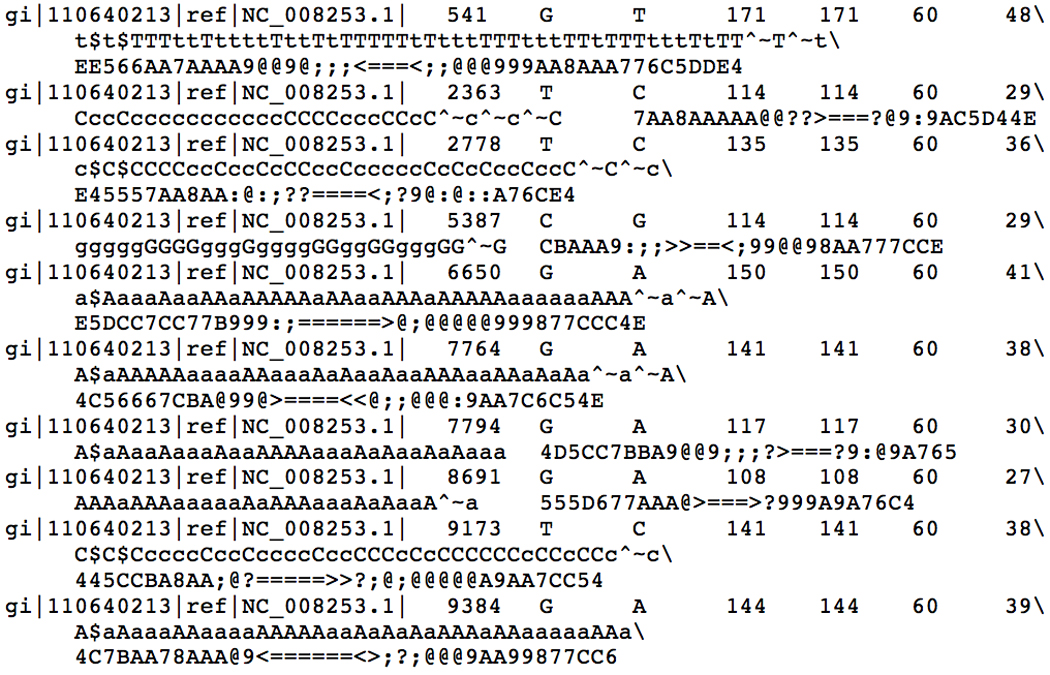
Output of the samtools consensus caller when calling SNPs from a simulated E. coli example dataset.
ALTERNATE PROTOCOL 3
RUNNING BOWTIE WITH VARIOUS COMMAND LINE OPTIONS
Table 11.7.1 shows a short list of important command line options that modify the behavior of the bowtie alignment tool. This protocol steps through a series of examples that illustrate some of these options. The protocol uses the E. coli index that comes with the Bowtie package.
Besides the options being demonstrated, these examples use Bowtie’s --suppress option, which suppresses unneeded output fields. Also, the summary output that is normally printed to “standard error” is omitted from the figures in this protocol. The purpose is to keep the figures as concise as possible.
Necessary Resources
Hardware
A Unix, Linux, Mac OS X or Windows 32-bit workstation.
Software
Bowtie 0.12.5 package (see Support Protocols 1 and 2 for download and installation instructions).
- Change to the directory containing Bowtie. For instance, if Bowtie is installed in a directory named /Users/joe/software/bowtie, type:
- cd /Users/joe/software/bowtie
- Align a test string to the E. coli genome using the −a and −v 2 options:
- ./bowtie −a −v 2 e_coli --suppress 1,3,5,6,7 −c ATGCATCATGCGCCAT
- The output should be similar to Figure 11.7.6. The --suppress option has suppressed all output fields besides strand (first column), offset (second column) and edit string (third column).
- Specifying −a, instructs bowtie to report all valid alignments subject to the alignment policy, which is −v 2 in this case. Here, bowtie finds 5 inexact hits in the E. coli genome; one hit (the second listed) has 1 mismatch, and the other 4 hits have 2 mismatches. Four are on the reverse reference strand and one is on the forward strand. Note that they are not listed in best-to-worst order.
- Align a test string to the E. coli genome using the −k 3 and −v 2 options:
- ./bowtie −k 3 −v 2 e_coli --suppress 1,3,5,6,7 −c ATGCATCATGCGCCAT
- The output should be similar to Figure 11.7.7. Specifying −k 3 instructs bowtie to report up to 3 valid alignments. In this case, a total of 5 valid alignments exist (see Figure 11.7.6); bowtie reports 3 out of those 5. −k can be set to any integer greater than 0.
- Align a test string to the E. coli genome using the −k 6 and −v 2 options:
- ./bowtie −k 6 −v 2 e_coli --suppress 1,3,5,6,7 −c ATGCATCATGCGCCAT
- The output should be similar to Figure 11.7.8. Specifying −k 6 instructs bowtie to report up to 6 valid alignments. In this case, a total of 5 valid alignments exist (see Figure 6), so bowtie reports all 5.
- Align a test string to the E. coli genome using just the −v 2 option:
- ./bowtie −v 2 e_coli --suppress 1,3,5,6,7 −c ATGCATCATGCGCCAT
- The output should be similar to Figure 11.7.9. Leaving the reporting options at their defaults causes bowtie to report the first valid alignment it encounters. Because --best was not specified, we are not guaranteed that bowtie will report the best alignment, and in this case it does not (the 1-mismatch alignment from Figure 11.7.8 would have been better). The default reporting mode is equivalent to −k 1.
- Align a test string to the E. coli genome using the --best, −a and −v 2 options:
- ./bowtie −a --best −v 2 e_coli --suppress 1,3,5,6,7 −c ATGCATCATGCGCCAT
- The output should be similar to Figure 11.7.10. The −a option causes all valid alignments to be printed and the --best option guarantees that Bowtie will find and print the alignments for a given read in best-to-worst order. In this case, the 1-mismatch alignment is printed first, as expected.
- Align a test string to the E. coli genome using the −a, −m 3 and −v 2 options:
- ./bowtie −a −m 3 −v 2 e_coli --suppress 1,3,5,6,7 −c ATGCATCATGCGCCAT
- There should be no alignment output. The −a option causes Bowtie to report all valid alignments, but the −m 3 option suppresses all alignments for reads with more than 3 valid alignments. Because this read has 5 valid alignments (see Figure 11.7.10), all alignments are suppressed and there is no alignment output.
- Align a test string to the E. coli genome using the −a and −m 5 and −v 2 options:
- ./bowtie −a −m 5 −v 2 e_coli --suppress 1,3,5,6,7 −c ATGCATCATGCGCCAT
- The output should be similar to Figure 11.7.11. The −a option causes Bowtie to report all valid alignments while the −m 5 option suppresses all alignments for reads with more than 5 valid alignments. Because this read has exactly 5 valid alignments, all alignments are reported.
Figure 11.7.6.

Bowtie output for previous example using −a and −v 2 options.
Figure 11.7.7.

Bowtie output for previous example using −k 3 and −v 2 options.
Figure 11.7.8.

Bowtie output for previous example using −k 6 and −v 2 options.
Figure 11.7.9.
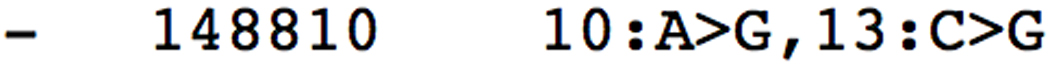
Bowtie output for previous example using just the −v 2 option.
Figure 11.7.10.

Bowtie output for previous example using the --best, −a and −v 2 options.
Figure 11.7.11.

Bowtie output for previous example using the −a and −m 5 and −v 2 options.
SUPPORT PROTOCOL 1
OBTAINING AND INSTALLING THE BOWTIE PACKAGE
The Bowtie binary packages are available from the Bowtie web site at http://bowtie-bio.sf.net. See the “Latest Release” section on the right-hand side of the Bowtie website for a link to the appropriate Sourceforge download page. All Bowtie packages are compressed in the zip format.
Necessary Resources
Hardware
A Unix, Linux, Mac OS X or Windows 32-bit workstation.
Software
Bowtie 0.12.5 package (see Support Protocol 1 for download and installation instructions)
A tool for extracting zip files
Download the desired Bowtie binary package from the Bowtie web site at http://bowtie-bio.sf.net.
Unzip the downloaded file to the desired installation path.
-
(Optional): For convenience and compatibility with other tools that rely on Bowtie, add the extracted directory (containing the bowtie and bowtie-build executables) to the search path.
Users unfamiliar with how to add directories to the search path should consult APPENDIX 1C.
SUPPORT PROTOCOL 2
BUILDING BOWTIE FROM SOURCE
The Bowtie source packages are available from the Bowtie web site at http://bowtie-bio.sf.net. See the “Latest Release” section on the right-hand side of the Bowtie website for a link to the appropriate Sourceforge download page. The source package filenames end with “−src.zip”. All Bowtie packages are compressed in the zip format.
Necessary Resources
Hardware
A Unix, Linux, Mac OS X or Windows 32-bit workstation.
Software
Bowtie 0.12.5 package (see Support Protocol 1 for download and installation instructions)
A tool for extracting zip files
Standard GNU development tools, including g++ and GNU make
Download the desired Bowtie binary package from the Bowtie web site at http://bowtie-bio.sf.net.
Unzip the downloaded file to the desired installation path.
- Change to the extracted directory and run the make command:
- cd (extracted directory)
- make
- The build process requires that standard development tools, including g++, GNU make, and find be installed, and libraries such as libpthread may also need to be installed. See the included MANUAL file for more details about how to build Bowtie.
- When the build process completes successfully, a set of binaries including bowtie, bowtie-build, and bowtie-inspect will be created in the extracted directory.
-
(Optional): For convenience and compatibility with other tools that rely on Bowtie, add the extracted directory (containing the bowtie and bowtie-build executables) to the search path.
Users unfamiliar with how to add directories the search path should consult APPENDIX 1C.
SUPPORT PROTOCOL 3
DOWNLOADING AND INSTALLING A PRE-BUILT BOWTIE INDEX
The Bowtie source packages are available from the Bowtie web site at http://bowtie-bio.sf.net. See the “Pre-built indexes” section on the right-hand side of the Bowtie website for download links. Indexes are compressed in the zip format.
This protocol steps through obtaining and using a pre-built index for the S. cerevisiae genome.
Necessary Resources
Hardware
A Unix, Linux, Mac OS X or Windows 32-bit workstation.
Software
Bowtie 0.12.5 package (see Support Protocol 1 for download and installation instructions)
A tool for extracting zip files
Download the pre-built index for the S. cerevisiae genome by visiting the Bowtie web site at http://bowtie-bio.sf.net and clicking the link labeled “S. cerevisiae” on the right-hand side of the website. Place the downloaded file in a temporary directory.
Unzip the downloaded file to the desired path.
- Run a simple Bowtie query against the new index as a test:
- bowtie (index install path)/s_cerevisiae −c ATATATATATATATA
- If the bowtie executable is not in the search path, specify the full path to bowtie instead. In this case, bowtie should print one alignment.
GUIDELINES FOR UNDERSTANDING RESULTS
The purpose of alignment is to determine reads’ point of origin with respect to the reference genome. Once points of origin are identified, downstream tools use that information, for example, to characterize differences between the subject and reference genome (e.g. when calling SNPs), or to relate the reads to annotations defined with respect to the reference genome (e.g. for digital gene expression). Alignment programs, together with appropriate reference sequences, serve this purpose because genomes of individuals of the same species tend to be highly similar. For example, two humans typically have on the order of 3–4 million single-nucleotide differences between them out of a total of 3 billion bases. However comparative strategies also have inherent drawbacks that should be kept in mind when interpreting Bowtie results.
Repetitive genome content affects results
Some genomes, including the human genome, have substantial repetitive content, i.e. sub-sequences that appear multiple times throughout the genome. Repeats come in several forms (e.g. simple repeats, tandem repeats, segmental duplications, interspersed repeats), and arise via various biological processes (e.g. slipped strand mispairing or retrotransposition). Repeats also affect alignments because reads originating from repetitive portions of the genome are difficult or impossible to unambiguously assign to a point of origin. Reads from repeats will tend to have many “valid” alignments, with no strong basis for preferring one over the others. Paired-end reads mitigate but do not necessarily eliminate this problem. Repetitive alignments in turn affect downstream analyses. For instance, if ambiguous alignments are included in the output from Bowtie, a SNP could yield false positives and false negatives purely owing to the repeat structure.
The simplest way to deal with alignment ambiguity is to use Bowtie’s −m and −M options to filter out and/or annotate ambiguous evidence as such. When ambiguous alignments are annotated in this way, downstream tools can choose to discount or ignore evidence from ambiguous alignments as appropriate. With the −m option, the user specifies a limit whereby alignments for reads that align to a number of locations exceeding the limit are excluded from the output. For instance, to suppress all alignments for reads that align to more than 3 locations on the reference, the user specifies −m 3. The −M option is similar to the −m option except that one random alignment from among those found is reported, but is marked as being ambiguous. See the MANUAL file included in the Bowtie package for details.
Sequence differences between subject and reference affect results
Though human genomes are close to 99.9% similar at the sequence level, the differences can have a significant effect on alignment. For instance, if the subject genome contains a cluster of 3 single-nucleotide variants within 10 bases of each other, and Bowtie is run with the −v 2 option (i.e. only alignments with up to 2 mismatches are valid), Bowtie is very unlikely to report the correct alignment for a read whose true point of origin spans the three variants. A similar example involves gaps, if the subject genome has a gap with respect to the reference genome (or vice versa), Bowtie is unlikely to report the correct alignment for a read whose true point of origin spans the gap. These potential effects must be taken into account when interpreting output from Bowtie.
COMMENTARY
Background Information
The Bowtie package enables ultrafast and memory-efficient alignment of large sets of sequencing reads to a reference sequence. Alignment poses a serious computational challenge because the increase in throughput provided by second-generation sequencing instruments is rapidly outpacing the growth in computer speeds. Alignment is also important for biological research because it typically constitutes both the first and the slowest step of comparative genomics pipelines.
Bowtie aligns reads with the aid of an index of the reference genome, and the indexing technique used in Bowtie is the key to its speed and memory efficiency. The Bowtie index is a refinement of the FM Index (Ferragina and Manzini 2000), which in turn uses the Burrows-Wheeler Transform (Burrows and Wheeler 1994). The user must have built or otherwise obtained an appropriate index before reads can be aligned to the reference, but once an index is built it can be queried any number of times. Alternatively, the user can download a pre-built index from the Bowtie website at http://bowtie-bio.sf.net.
Critical Parameters and Troubleshooting
The most often-used Bowtie options are discussed in the protocols above, but bowtie and bowtie-build both support dozens of options that help the user achieve good results for a breadth of inputs. This section describes some additional families of options that are useful under certain circumstances; see the MANUAL file included in the Bowtie package for a complete listing of options.
If either sensitivity (i.e. percent of reads that align) and/or speed are an issue, try adjusting the alignment policy. Alignment policy is controlled either by the −v option (in end-to-end mode) or by the −n, −l and −e options (in seeded mode). For greater speed but lower sensitivity, try the following adjustments as appropriate: adjust −v down, −n down, −l up, −e down. For lower speed but greater sensitivity, make the opposite adjustments.
For greater speed without sacrificing sensitivity, try the −p option. Specifying a value for −p greater than 1 is a convenient way to allow Bowtie to use more than one computer processor. When multiple processors are available and Bowtie is run with −p equal to the number of available processors, Bowtie will take proportionally less time to complete.
If you receive an error message about memory having been exhausted, try adjusting the --offrate option up, first to 6, then higher. If that does not work, try running on a computer with more memory.
The −C option, when passed to bowtie, enables alignment of colorspace reads, such as those produced by the ABI SOLiD instrument. When specified to bowtie-build, the −C option causes bowtie-build to generate a colorspace index, rather than a normal index (also called a base space or nucleotide space index). When the −C option is passed to bowtie, the specified index must be a colorspace index. Likewise, if the −C option is not passed to bowtie, the specified index must be a normal index.
For alignment of paired-end or mate-paired reads, use the −1 and −2 options to specify the paired input files. See the --ff, --fr, --rf options for information on read orientations. Note that the default for non-colorspace reads is --fr, since this matches the output of the Illumina instruments’ most commonly used paired-end protocol. Likewise, --ff is the default for colorspace reads, since this matches the output of the ABI SOLiD instrument’s most commonly used protocol. Also see the −I and −X options, which place bounds on the permitted fragment size.
Finally, the --al, --un and --max options can be a convenient way to prepare for the use of a downstream tool. Each option takes a filename argument. If all three are specified, Bowtie, in addition to printing alignment output as usual, will print each read to one of the three specified files according to the alignment result. If one or more alignments were reported for the read, the read is printed to the file specified with the --al option. If no alignments were found for the read, the read is printed to the file specified with the --un option. If alignments were found, but were suppressed due to the −m or −M options, the read is printed to the file specified with the --max option. These files can then be used as input to another analysis; e.g., the reads that failed to align could then be passed to a spliced aligner or to another invocation of Bowtie using a different index.
Footnotes
INTERNET RESOURCES
The project’s home page where the latest version of the package can be downloaded and where announcements are made.
LITERATURE CITED
- Burrows M, Wheeler DJ. A block sorting lossless data compression algorithm. Palo Alto, CA: Digital Equipment Corporation; 1994. Technical Report 124. [Google Scholar]
- Ferragina P, Manzini G. Opportunistic data structures with applications; Proceedings of the 41st Annual Symposium on Foundations of Computer Science; IEEE Computer Society; 2000. [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. See above. Describes the indexing technique underlying the tool and compares to other alignment tools.
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]