

Abstract
T cell lines with defined cytokine profiles are an invaluable tool for assessing the control of immune responses both in vitro and in vivo. Production of such cell lines can be complex and time-consuming. Here we present a powerful technique to assay the cytokines produced by T cells activated polyclonally or with specific antigens. This paper presents a detailed methodology for the identification and isolation of cytokine-producing T cells activated with the artificial superantigen, CytoStim, or viral and fungal antigens. These cells can be analysed for different cytokines simultaneously, or cultured further to rapidly establish T cell lines making known cytokine types. We highlight the enumeration, isolation and phenotype of interleukin-17-producing T cells, and the rapid generation of virus-specific Th1 T cell lines.
Keywords: cytokines, magnetic isolation, memory, T cells
Introduction
Understanding T cell orchestration of immune responses has been greatly advanced by the classification of T cells based on the cytokines they secrete. Characterization of subsets such as T helper type 1 (Th1), Th2 and Th17 have accelerated understanding of the control of inflammatory and humoral responses but have also highlighted the incredible plasticity of these subsets ([1] for recent reviews see [2,3]). Manipulation of T cell responses in vivo, by the infusion of defined T cell populations, has relied to date upon in-vitro-generated T cell lines. These have been generated successfully, e.g. for Th17 [4], Th1 [5], but this success relies upon the generation of lines from a small number of precursors through prolonged expansion in the presence of appropriate antigen and cytokines. Production of such T cell lines is often complex, expensive and runs the risk of producing a line that is less than representative of the original antigen-specific precursors, particularly if they have a largely effector phenotype (see reference [6] for a discussion of current approaches to this issue using conventional methodology).
An alternative methodology is to stimulate bulk T cells polyclonally, or with specific antigen, and to identify and isolate the cells which respond by producing cytokine. Cytokine secretion assays work by building an antibody matrix on the cell surface to capture secreted cytokine. The captured cytokines thus become a surface antigen and can be detected and used for cell isolation with anti-cytokine antibodies [7,8]. The cytokine-producing cells isolated are the small number of precursors fated to be grown out through repeat stimulation to produce T cell lines. By isolating these cells without any influence of long-term culture or the need to induce a phenotype with other stimuli, it is possible to work with these specific T cell subsets in their most natural state, whether for simple phenotyping or generation of T cell lines.
In this technology focus we present examples of how cytokine secretion can be used to identify and isolate different T cell subsets rapidly, and the subsequent behaviour of these T cells when used to generate T cell lines.
We present a highly detailed methodology for the use of this technique. In the specimen results section we focus upon specific examples of how this technology can be applied:
Identification and isolation of Th17 T cells – human and mouse.
Rapid generation of virus-specific Th1 human T cell lines using interferon (IFN)-γ.
Method overview
The cytokine secretion assay involves the following steps (Fig. 1): (i) T cells are stimulated with specific antigen or polyclonal T cell receptor (TCR)-stimulus; (ii) a cytokine-specific catch reagent is added to the cells. This is composed of the cytokine-specific ‘catch’ antibody, conjugated with a CD45-specific monoclonal antibody, labelling all leucocytes evenly with the catch reagent; (iii) the cells are incubated for 45 min at 37°C to allow cytokine secretion, and the secreted cytokine binds to the catch reagent on the secreting cells; and (iv) bound cytokine is labelled subsequently with a second cytokine-specific fluorochrome-conjugated antibody for sensitive analysis by flow cytometry. Optionally, the caught cytokine is magnetically labelled further with specific antibody conjugated to super-paramagnetic particles for enrichment by magnetic cell sorting (MACS®).
Fig. 1.
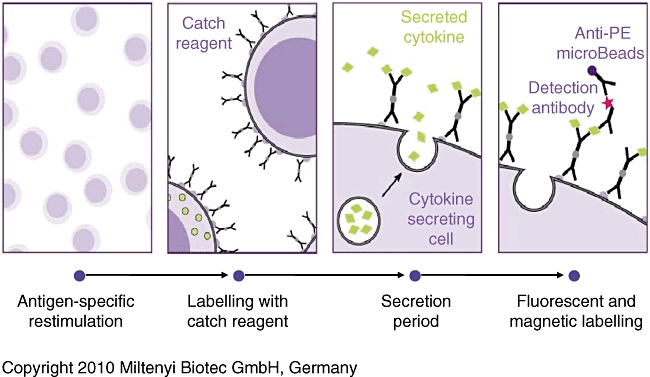
Summary of principal steps to label and isolate T cells using the cytokine secretion assay system.
Specimen results
Brief materials and methods
Human blood was collected following informed consent under local ethical guidelines, and mouse spleen cells were harvested from animals licensed under appropriate local regulations.
Human peripheral blood mononuclear cells (PBMC) were stimulated variously with CytoStim for 3 h (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany); Candida albicans extract (Greer Source Materials, Lenoir, NC, USA) 16 h; cytomegalovirus (CMV) lysate (Dade-Behring, Marburg, Germany) for 16 h; PepTivator CMV pp65 (Miltenyi Biotec) for 4 h and pp65 NLV(495–503) human leucocyte antigen (HLA)-A2-restricted peptide (Miltenyi Biotec) for 3 h. CD40 monoclonal antibody (mAb) functional grade (Miltenyi Biotec) was added to cultures if CD154 expression was analysed (has no T cell stimulatory effect). After stimulation cells were processed through the interleukin (IL)-17-phycoerythrin (PE), IL-2-allophycocyanin (APC) and IFN-γ-PE and APC cytokine secretion assay enrichment and detection kits (Miltenyi Biotec). Mouse splenocytes were stimulated with phorbol myristate acetate (PMA)/ionomycin for 3–6 h and processed through the mouse IL-17 secretion assay detection kit. Cells were isolated by MiniMACS magnet and two consecutive MS columns and stained with CD154 antibodies (human only) and appropriate phenotyping markers. Cells cultured into lines (see Rauser et al. [9] for method) were also stained with HLA-restricted tetramers for various CMV pp65 peptides in addition to phenotyping antibodies. Flow cytometry was carried out using BD FACS Calibur and Miltenyi Biotec MACSQuant analysers.
IL-17-producing cells
Human IL-17-producing cells were detected readily following 3 h Cytostim stimulation, typically forming 0·1% of viable T cells (Fig. 2a). The production of IL-17 was found only in CD154+ activated T cells, and confined almost exclusively to the CD4 subset (Fig. 2a). IL-17 was produced by 0·04–2% of human CD4 T cells (n = 21), thus there was a large amount of donor variability. In accordance with previously reported in vitro-generated IL-17-producing T cells lines [10], IL-17-producing cells in PBMC were >90% positive for the C-type lectin-like receptor CD161 (Fig. 2b). Human IL-17-secreting cells could be isolated readily from Cytostim-stimulated PBMC and enriched to very high purities of more than 90% (Fig. 2a). Such isolated cells are excellent for determining the ‘natural’ delineation of immune responses, and cells co-processed with IL-17 and IL-2 or IFN-γ secretion assays neatly illustrate the separation of Th1 and Th17 responses with mutually exclusive production of IFN-γ and IL-17 (Fig. 2c). Conversely, three populations of cells were seen when co-processed with IL-2 with a distinct IL-2+ IL-17+ population (Fig. 2c).
Fig. 2.
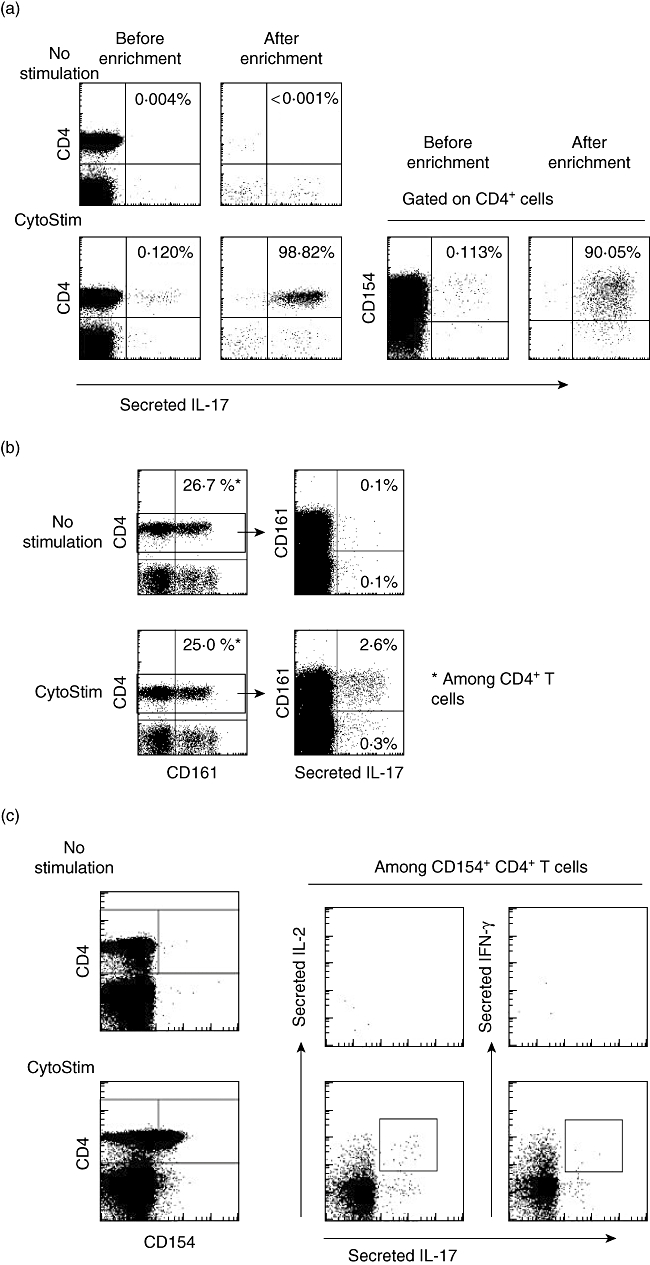
Human peripheral blood mononuclear cells (PBMC) stimulated with Cytostim for 3 h or unstimulated controls, assayed with the interleukin (IL)-17 secretion assay. (a) Stimulated and unstimulated cells pre- and post-isolation over two MS columns on a mini-MACS magnet. IL-17+ cells are >90% CD4+ CD154+. (b). Cells stained with CD4 and CD161 confirm that >90% of IL-17+ cells are CD161+. (c) Cells co-processed with interferon (IFN)-γ or IL-2 secretion assays show that there is a significant population of IL2+ IL-17+ cells (box) but not of IFN-γ+ IL-17+ cells. Note that IL-2 and IFN-γ are produced in significantly larger quantities than IL-17.
In stark contrast to human cells, IL-17 was made by multiple different cell types in mouse spleen (BALB/c) – CD4+, CD8+, γ/δ TCR+ and natural killer (NK) T cells (Fig. 3a). IL-17 formed a major part of the cytokine responses of γ/δ and NK T cells at 18·8% and 6·4%, respectively. The peak levels of mouse IL-17 secretion were reached extremely quickly, with maximal numbers of IL-17 producing CD4+ T cells and maximum mean fluorescence intensity (MFI) of cytokine produced by 3–4 h (Fig. 3b). The kinetics of IL-17 production and amount of cytokine produced vary markedly from mouse strain to strain and this should be checked before embarking on a study. The housing conditions of the mice are also important; for example, specific pathogen-free (SPF) mice make no detectable IL-17 (data not shown).
Fig. 3.
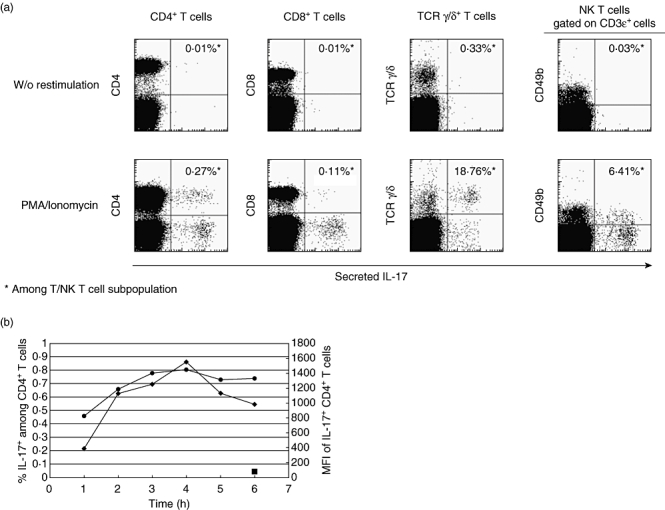
BALB/c spleen cells stimulated with phorbol myristate acetate (PMA) and ionomycin for 3–6 h, or unstimulated controls. (a) Interleukin (IL)-17 is secreted by CD4+, CD8+γ/δ T cell receptor (TCR)+ T cells and natural killer (NK) T cells at 3 h. (b) Kinetics of IL-17 secretion by CD4+ cells. The maximum percentage positive cells (•) and maximum mean fluorescence intensity (♦) are both reached within 3–4 h of stimulus.  : Unstimulated controls.
: Unstimulated controls.
One of the few well-defined antigen-specific Th17 responses in humans is against C. albicans[11]. Although Candida-specific T cells are relatively rare – typically, <0·04% of CD4+ cells make IL-17 when stimulated with Candida lysate (Fig. 4) – it was possible to enrich these cells easily to >84% purity (Fig. 4). These isolated cells can be cultured easily to establish true human Th17 T cell lines rapidly.
Fig. 4.
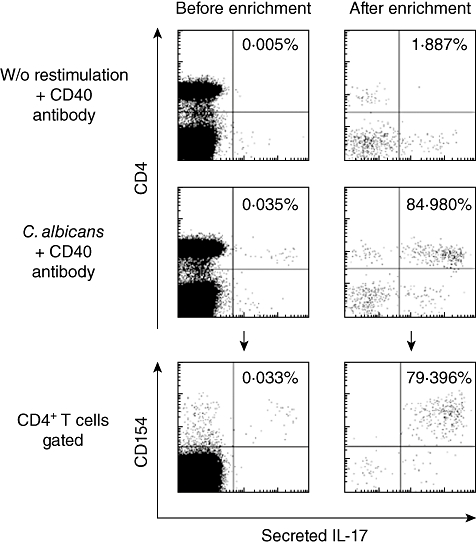
Human peripheral blood mononuclear cells (PBMC) stimulated with Candida albicans lysate for 16 h or unstimulated controls, assayed with the interleukin (IL)-17 secretion assay. The small number of IL-17+ cells can be enriched easily more than 2000 times and are confined to the activated CD154+ cells.
Rapid generation of virus-specific Th1 human T cell lines using IFN-γ
CMV+ donors carry a high precursor frequency of CMV-specific T cells, and CMV-reactive T cells lines are already in use to treat infection in stem cell transplant patients [5]. Here we stimulated PBMC with CMV antigen, isolated the antigen-specific cells using IFN-γ secretion and expanded the T cells into T cell lines
CMV-specific cells isolated
Human PBMC from CMV+ donors were stimulated with CMV lysate antigen (Dade Behring) for 16 h. For some HLA-A2+ donors, pp65 NLV(495–503) peptide was added during the last 3 h of the protein stimulus. IFN-γ selection isolated a mean of 7·7 × 104 CMV-reactive CD4+ T cells and 2·9 × 104 CD8+ T cells per 1 × 108 starting PBMC; adding the pp65 NLV peptide boosted the mean number of CD8+ T cells to a mean of 3·7 × 104 (Fig. 5a). Culturing these isolated T cells as described previously [9] for one round of expansion (2 weeks) led to a 2-log overall expansion rate, with slightly better proliferation of CD4 T cells (CD4 cells mean 2·3 log expansion versus CD8 cells 1·8-log expansion n = 20 Fig. 5b); also see [9]. Thus an average of 1 × 105 total CMV-reactive T cells isolated from 1 × 108 PBMC can be expanded to more than 1 × 107 total specific cells in 2 weeks – this is already similar to the total doses of cells currently given therapeutically [5].
Fig. 5.
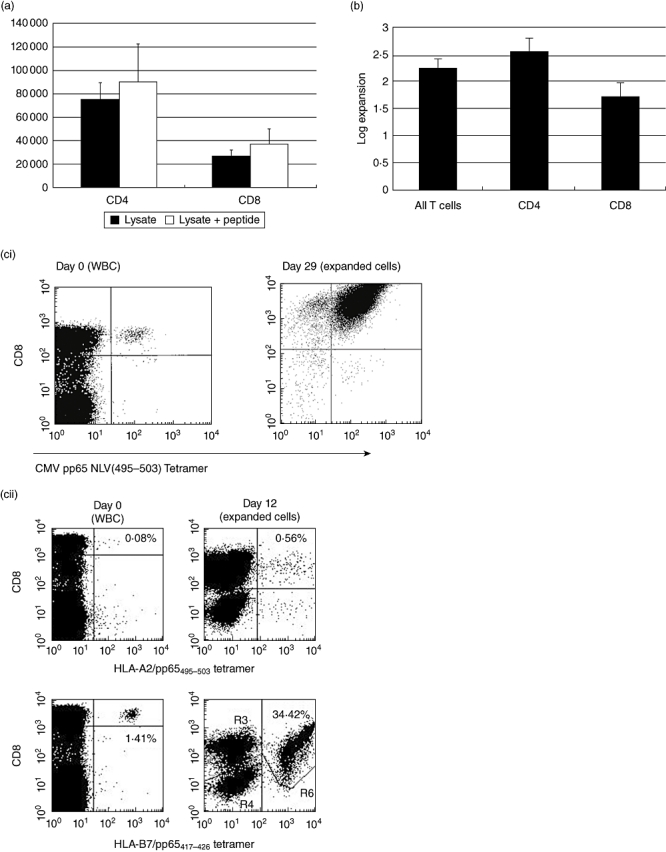
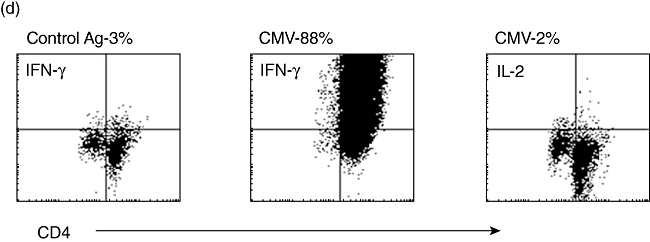
(a) Mean [±standard error of the mean (s.e.m.)] numbers of CD4 and CD8 T cells isolated using the interferon (IFN)-γ secretion assay per 108 starting human peripheral blood mononuclear cells (PBMC) after stimulus with cytomegalovirus (CMV)-lysate for 16 h (n = 20). Addition of the human leucocyte antigen (HLA)-A2-specific NLV(495–503) peptide from pp65 increases the numbers of CD8+ cells isolated, with an apparent bystander effect on CD4+ cells (n = 8). (b) Mean (± s.e.m.) expansion rate of T cells isolated using the IFN-γ secretion assay and expanded as per Rauser et al. [9]. CD4+ cells show an advantage in expansion rates over CD8+ cells. (c) (i) HLA-A2+ human PBMC stimulated with the HLA-A2 restricted NLV(495–503) peptide from pp65 before isolation of positive cells using the IFN-γ secretion assay (day 0–0·04% of the start total of 108 PBMC – 40 000+ cells). The isolated and expanded cells after two rounds of expansion are >99% NLV(495–503) tetramer+ and have reached 108 cells in total (day 29). (ii) HLA-A2+ HLA-B7+ human PBMC stimulated with pp65 PepTivator peptide pool. Pre-stimulus B7-restricted CD8- T cells heavily outnumber A2-restricted cells (day 0). After stimulus, isolation using the IFN-γ secretion assay and one round of expansion (day 12) the very heavy bias towards the B7-restricted cells is maintained, with 34·42% of all cells B7-restricted. (d) CD4+ T cells stimulated with CMV lysate, isolated using the IFN-γ secretion assay, and expanded for 12 days post-isolation. Restimulated with autologous dendritic cells and CMV or control lysate and assayed for intracellular IL-2 or IFN-γ expression after 6 h. The vast majority make IFN-γ to CMV-restimulation but make no IL-2, confirming the effector-memory status of these helper cells.
The specificity of CD8+ cells can be checked easily by major histocompatibility complex (MHC)-tetramer staining, but can be influenced heavily by the HLA-type of the donor – here we illustrate two HLA-A2+ T cell lines made following pp65 stimulus, but one donor is also HLA-B7+. In the HLA-B7- donor the cells produced are >99% positive for the dominant NLV(495–503) antigen (Fig. 5ci), but are almost completely absent in the HLA-B7+ donor, where most cells are specific for the B7-restricted TPR(417–426) peptide (Fig. 5cii). Thus care must be taken in understanding the immunodominance of different antigens in different HLA-types. CD4+ T cells are best assayed by antigen-specific cytokine production – here we illustrate CD4+ T cells restimulated with autologous dendritic cells and CMV-lysate – the effector memory phenotype for these cells is illustrated graphically, as 88% of the cells make IFN-γ in response to restimulation but only 2% make IL-2 (Fig. 5d).
Methodology
This section describes the protocol for cytokine detection and enrichment in detail. In this protocol, there are a number of critical steps, and failure to follow these will render results impossible to interpret. Critical steps and common areas that require troubleshooting are highlighted
Detailed methodology
Prepare mononuclear cell suspension
Prepare human PBMC or mouse spleen/lymph node (LN) cells.
After isolating cells as normal, wash cells into culture medium by adding medium, centrifuge at 300 g for 10 min, remove supernate.
Critical step – foreign protein such as fetal calf serum (FCS) leads to higher background cytokine production in the non-stimulated control – use human AB serum or mouse serum.
Resuspend cells in culture medium at 1 × 107 cells/ml and 5 × 106 cells/cm2 (e.g. 106 cells/100 µl in a 96-well plate, 107 cells/1 ml in a 24-well plate or 108 cells/10 ml in a 20 cm2 flask).
Critical step – this high cell density is essential for thorough and complete activation of all T cells in the culture.
Stimulation with antigen
If cells are to be stimulated for a long time-period (e.g. 16 h with protein antigen) then proceed directly to antigen stimulation. If cells are to be stimulated for a short time-period (e.g. 3 h with peptide), the cells may be stimulated immediately, or the cells may be cultured unstimulated overnight at 37°C, 5–7% CO2. Cells can then be stimulated with antigen the following morning.
Antigens. Add antigen, e.g. peptide (commonly 1–10 µg/ml) or protein (commonly 1–100 µg/ml), mix well and incubate at 37°C, 5–7% CO2 for either 3–6 h (peptide) or 6–16 h (protein). Ensure that a suitable negative control culture is also established, e.g. with no antigen, control peptide or control protein.
Mitogens. As a general rule mitogens may not be used, as the non-physiological stimulus can swamp the secretion assay reagent. The only example where mitogens are an appropriate stimulus is for mouse IL-17 assays with very low precursor frequencies where cells can be stimulated with PMA (10 ng/ml) and ionomycin (1 ug/ml) for 3 h.
Polyclonal stimuli. In order to elaborate as complete a cytokine response a possible, polyclonal stimuli such as CytoStim (Miltenyi Biotec GmbH), CD3/CD28 or Staphylococcal enterotoxin B (SEB, e.g. Sigma, St Louis, MO, USA) 1 µg/ml may be used. CytoStim is an artificial superantigen which binds simultaneously to an invariant part of the TCR complex and to accessory cells, thus cross-linking T cells and APC, producing a strong T cell stimulus without any TCR Vbeta restrictions. CytoStim-stimulated T cells may be isolated using the secretion assay system and used to generate T cell lines [12] but investigators need to establish specific conditions needed for the cells they are investigating.
Anti-CD40 mAb. Co-staining of cells in the analysis phase with anti-CD154 is very informative in confirming that cytokine-producing cells are activated. If this is desired, anti-CD40 mAb (e.g. Miltenyi Biotec Clone HB14) must be added to block CD40-induced down-regulation of CD154 on the T cells. Anti-CD40 does not have an additive stimulus effect on T cells.
Troubleshooting– it is necessary to establish the optimal stimulation time for your antigen and the cytokine being examined: short (3–6 h) periods for peptide stimulation are usually sufficient, while activation with proteins takes longer (6–16 h). Protein and peptides may be combined: add the peptide to the culture during the last 3–6 h of the protein stimulation.
Critical step– when assaying two cytokines together, a good knowledge of the kinetics of the production of both is required, and a compromise may need to be struck.
Label cells with cytokine catch reagent
After stimulation, the cells should be transferred to a suitable container, e.g. tube to allow sufficient washing and cooling throughout the process. This depends upon the cell number being analysed, and the expected antigen frequency. Up to 1 × 107 cells with an antigen frequency of <5% can be processed in 15-ml tubes. Larger volumes should be scaled up accordingly. Ensure maximum cell recovery by washing the cell culture vessel used for stimulation thoroughly with cold buffer. If necessary use a cell scraper to collect all cells.
Fill tube containing the cells with ice-cold buffer, centrifuge at 300 g for 10 min at 4°C. Remove supernatant completely.
Catch reagent. This is prepared by adding 20 µl of catch reagent stock to 80 µl of ice-cold culture medium; 100 µl of this stock solution is required per 1 × 107 cells. For example, for 5 × 107 dilute 100 µl of reagent with 400 µl medium. Store on ice until used. If two different cytokines are to be analysed at the same time add 10 µl of each catch reagent per 80 µl of medium. It is essential to pre-make the mix for dual cytokines so there is no competitive binding of catch reagents to CD45.
Remove buffer supernatant completely. Resuspend cells in 100 µl of diluted cytokine catch reagent per 1 × 107 cells, mix well and incubate for 5 min on ice.
Critical step– the only thing stopping the cells from making cytokines at this point is keeping them ice-cold.
Allow cells to secrete cytokines
Add warm (37°C) culture medium to dilute the cells to 105−106 cells/ml depending on the expected frequency of cytokine-secreting cells (among all cells): <1–5%: 1–2 × 106 cells/ml; 5–20%: 1–2 × 105 cells/ml; >20–50%: <105 cells/ml.
Critical step – the tube for the secretion phase must have sufficient volume to allow the addition of at least an equivalent volume of cold buffer to stop the reaction at the end of the secretion phase. This may mean that the cell sample has to be divided among several tubes for the secretion phase. For example, 5 × 107 cells, secretion volume 50 ml. Use 2 × 50 ml tubes, 25 ml each during secretion phase. This will allow the addition of 25 ml cold buffer at the end of the secretion phase.
Incubate cells for 45 min at 37°C under slow continuous agitation/rotation or mix tube every 5–10 min to avoid sedimentation of the cells.
Label cells with cytokine detection antibody
Stop the secretion reaction by adding a minimum of 1 vol of ice-cold buffer to the tube. Place tube on ice and incubate for 10 min to ensure that the sample is completely chilled. Centrifuge at 300 g for 10 min at 4°C. Remove supernatant completely.
Critical troubleshooting! This step is the primary cause of non-specific positive results with the secretion assay. Centrifuging cells into a pellet when they are still warm will contaminate the assay. Keep the cells ice-cold to stop secretion of cytokines after the secretion period. Ensure that the wash is in buffer, as the ethylenediamine tetraacetic acid (EDTA) helps to stop the reaction
Repeat washing step in ice-cold buffer. During the second wash prepare the cytokine detection antibody. This is diluted by adding 20 µl of cytokine detection antibody stock to 80 µl of ice-cold buffer; 100 µl of this stock solution is required per 1 × 107 cells. For example, for 5 × 107 dilute 100 µl of reagent with 400 µl buffer. Store on ice until used. For detection of two cytokines, add 10 µl of each detection antibody per 80 µl buffer.
Resuspend cells in 100 µl cytokine detection stock solution antibody per 1 × 107 cells. Mix well and incubate for 10 min on ice. If only analysis is required, additional staining reagents such as anti-CD4 or anti-CD8 (typically 1–5 µg/ml) may also be added at this point. Alternatively (e.g. if the cells are to be cultured after isolation), aliquots may be stained after the secretion assay process is finished.
Wash cells by adding >10 vol of ice-cold buffer, centrifuge at 300 g for 10 min at 4°C, remove supernatant completely. During this wash, if magnetic separation is to be performed, prepare the anti-PE or APC microbeads. This is prepared by adding 20 µl of microbead stock to 80 µl of ice-cold cell buffer; 100 µl of this stock solution is required per 1 × 107 cells. For example, for 5 × 107 dilute 100 µl of reagent with 400 µl buffer. Store on ice until used.
Critical step– if separating two cytokine populations consecutively, add only one anti-fluorochrome microbead at this point. The second microbead can be added after the first separation: repeat the steps described here.
Completely remove supernate. Resuspend in 500 µl buffer. For magnetic labelling, add 100 µl diluted anti-PE or APC microbeads per 1 × 107 cells, mix well and incubate for 15 min at 8°C (i.e. in the refrigerator, not on ice).
Wash cells by adding >10 vol of cold buffer, centrifuge at 300 g for 10 min at 4°C, remove supernatant completely.
Resuspend cells in a minimum of 500 µl of cold buffer (for 107 cells or less); for higher cell numbers use a dilution of 108 cells/ml. Take an aliquot for flow cytometry analysis and cell count before starting enrichment. Store sample on ice until separation.
Critical step– it is essential to have an unseparated sample to work out the start frequency and subsequent recovery of cells.
Prepare two MS columns per sample by rinsing with 500 µl of cold buffer. Place the first column into the magnetic field of a suitable MACS Separator (e.g. MiniMACS).
Apply the magnetically labelled cells to the column, allow the cells to pass through and wash with 3 × 500 µl of cold buffer.
Remove the first column from the separator, place the second column into the separator, and put the first column on top of the second one. Pipette 1 ml of cold buffer on to the first column, firmly flush out the retained cells using the plunger directly onto the second one.
Allow the cells to pass through and wash with 3 × 500 µl of cold buffer.
Remove the column from the separator, place the column in a suitable collection tube. Pipette 500 µl of cold buffer on top of the column, firmly flush out the retained cells using the plunger.
Troubleshooting – it is essential to use two columns. Each column can enrich the cells about 100 times. Thus, because of the low frequency of cytokine-producing cells, two columns are required to obtain the best purity.
If cells are to be cultured: if cells are to be cultured directly after isolation, cells can also be eluted with culture medium. In this case, replace the last buffer wash with a medium wash, and then elute the cells with medium. If medium is to be used, ensure that it does not contain any particles, e.g. from serum. If in doubt, filter medium before use. If medium elution is used, cells for flow cytometry should be washed free of any phenol red.
Resuspend cells in 500 µl of cold buffer and proceed with flow cytometric analysis (see below).
Flow cytometry analysis
Critical point
Do not use any PE- or APC-based tandem fluorochromes to stain cells sorted with anti-PE or APC beads, as they will be bound and stain non-specifically.
Exclude unwanted cells
All cytokine assays are low-frequency analyses. To properly identify cytokine producing cells, both positive staining with, e.g. CD4 or CD8 is required and also exclusion of unwanted cells from the analysis is vital. Exclusion of the dead cells (lymphocyte gating alone is not enough) using propridium iodide (PI), 7-AAD or other vital dyes will virtually eliminate non-specific background staining. It may also be necessary to exclude cells that tend to non-specifically bind fluorochromes, e.g. CD14 (human) or B220 (CD45R) (mouse). These cells can then be excluded from the analysis.
Do not use CD3 or TCR staining
When T cells are activated by antigen, CD3 and TCR are rapidly down-regulated. It is therefore not recommended to use CD3 or TCR antibodies for the analysis of the secretion assay. Although CD3 may not appear to be down-regulated in the whole population in comparison between control and stimulated samples, the small percentage of the cells that have reacted have done so. Using CD3 would therefore exclude the activated T cells. CD4 and CD8 may also be down-regulated partially after activation, but not to the same extent as CD3. However, care should be taken to ensure that activated cells are not excluded from the analysis.
General reagents
Cells
The secretion assay system is designed to be used with mononuclear cell preparations from, e.g. peripheral blood, leukapheresis (steady state) or spleen. Use with any other T cell preparation will require the presence of antigen presenting cells appropriate to the antigen for the assay to function.
Cytokine secretion assays
An up-to-date range of the cytokine assays available is available at: http://www.miltenyibiotec.co.uk/en/NN_67_Cytokine_producing_cells.aspx for human cells, and at: http://www.miltenyibiotec.co.uk/en/NN_98_Cytokine_producing_cells.aspx for mouse cells.
Buffer
Phosphate-buffered saline (PBS) pH 7·2, containing 0·5% (w/v) bovine serum albumin (BSA) and 2 mm EDTA, must be used ice-cold. For clinically orientated studies where bovine material is undesirable, 0·5% human serum albumin or AB serum may be substituted for BSA. Note that no bovine material should be used in culture medium.
0·5 m EDTA stock solution: dissolve 56 g sodium hydroxide (NaOH) in 900 ml distilled water. Add 146·2 g EDTA, adjust pH to 7·5, fill up to 1 l. Prepare buffer with, e.g. 4 ml of 0·5 m EDTA stock solution per 1 l of buffer.
Culture medium
Any standard medium may be used, e.g. RPMI-1640 containing 10% AB or autologous serum for human cells or mouse serum for murine cells. Medium is required both ice-cold and at 37°C for this procedure, and enough medium of each temperature must be available at the beginning. Never use FCS, as this gives high non-specific ‘background’ responses.
The use of complete ‘serum-free’ media, e.g. X-vivo series, is not recommended for stimulation with protein antigens as the lack of serum makes protein processing and presentation times unreliable.
No antibiotics are used throughout these experiments.
Culture medium for cell line culture
Isolated cells may be cultured in RPMI-1640 containing 10% AB or autologous serum for human cells or mouse serum for murine cells, or serum-free media, e.g. X-vivo15, which may require to be supplemented with appropriate serum. Improved performance may be seen by using HEPES buffered basic media and supplements such as mercaptoethanol, but this needs to be determined by the user for the specific T cells being grown.
Disclosures
All authors are employees of Miltenyi Biotec GmbH.
References
- 1.Mosmann TR, Cherwinski H, Bond MW, et al. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–57. [PubMed] [Google Scholar]
- 2.Wan YY. Multi-tasking of helper T cells. Immunology. 2010;130:166–71. doi: 10.1111/j.1365-2567.2010.03289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Connor RA, Taams LS, Anderton SM. Translational mini-review series on Th17 cells: CD4 T helper cells: functional plasticity and differential sensitivity to regulatory T cell-mediated regulation. Clin Exp Immunol. 2010;159:137–47. doi: 10.1111/j.1365-2249.2009.04040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O'Connor RA, Prendergast CT, Sabatos CA, et al. Th1 cells facilitate the entry of Th17 cells to the central nervous system during experimental autoimmune encephalomyelitis. J Immunol. 2008;181:3750–54. doi: 10.4049/jimmunol.181.6.3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peggs KS, Verfuerth S, Pizzey A, et al. Cytomegalovirus-specific T cell immunotherapy promotes restoration of durable functional antiviral immunity following allogeneic stem cell transplantation. Clin Infect Dis. 2009;49:1851–60. doi: 10.1086/648422. [DOI] [PubMed] [Google Scholar]
- 6.Berger C, Jensen MC, Lansdorp PM, et al. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manz R, Assenmacher M, Pfluger M, et al. Analysis and sorting of live cells according to secreted molecules, relocated to a cell-surface affinity matrix. Proc Natl Acad Sci USA. 1995;92:1921–25. doi: 10.1073/pnas.92.6.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Campbell JDM. Detection and enrichment of antigen-specific CD4+ and CD8+ T cells based on cytokine secretion. Methods. 2003;31:150–59. doi: 10.1016/s1046-2023(03)00125-7. [DOI] [PubMed] [Google Scholar]
- 9.Rauser G, Einsele H, Sinzger C, et al. Rapid generation of combined CMV-specific CD4+ and CD8+ T-cell lines for adoptive transfer into recipients of allogeneic stem cell transplants. Blood. 2004;103:3565–72. doi: 10.1182/blood-2003-09-3056. [DOI] [PubMed] [Google Scholar]
- 10.Maggi L, Santarlasci V, Capone M, et al. CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. Eur J Immunol. 2010;40:2174–81. doi: 10.1002/eji.200940257. [DOI] [PubMed] [Google Scholar]
- 11.Acosta-Rodriguez EV, Rivino L, Geginat J, et al. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639–46. doi: 10.1038/ni1467. [DOI] [PubMed] [Google Scholar]
- 12.Mazza G, Sabatos-Payton CA, Protheroe RE, et al. Isolation and Characterization of interleukin-10 secreting T cells from human peripheral blood. Hum Immunol. 2010;71:225–34. doi: 10.1016/j.humimm.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]