

Abstract
Vesicles released by cells have been described using various names, including exosomes, microparticles, microvesicles and ectosomes. Here we propose to differentiate clearly between ectosomes and exosomes according to their formation and release. Whereas exosomes are formed in multi-vesicular bodies, ectosomes are vesicles budding directly from the cell surface. Depending upon the proteins expressed, exosomes activate or inhibit the immune system. One of the major properties of exosomes released by antigen-presenting cells is to induce antigen-specific T cell activation. Thus, they have been used for tumour immunotherapy. By contrast, the major characteristics of ectosomes released by various cells, including tumour cells, polymorphonuclear leucocytes and erythrocytes, are the expression of phosphatidylserine and to have anti-inflammatory/immunosuppressive activities similarly to apoptotic cells.
Keywords: ectosomes, exosomes, inflammation, immunity, vesicles
Microvesicles
A direct membrane contact is the most evident way for two cells to communicate. In the immune system this is indeed the central mechanism by which T cells are activated by antigen-presenting cells. However, soluble mediators are also an essential link for the right information to be transmitted. Such mediators include cytokines, chemokines, hormones and smaller molecules, such as bioactive lipids and nitric oxide. Recent work has emphasized a third mechanism by which cells communicate, i.e. microvesicles released by one cell, which transmit essential information to target cells (reviewed in [1–3]).
Various eukaryotic cell types release membrane-derived microvesicles under specific physiological or pathological conditions. Interestingly, this phenomenon seems conserved during evolution, as bacteria are even described to release microvesicles that are important components of biofilms, and is a major signal trafficking system [4].
Vesiculation is a physiological mechanism that is used in cell growth, activation and protection. For example, for mineral formation in cartilage, bone and predentin, calcification is initiated by matrix vesicles released by chondrocytes, osteoblasts and odontoblasts [5]. Vesicles released by activated monocytes expose tissue factor and enhance coagulation [6]. Vesicle shedding is also an important defence mechanism protecting against complement attack, by allowing the removal of the C5b-9 attack complex from the cell surface by a calcium (Ca++)-dependent elimination as shown for many cell types including platelets, polymorphonuclear leucocytes (PMN), erythrocytes and oligodendrocytes [7]. Specific vesiculation is triggered or enhanced in pathological conditions such as inflammation, injury, vascular dysfunction or cancer [1].
A major problem in the microvesicle literature is the somewhat confusing nomenclature. Various names have been used, including particles, microparticles, vesicles, microvesicles, nanovesicles, exosomes, dexosomes, argosomes, ectosomes, etc. [1,2]. We will make no attempt in this review to be complete, but rather to highlight some important aspects, in order to try to define more clearly the role of ectosomes in human biology. Many excellent reviews are cited.
Whereas their formation, size and biological function may be different, one common point between all vesicles is the fact that they bud from a membrane, whether this occurs at the cell surface or in a vesicular compartment inside the cell (Fig. 1). Johnstone et al. [8] initially named exosomes, vesicles produced by intracellular budding into the late endosomal compartment. Evidently, every vesicle brings many of the specificities of the originating cell; however, there are some general properties characterizing those released directly from the cell surface which justifies a specific name, i.e. ectosomes[9], or ‘shedding’ microvesicles [3]. Before reviewing the evidence for this new nomenclature, it is worthwhile to describe exosomes briefly as they were defined initially, then misused as a generic name for every type of vesicle.
Fig. 1.
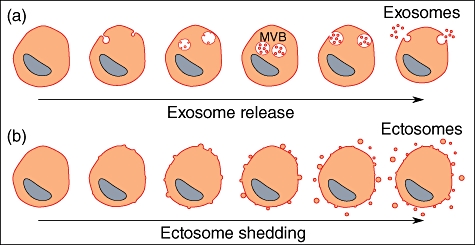
Exosomes and ectosomes are microvesicles budding from a membrane. (a) Exosomes are produced by inward budding into the late endosomal compartment, called multi-vesicular bodies (MVB). When MVB fuse with the cell membrane, exosomes are released as preformed vesicles. (b) Ectosomes are small membrane vesicles shed by many cells by budding directly from the cell membrane.
Exosomes
Exosomes are defined as small membrane vesicles formed by inward budding of endosomal membranes, called multi-vesicular bodies [1] (Fig. 1a). Recently the machinery responsible for exosome formation has been defined more clearly and includes a set of protein complexes (endosomal sorting complex required for transport: ESCRT) and an alternative pathway involving the sphingolipid ceramid [10,11]. When multi-vesicular bodies fuse with the plasma membrane, the preformed exosomes are released extracellularly. Many haematopoietic cells, including reticulocytes, platelets and leucocytes, produce and release exosomes. For reticulocytes, exosomes mediate the clearance of obsolete proteins such as the transferrin receptor [8]. Exosomes released by mature dendritic cells (DC) and B lymphocytes bind firmly to follicular DC and have the function of presenting antigen–major histocompatibility complex (MHC) class II complexes to T lymphocytes, and therefore have strong immunostimulatory activities. To identify them more clearly, DC-derived exosomes have been called ‘dexosomes’ by many authors (reviewed in [2,12–14]).
From the foregoing it is evident that (d)exosomes have been and are considered as potential candidates for cancer vaccines [15,16], and several clinical trials have been initiated [17,18]. There was then the hope that vesicles released by tumour cells – called exosomes by the authors – would have similar properties to dexosomes. Indeed, vesicles/exosomes of tumour cells express specific antigens, which are clear targets for vaccines, and are enriched in heat shock proteins known to favour antigen-presenting cell activation by delivering danger signals [12]. However, it was already known for some years that ‘shed vesicles’ released by cancer cells could also exhibit immune suppressive properties and more data have accumulated recently [19–21]. The down-regulation of the immune system was related to specific molecules expressed by the vesicles, such as Fas ligand (FasL) or transforming growth factor (TGF)-β1 [19,20]. Valenti et al. [22] showed the inhibition of DC differentiation by ‘exosomes’ released by melanoma and colon carcinoma cell lines by yet another, undefined mechanism. Thus, each type of tumour-derived vesicles might have unique inhibitory properties related to the cells producing them. In addition, for most of these data we are confronted with the unclear definition of exosomes as used by many authors in the literature. Collecting vesicles from the supernatant of tumour cell cultures or from plasma/ascites of patients with tumours does not provide sufficient information about their origin, despite some structural analogies with dexosomes [19]. The well-characterized immune presentation properties of dexosomes is related to their formation in multi-vesicular bodies, where the antigen is fixed onto the MHC molecules before the dexosome is released. Vesicles released by tumour cells, although expressing specific antigens, do not present them in the same correct context, and because they may be, to a large extent, shed directly from the cell surface, these vesicles do not correspond to the structurally organized exosomes which are formed inside the cell, or exceptionally in equivalent specialized cell membrane domains [23].
Ectosomes
As well as the release of preformed vesicles, many cells shed small membrane vesicles, which bud directly from the cell membrane (Fig. 1b) [3]. Stein and Luzio [9] coined the term ‘ectocytosis’ for the release of right-side-out-orientated vesicles with cytosolic content (ectosomes) from the surface of PMN attacked by complement. However, ectocytosis corresponded not only to the removal of the C5b-9 complex, but also to a specific sorting of membrane proteins into the shed ectosomes. Enrichment in cholesterol and diacylglycerol in the membrane attested to a specific sorting of lipids. The exposure of phosphatidylserine (PS) in the outer leaflet of the membrane is also a specific characteristic [24].
Although ectocytosis describes the same phenomenon in all cell types, the stimuli inducing cell-membrane budding can differ from one cell to another. Endothelial and circulating blood cells release ectosomes when exposed to specific stimuli such as complement attack [7]. Monocyte ectocytosis is induced by bacterial cell wall components including lipopolysaccharides (LPS), and platelets release ectosomes by activation through thrombin [6]. Fibroblasts release ectosomes in response to stress relaxation when cultured in a three-dimensional collagen matrix [25]. Many cancerous cells have an activated phenotype with highly active ectocytosis in the absence of any stimulus [1,26,27]. Although the shedding of ectosomes is enhanced when cells are activated, ectocytosis is an ongoing process in vivo for many cells (see osteoblasts [5]). Background levels of microvesicles originating from circulating and endothelial cells are found in blood [6], and similarly ectosomes originating from glomerular epithelial cells are found in urine [28,29].
It is known that different cells can produce both ectosomes and exosomes (e.g. platelets, DC). In a landmark paper, Heijnen et al. described the characteristics of these two type of vesicles released by platelets [30]. When activated with a thrombin receptor agonist, platelets release two distinct populations of vesicles. The first, corresponding to exosomes, were sized between 40 and 100 nm, bound annexin V poorly and were enriched in the tetraspanin protein CD63 – known to be a marker for late endocytic and multi-vesicular compartments. The second, being shed from the cell surface, were sized between 100 and 1000 nm, expressed many platelet surface proteins and bound annexin V, readily indicated an enrichment in PS. Interestingly, the properties of the two types of vesicles also differed; for instance, exosomes did not allow the prothrombinase complex to form, whereas factor X and prothrombin bound to the PS of shed vesicles (= ectosomes). These distinctions between the two types of vesicles were taken up by many authors who worked with vesicles released by cells (size and expression of specific markers to distinguish between intracellular-produced vesicles/exosomes versus vesicles shed from the cell surface/ectosomes) [31,32]. The presence of only trace amounts of PS on exosomes of reticulocytes had already been observed by Johnstone, although some PS is certainly present, as lactadherin/milk fat globule-EGF factor 8 (MFG-E8) is hooked onto PS on specific tumour and other exosomes [33]. Of interest is the observation by Heijnen et al., that exosomes were too small to be characterized by flow cytometry unless prebound to larger beads before flow cytometric analysis [30,34]. Electron microscopy is the major tool to characterize them.
For clarity, we will refer to microvesicles when the origin is uncertain, and to ectosomes only when the evidence is sufficient to exclude the presence of significant numbers of exosomes. Recent data suggest that ectosomes from polymorphonuclear leucocytes, erythrocytes and possibly tumour cells have, beside their specific properties, similar biological effects on inflammation and immune response.
Ectosomes released by PMN
Beside complement attack, the stimulation of PMN with N-formyl-methionyl-leucyl-phenylalanine (fMLP) or C5a induces the release of ectosomes within a few minutes [35]. Using electron microscopy it is possible to visualize the formation of buds on activated PMN and the newly formed ectosomes (diameter of 50–200 nm). They express a selective set of proteins originating not only from the cell membrane but also from intracellular compartments [e.g. elastase, myeloperoxidase/myeloperoxidase (MPO)], probably by binding of the soluble proteins released back to the ectosomes [36,37]. They also acquire proteins from plasma [38]. Gasser and Schifferli [39] demonstrated that they block the inflammatory response of human monocyte-derived macrophages to Zymosan A and LPS by inhibiting the release of tumour necrosis factor (TNF)-α, and reducing the release of interleukin (IL)-8 and IL-10. These results were unexpected, as PMN have a major role in defence against pathogens and in inflammatory processes. In addition, previous data suggested the opposite, i.e. microvesicles released by PMN were proinflammatory [40]. However, Mesri and Altieri [40] analysed a mixture of microvesicles released by PMN after a very long incubation time (overnight), a time-point at which PMN undergo apoptosis, necrosis and release many intracellular fragments, which might have contained proinflammatory stimuli. In addition, Gasser and Schifferli [39] noticed that PMN ectosomes induced an immediate release of TGF-β1, which is a known anti-inflammatory/repair cytokine.
The observation that ectosomes released by PMN can deactivate macrophages arriving at the site of injury suggests that they may play a central role in the control of local inflammation. Gout is a disease caused by the deposition of monosodium urate monohydrate (MSU) crystals in articular and periarticular tissues. The massive infiltration of PMN into the joints leads to dramatic clinical signs and symptoms. The central role of activation of the inflammasome by MSU has been elucidated in recent years [41]. However, even in the absence of clinical intervention, acute gouty arthritis undergoes self-resolution within a few days [42]. The underlying mechanism responsible for the resolution of the inflammation still remains poorly understood. Recent observations imply that macrophages attracted by PMN to the site of inflammation might play a major role in its resolution [43]. Indeed, Yagnik and colleagues [44] have demonstrated that macrophages do not release proinflammatory cytokines such as IL-1β, IL-6 and TNF-α, but in contrast release the anti-inflammatory cytokine TGF-β1 in the presence of MSU crystals as well as in human cantharidin-induced skin blisters. These results are similar to those obtained by exposing macrophages to PMN ectosomes [39]. Whether or not they are involved in the resolution of inflammation in acute gout remains to be tested.
The redistribution of PS to the surface is a hallmark of ectosomes, including those released by PMN [37]. It may explain some of the main properties of ectosomes. Under normal conditions, most of the PS and phosphatidylethanolamine are maintained by an active adenosine-5'-triphosphate (ATP)-dependent transport to the inner leaflet of the cell membrane, an aminophospholipid translocase. At the time of cell activation PS is externalized to the outer leaflet by several mechanisms, which include the blockade of the translocase and the activation of a Ca++-dependent scramblase or ‘scramblase activities’, as there is still debate about all possible reactions leading to scrambling. The exposure of PS on the activated PMN cell surface is patchy and transient, less than 1 h [45], whereas all the ectosomes released express PS as if the patches of PS exposure might be related to the budding of the cell membrane and ectosome shedding. This reversible expression of PS by living cells contrasts with the mechanisms leading to PS expression by dying/apoptotic cells [46]. When polarized neutrophils are studied, PS localizes predominantly to the uropod, where the release of vesicles (= ectosomes) has been observed [45]. Interestingly (d)exosomes express little PS [12].
The exposure of PS on ectosomes is a reminder of its known high expression on apoptotic cells. The clearance of apoptotic cells by phagocytes such as macrophages and DC occurs in a non-inflammatory manner, and there is growing evidence that their phagocytosis results in powerful anti-inflammatory or even immunosuppressive effects [47]. Multiple molecules on apoptotic cells drive this ‘death and silent clearance’ by phagocytic cells. Essential is the expression of PS, which might dock directly on the specific PS receptors [48] or, thanks to soluble proteins such as growth arrest-specific gene 6 (GAS6) or MFG-E8, bind receptors involved in phagocytosis and inhibition of the immune response (TAM receptors) [49]. A central role for PS in the immunomodulation of macrophages and DC has been emphasized by experiments using PS-expressing liposomes [50,51]. Thus, it would not be surprising that PS plays a similar central role for ectosomes to inhibit macrophages and DC. Experiments using annexin V as PS blocking agent would support this hypothesis [52].
Like apoptotic cells, ectosomes of PMN have been shown to inhibit/modify DC maturation and function [52]. Thus, ectosomes may represent a host factor influencing DC maturation at the site of injury, thereby possibly impacting upon downstream DC-dependent immunity. Returning to human diseases characterized by massive PMN infiltration, such as gout or myocardial infarction, it is noteworthy that no autoimmunity is induced despite strong local inflammation. In contrast to apoptotic cells, ectosomes have the particularity to be involved very early in inflammation, a time-point which might be crucial for determining later aspects of the cascade responsible for acquired immunity. In that sense, ectosomes might be involved not only in the termination of inflammation, but also in the initial control of the immune response, even perhaps in helping monocyte/macrophages to convert into a repair phenotype. PMN ectosomes are not only released in vitro; there is good evidence for their release in vivo in humans at sites of injury, as in synovial fluids and skin blisters [35,53]. In addition, during sepsis, ‘microvesicles’ of PMN origin have been found in the circulation [54]. These data indicate that significant ectocytosis occurs in vivo. However, PS is certainly not the only molecule involved; recently it has been shown that ectosomes of PMN may also inhibit inflammation by the expression of annexin 1 [55].
Ectosomes released by erythrocytes
Another situation during which humans are exposed to a large amount of ectosomes is blood transfusion. Stored blood contains ‘microparticles’ formed by ectocytosis from erythrocytes. The number of such particles increases with storage time, and it should be realized that high quantities are transfused with erythrocytes. Erythrocytes release them during in vitro (ATP depletion) and in vivo ageing [56–58]. Whereas erythrocyte ectosomes are devoid of spectrin they are enriched in several membrane proteins, including glycophosphatidyl-inositol (GPI)-linked proteins such as decay accelerating factor (DAF) and acetylcholine esterase, and in PS [58,59], similar to those released by PMN. They blocked the activation and release of proinflammatory cytokines from macrophages exposed to LPS or Zymosan A [60]. PS expression might not be the only molecule involved; however, CD47, known to inhibit the clearance of whole erythrocytes [61,62], was evidently insufficient to block the uptake of erythrocyte ectosomes by macrophages (Fig. 2). They were also shown to have long-lasting effects on macrophages, i.e. they did not only inhibit macrophages transiently, but modified their phenotypic profile [60]. Whether the ectosomes transfused with erythrocytes may account for some of the putative immunosuppressive properties attributed to blood transfusions remains to be further defined. Recent clinical study has shown that transfusions of erythrocytes might be responsible for a diminished survival in cancer patients [63]. In addition, the risk of complications after cardiac surgery (sepsis/mortality) increases with the duration of erythrocyte storage before transfusion, i.e. with the number of ectosomes transfused [64].
Fig. 2.

Confocal microscopy of erythrocyte ectosomes phagocytosed by human macrophages. Human monocyte-derived macrophages were incubated with fluorescently labelled ectosomes for 30 min, fixed and analysed by confocal laser microscopy. (a) Macrophages bind and ingest erythrocyte-derived ectosomes in absence of cytochalasin D (CytD). (b) Alternatively, macrophages were preincubated with CytD prior to the addition of labelled ectosomes showing absence of intracellular fluorescence. Phagocytosis of erythrocyte ectosomes was blocked.
Vesicles released by tumour cells: ectosomes or exosomes?
The answer to this question is certainly both. There is, however, evidence that many vesicles released by tumour cells are ectosomes, even when they are not described as such. For instance, Koppler et al. [65] have analysed the characteristics of microvesicles released by the human gastric carcinoma cell line Kato. These vesicles interfered with the activation of monocytes by LPS [decreased release of granulocyte–macrophage colony-stimulating factor (GM-CSF), TNF-α and increased release of IL-10], suggesting similar anti-inflammatory/immunosuppressive properties than ectosomes from PMN. Although the formation and release of these vesicles was not studied, it is noteworthy that they expressed PS and were pelleted by 15 000 g for 10 min only, suggesting that they were larger vesicles than exosomes. A different inhibitory profile was shown for microvesicles released by melanoma and colon carcinoma cell lines [22]. These vesicles interfered with the differentiation of normal human monocytes to DC, producing a subset of immunosuppressive cells. In addition, the vesicles isolated from the plasma of patients with melanoma had similar properties. The studied vesicles had a size of 100–200 nm, and from previous work showed many of the characteristics of exosomes (CD63 expression) [66], suggesting that there might be a size overlap between exo- and ectosomes. They most probably inhibit immunity by the expression of a series of proteins [FasL, TNF-related apoptosis-inducing ligand (TRAIL) and lactadherin/MFG-E8] fixed onto PS [33,66,67]. Multiple other data suggest that tumour releases exosomes. However, ectocytosis by various tumour cells is also well documented [1,26,27]. In addition, there is evidence that tumour ectosomes express PS as they bind to, and their function is inhibited by, annexin V [34,68]. Finally, Lima et al. [69] showed that vesicles shed by a melanoma cell line could be pelleted at 14 000 g, were approximately 200 nm in size, expressed PS and that their in vitro and in vivo down-regulation of inflammation/immune response could be inhibited by annexin V, i.e. by blockade of PS-mediated functions. Thus, many vesicles released by tumour cells have structural and functional characteristics of ectosomes. In sum, tumour cells are releasing vesicles by different routes (shedding from the cell surface, release of exosomes from multivesicular bodies), and the data in the literature do not take these differences into consideration systematically.
Other microvesicles
Microvesicles derived from activated endothelial cells, platelets and monocytes have been described to induce procoagulant activity due, to a large extent, to the expression of PS [6,70], the hallmark of ectosomes. The formation of a clot may enhance inflammation [6]; however, under most circumstances a vascular injury with thrombosis does not produce excessive inflammation or activation of cells responsible for an immune response. PS expression might be important in this control.
Differences between ectosomes
Whereas PS confers many general properties to ectosomes (procoagulant/anti-inflammatory), there are also significant differences related to the specificity of the ectosome released. First, their origin is evidently essential. For instance, there are differences in the inhibitory profiles of PMN versus erythrocyte ectosomes [39,60]. TGF-β1 is released by macrophages exposed to PMN ectosomes, but not when exposed to those of erythrocytes. Secondly, the site of release determines its function: ectosomes released by platelets in blood vessels will be active mainly as ‘procoagulant factors’, whereas those of PMN released in tissues will essentially regulate local inflammation. Thirdly, the continuous release of ectosomes by tumour cells may act as efficient immunosuppressors, whereas a burst of ectosome shedding by PMN will affect only local immunity. Many other factors will determine the functions of ectosomes. The unravelling of their biological roles is only beginning.
Conclusion
Ectocytosis describes the direct budding of vesicles from the cell membrane, and is different from exocytosis, which indicates the release of preformed vesicles. The resulting ectosomes express PS, whereas there is little expression of PS on exosomes [31,32]. Despite these clear differences, it is often difficult to distinguish between the two types of vesicles, particularly when they are harvested from in vivo materials, or even from supernatants of cell cultures. The hope that exosomes might present tumour antigens has led many research groups to collect ‘vesicles’ from tumour cells, defining them as exosomes, without controlling for ectocytosis. The debate on the terminology is not over, as was evident at a workshop in Montreal discussed by Johnstone [1]. The debate is, however, not about terminology, but rather about putative functions of ‘vesicles’, as exosomes have various effects on the immune system with, for example, (d)exosomes enhancing the immune response, and thus are worth vaccine trials, whereas ectosomes have generic functions in the opposite direction, due in particular to the expression of PS, i.e. to down-regulate inflammation and immunity.
Disclosure
The authors have no conflict of interest.
References
- 1.Johnstone RM. Exosomes biological significance: a concise review. Blood Cells Mol Dis. 2006;36:315–21. doi: 10.1016/j.bcmd.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 2.Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–93. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 3.Cocucci E, Racchetti G, Meldolesi J. Shedding microvesicles: artefacts no more. Trends Cell Biol. 2009;19:43–51. doi: 10.1016/j.tcb.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Mashburn LM, Whiteley M. Membrane vesicles traffic signals and facilitate group activities in a prokaryote. Nature. 2005;437:422–5. doi: 10.1038/nature03925. [DOI] [PubMed] [Google Scholar]
- 5.Anderson HC. Matrix vesicles and calcification. Curr Rheumatol Rep. 2003;5:222–6. doi: 10.1007/s11926-003-0071-z. [DOI] [PubMed] [Google Scholar]
- 6.Freyssinet JM. Cellular microparticles: what are they bad or good for? J Thromb Haemost. 2003;1:1655–62. doi: 10.1046/j.1538-7836.2003.00309.x. [DOI] [PubMed] [Google Scholar]
- 7.Pilzer D, Gasser O, Moskovich O, Schifferli JA, Fishelson Z. Emission of membrane vesicles: roles in complement resistance, immunity and cancer. Springer Semin Immunopathol. 2005;27:375–87. doi: 10.1007/s00281-005-0004-1. [DOI] [PubMed] [Google Scholar]
- 8.Johnstone RM, Adam M, Hammond JR, Orr L, Turbide C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes) J Biol Chem. 1987;262:9412–20. [PubMed] [Google Scholar]
- 9.Stein JM, Luzio JP. Ectocytosis caused by sublytic autologous complement attack on human neutrophils. The sorting of endogenous plasma-membrane proteins and lipids into shed vesicles. Biochem J. 1991;274(Pt 2):381–6. doi: 10.1042/bj2740381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trajkovic K, Hsu C, Chiantia S, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319:1244–7. doi: 10.1126/science.1153124. [DOI] [PubMed] [Google Scholar]
- 11.Marsh M, van Meer G. Cell biology. No ESCRTs for exosomes. Science. 2008;319:1191–2. doi: 10.1126/science.1155750. [DOI] [PubMed] [Google Scholar]
- 12.Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569–79. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 13.Fevrier B, Raposo G. Exosomes: endosomal-derived vesicles shipping extracellular messages. Curr Opin Cell Biol. 2004;16:415–21. doi: 10.1016/j.ceb.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 14.Iero M, Valenti R, Huber V, et al. Tumour-released exosomes and their implications in cancer immunity. Cell Death Differ. 2008;15:80–8. doi: 10.1038/sj.cdd.4402237. [DOI] [PubMed] [Google Scholar]
- 15.Wolfers J, Lozier A, Raposo G, et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001;7:297–303. doi: 10.1038/85438. [DOI] [PubMed] [Google Scholar]
- 16.Andre F, Schartz NE, Movassagh M, et al. Malignant effusions and immunogenic tumour-derived exosomes. Lancet. 2002;360:295–305. doi: 10.1016/S0140-6736(02)09552-1. [DOI] [PubMed] [Google Scholar]
- 17.Navabi H, Croston D, Hobot J, et al. Preparation of human ovarian cancer ascites-derived exosomes for a clinical trial. Blood Cells Mol Dis. 2005;35:149–52. doi: 10.1016/j.bcmd.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Le Pecq JB. Dexosomes as a therapeutic cancer vaccine: from bench to bedside. Blood Cells Mol Dis. 2005;35:129–35. doi: 10.1016/j.bcmd.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 19.Taylor DD, Gercel-Taylor C. Tumour-derived exosomes and their role in cancer-associated T-cell signalling defects. Br J Cancer. 2005;92:305–11. doi: 10.1038/sj.bjc.6602316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clayton A, Mitchell JP, Court J, Mason MD, Tabi Z. Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res. 2007;67:7458–66. doi: 10.1158/0008-5472.CAN-06-3456. [DOI] [PubMed] [Google Scholar]
- 21.Clayton A, Mitchell JP, Court J, Linnane S, Mason MD, Tabi Z. Human tumor-derived exosomes down-modulate NKG2D expression. J Immunol. 2008;180:7249–58. doi: 10.4049/jimmunol.180.11.7249. [DOI] [PubMed] [Google Scholar]
- 22.Valenti R, Huber V, Filipazzi P, et al. Human tumor-released microvesicles promote the differentiation of myeloid cells with transforming growth factor-beta-mediated suppressive activity on T lymphocytes. Cancer Res. 2006;66:9290–8. doi: 10.1158/0008-5472.CAN-06-1819. [DOI] [PubMed] [Google Scholar]
- 23.Booth AM, Fang Y, Fallon JK, Yang JM, Hildreth JE, Gould SJ. Exosomes and HIV Gag bud from endosome-like domains of the T cell plasma membrane. J Cell Biol. 2006;172:923–35. doi: 10.1083/jcb.200508014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zwaal RF, Schroit AJ. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood. 1997;89:1121–32. [PubMed] [Google Scholar]
- 25.Lee TL, Lin YC, Mochitate K, Grinnell F. Stress-relaxation of fibroblasts in collagen matrices triggers ectocytosis of plasma membrane vesicles containing actin, annexins II and VI, and beta 1 integrin receptors. J Cell Sci. 1993;105:167–77. doi: 10.1242/jcs.105.1.167. [DOI] [PubMed] [Google Scholar]
- 26.Dolo V, Ginestra A, Cassara D, et al. Selective localization of matrix metalloproteinase 9, beta1 integrins, and human lymphocyte antigen class I molecules on membrane vesicles shed by 8701-BC breast carcinoma cells. Cancer Res. 1998;58:4468–74. [PubMed] [Google Scholar]
- 27.Al-Nedawi K, Meehan B, Micallef J, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10:619–24. doi: 10.1038/ncb1725. [DOI] [PubMed] [Google Scholar]
- 28.Pascual M, Steiger G, Sadallah S, et al. Identification of membrane-bound CR1 (CD35) in human urine: evidence for its release by glomerular podocytes. J Exp Med. 1994;179:889–99. doi: 10.1084/jem.179.3.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lescuyer P, Pernin A, Hainard A, et al. Proteomics analysis of a podocyte vesicle-enriched fraction from normal human and pathological urines. Proteomics Clin Appl. 2008;2:1008–18. doi: 10.1002/prca.200800033. [DOI] [PubMed] [Google Scholar]
- 30.Heijnen HF, Schiel AE, Fijnheer R, Geuze HJ, Sixma JJ. Activated platelets release two types of membrane vesicles: microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood. 1999;94:3791–9. [PubMed] [Google Scholar]
- 31.Al-Nedawi K, Meehan B, Rak J. Microvesicles: messengers and mediators of tumor progression. Cell Cycle. 2009;8:2014–18. doi: 10.4161/cc.8.13.8988. [DOI] [PubMed] [Google Scholar]
- 32.Bianco F, Perrotta C, Novellino L, et al. Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J. 2009;28:1043–54. doi: 10.1038/emboj.2009.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeelenberg IS, Ostrowski M, Krumeich S, et al. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. 2008;68:1228–35. doi: 10.1158/0008-5472.CAN-07-3163. [DOI] [PubMed] [Google Scholar]
- 34.Keller S, Konig AK, Marme F, et al. Systemic presence and tumor-growth promoting effect of ovarian carcinoma released exosomes. Cancer Lett. 2009;278:73–81. doi: 10.1016/j.canlet.2008.12.028. [DOI] [PubMed] [Google Scholar]
- 35.Hess C, Sadallah S, Hefti A, Landmann R, Schifferli JA. Ectosomes released by human neutrophils are specialized functional units. J Immunol. 1999;163:4564–73. [PubMed] [Google Scholar]
- 36.Hess C, Sadallah S, Schifferli JA. Induction of neutrophil responsiveness to myeloperoxidase antibodies by their exposure to supernatant of degranulated autologous neutrophils. Blood. 2000;96:2822–7. [PubMed] [Google Scholar]
- 37.Gasser O, Hess C, Miot S, Deon C, Sanchez JC, Schifferli JA. Characterisation and properties of ectosomes released by human polymorphonuclear neutrophils. Exp Cell Res. 2003;285:243–57. doi: 10.1016/s0014-4827(03)00055-7. [DOI] [PubMed] [Google Scholar]
- 38.Gasser O, Schifferli JA. Microparticles released by human neutrophils adhere to erythrocytes in the presence of complement. Exp Cell Res. 2005;307:381–7. doi: 10.1016/j.yexcr.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 39.Gasser O, Schifferli JA. Activated polymorphonuclear neutrophils disseminate anti-inflammatory microparticles by ectocytosis. Blood. 2004;104:2543–8. doi: 10.1182/blood-2004-01-0361. [DOI] [PubMed] [Google Scholar]
- 40.Mesri M, Altieri DC. Endothelial cell activation by leukocyte microparticles. J Immunol. 1998;161:4382–7. [PubMed] [Google Scholar]
- 41.Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–65. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 42.Akahoshi T, Murakami Y, Kitasato H. Recent advances in crystal-induced acute inflammation. Curr Opin Rheumatol. 2007;19:146–50. doi: 10.1097/BOR.0b013e328014529a. [DOI] [PubMed] [Google Scholar]
- 43.Dalbeth N, Haskard DO. Inflammation and tissue damage in crystal deposition diseases. Curr Opin Rheumatol. 2005;17:314–18. doi: 10.1097/01.bor.0000157041.12116.69. [DOI] [PubMed] [Google Scholar]
- 44.Yagnik DR, Evans BJ, Florey O, Mason JC, Landis RC, Haskard DO. Macrophage release of transforming growth factor beta1 during resolution of monosodium urate monohydrate crystal-induced inflammation. Arthritis Rheum. 2004;50:2273–80. doi: 10.1002/art.20317. [DOI] [PubMed] [Google Scholar]
- 45.Frasch SC, Henson PM, Nagaosa K, Fessler MB, Borregaard N, Bratton DL. Phospholipid flip-flop and phospholipid scramblase 1 (PLSCR1) co-localize to uropod rafts in formylated Met-Leu-Phe-stimulated neutrophils. J Biol Chem. 2004;279:17625–33. doi: 10.1074/jbc.M313414200. [DOI] [PubMed] [Google Scholar]
- 46.Balasubramanian K, Mirnikjoo B, Schroit AJ. Regulated externalization of phosphatidylserine at the cell surface: implications for apoptosis. J Biol Chem. 2007;282:18357–64. doi: 10.1074/jbc.M700202200. [DOI] [PubMed] [Google Scholar]
- 47.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–75. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 48.Bratton DL, Henson PM. Apoptotic cell recognition: will the real phosphatidylserine receptor(s) please stand up? Curr Biol. 2008;18:R76–9. doi: 10.1016/j.cub.2007.11.024. [DOI] [PubMed] [Google Scholar]
- 49.Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol. 2008;8:327–36. doi: 10.1038/nri2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen X, Doffek K, Sugg SL, Shilyansky J. Phosphatidylserine regulates the maturation of human dendritic cells. J Immunol. 2004;173:2985–94. doi: 10.4049/jimmunol.173.5.2985. [DOI] [PubMed] [Google Scholar]
- 51.Shi D, Fu M, Fan P, et al. Artificial phosphatidylserine liposome mimics apoptotic cells in inhibiting maturation and immunostimulatory function of murine myeloid dendritic cells in response to 1-chloro-2,4-dinitrobenze in vitro. Arch Dermatol Res. 2007;299:327–36. doi: 10.1007/s00403-007-0770-9. [DOI] [PubMed] [Google Scholar]
- 52.Eken C, Gasser O, Zenhaeusern G, Oehri I, Hess C, Schifferli JA. Polymorphonuclear neutrophil-derived ectosomes interfere with the maturation of monocyte-derived dendritic cells. J Immunol. 2008;180:817–24. doi: 10.4049/jimmunol.180.2.817. [DOI] [PubMed] [Google Scholar]
- 53.Sadallah S, Lach E, Lutz HU, Schwarz S, Guerne PA, Schifferli JA. CR1, CD35 in synovial fluid from patients with inflammatory joint diseases. Arthritis Rheum. 1997;40:520–6. doi: 10.1002/art.1780400318. [DOI] [PubMed] [Google Scholar]
- 54.Nieuwland R, Berckmans RJ, McGregor S, et al. Cellular origin and procoagulant properties of microparticles in meningococcal sepsis. Blood. 2000;95:930–5. [PubMed] [Google Scholar]
- 55.Dalli J, Norling LV, Renshaw D, Cooper D, Leung KY, Perretti M. Annexin 1 mediates the rapid anti-inflammatory effects of neutrophil-derived microparticles. Blood. 2008;112:2512–19. doi: 10.1182/blood-2008-02-140533. [DOI] [PubMed] [Google Scholar]
- 56.Dumaswala UJ, Greenwalt TJ. Human erythrocytes shed exocytic vesicles in vivo. Transfusion. 1984;24:490–2. doi: 10.1046/j.1537-2995.1984.24685066807.x. [DOI] [PubMed] [Google Scholar]
- 57.Dumaswala UJ, Dumaswala RU, Levin DS, Greenwalt TJ. Improved red blood cell preservation correlates with decreased loss of bands 3, 4.1, acetylcholinestrase, and lipids in microvesicles. Blood. 1996;87:1612–16. [PubMed] [Google Scholar]
- 58.Pascual M, Lutz HU, Steiger G, Stammler P, Schifferli JA. Release of vesicles enriched in complement receptor 1 from human erythrocytes. J Immunol. 1993;151:397–404. [PubMed] [Google Scholar]
- 59.de Jong K, Beleznay Z, Ott P. Phospholipid asymmetry in red blood cells and spectrin-free vesicles during prolonged storage. Biochim Biophys Acta. 1996;1281:101–10. doi: 10.1016/0005-2736(96)00026-0. [DOI] [PubMed] [Google Scholar]
- 60.Sadallah S, Eken C, Schifferli JA. Erythrocyte-derived ectosomes have immunosuppressive properties. J Leukoc Biol. 2008;84:1316–25. doi: 10.1189/jlb.0108013. [DOI] [PubMed] [Google Scholar]
- 61.Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FP. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051–4. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 62.Subramanian S, Tsai R, Discher DE. The ‘metabolon,’ CD47, and the ‘phagocytic synapse’: molecular co-localization and species divergence. Transfus Clin Biol. 2006;13:31–8. doi: 10.1016/j.tracli.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 63.Jensen LS, Puho E, Pedersen L, Mortensen FV, Sorensen HT. Long-term survival after colorectal surgery associated with buffy-coat-poor and leucocyte-depleted blood transfusion: a follow-up study. Lancet. 2005;365:681–2. doi: 10.1016/S0140-6736(05)17949-5. [DOI] [PubMed] [Google Scholar]
- 64.Koch CG, Li L, Sessler DI, et al. Duration of red-cell storage and complications after cardiac surgery. N Engl J Med. 2008;358:1229–39. doi: 10.1056/NEJMoa070403. [DOI] [PubMed] [Google Scholar]
- 65.Koppler B, Cohen C, Schlondorff D, Mack M. Differential mechanisms of microparticle transfer toB cells and monocytes: anti-inflammatory propertiesof microparticles. Eur J Immunol. 2006;36:648–60. doi: 10.1002/eji.200535435. [DOI] [PubMed] [Google Scholar]
- 66.Andreola G, Rivoltini L, Castelli C, et al. Induction of lymphocyte apoptosis by tumor cell secretion of FasL-bearing microvesicles. J Exp Med. 2002;195:1303–16. doi: 10.1084/jem.20011624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huber V, Fais S, Iero M, et al. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: role in immune escape. Gastroenterology. 2005;128:1796–804. doi: 10.1053/j.gastro.2005.03.045. [DOI] [PubMed] [Google Scholar]
- 68.Al-Nedawi K, Meehan B, Kerbel RS, Allison AC, Rak J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc Natl Acad Sci USA. 2009;106:3794–9. doi: 10.1073/pnas.0804543106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lima LG, Chammas R, Monteiro RQ, Moreira ME, Barcinski MA. Tumor-derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine-dependent manner. Cancer Lett. 2009;283:168–75. doi: 10.1016/j.canlet.2009.03.041. [DOI] [PubMed] [Google Scholar]
- 70.Sabatier F, Roux V, Anfosso F, Camoin L, Sampol J, Dignat-George F. Interaction of endothelial microparticles with monocytic cells in vitro induces tissue factor-dependent procoagulant activity. Blood. 2002;99:3962–70. doi: 10.1182/blood.v99.11.3962. [DOI] [PubMed] [Google Scholar]