

Abstract
B and T lymphocyte attenuator (BTLA) is an immunoglobulin superfamily member surface protein expressed on B and T cells. Its ligand, herpesvirus entry mediator (HVEM), is believed to act as a monomeric agonist that signals via the CRD1 of HVEM to inhibit lymphocyte activation: HVEM is also the receptor for lymphotoxin-α and LIGHT, which both bind in the CRD2 and CRD3 domains of the HVEM molecule, and for CD160 which competes with BTLA. We have shown that recombinant HVEM and a panel of different monoclonal antibodies specifically bind murine BTLA on both B and T cells and that some antibodies inhibit anti-CD3ε-induced T cell proliferation in vitro, but only when constrained appropriately with a putatively cross-linking reagent. The antibodies had no significant effect on in vitro T cell proliferation in a mixed lymphocyte reaction (MLR) assay nor on in vitro DO11.10 antigen-induced T cell proliferation. None of these antibodies, nor HVEM-Fc, had any significant effect on in vitro B cell proliferation induced by anti-immunoglobulin M antibodies (±anti-CD40) or lipopolysaccharide. We further elucidated the requirements for inhibition of in vitro T cell proliferation using a beads-based system to demonstrate that the antibodies that inhibited T cell proliferation in vitro were required to be presented to the T cell in a cis, and not trans, format relative to the anti-CD3ε stimulus. We also found that antibodies that inhibited T cell proliferation in vitro had no significant effect on the antibody captured interleukin-2 associated with the in vivo activation of DO11.10 T cells transferred to syngeneic recipient BALB/c mice. These data suggest that there may be specific structural requirements for the BTLA molecule to exert its effect on lymphocyte activation and proliferation.
Keywords: activation, B lymphocyte, BTLA, cross-linking, inhibition, T lymphocyte
Introduction
B and T lymphocyte attenuator (BTLA) is a recently described molecule that is expressed on B and T lymphocytes and at lower levels on dendritic cells, splenic macrophages and natural killer (NK) cells [1,2]. It has been reported to be absent on naive T cells, up-regulated on activated T cells and maintained on polarized T helper type 1 (Th1), but not Th2 cells, in both mice and humans [3]. It has an immunoglobulin superfamily domain in its extracellular region and the classical immunoreceptor tyrosine-based inhibitory motif (ITIM) sequences in its intracellular region [1]. Recent data have demonstrated that BTLA binds uniquely as a monomer to the herpesvirus entry mediator (HVEM) molecule in the most membrane distal cysteine-rich domain 1 (CRD1) of HVEM and that HVEM signals unidirectionally through BTLA to inhibit T cell proliferation, possibly by recruiting intracellular SHP-1 and SHP-2 [2–5].
HVEM is also the receptor for both LIGHT and lymphotoxin-α, which bind in the CRD2 and CRD3 domains, and for CD160, which has been reported to compete with BTLA for binding to HVEM [6].
Functionally, several investigators have provided evidence that signalling through BTLA acts to inhibit T lymphocyte proliferation using a transfected cell co-culture system, plate-immobilized HVEM ligand or monoclonal antibodies specific for mBTLA [3,7–9]. With the exception of the reported slightly greater in vitro proliferation of purified B cells from the BTLA knock-out mice to anti-immunoglobulin M (IgM), little work has been conducted on the functional role of BTLA on B cells, despite the demonstrably high levels of BTLA expression on B cells [1,2,4].
Identification of the HVEM : BTLA axis has allowed new insights into some long-standing puzzling in vivo observations. HVEM knock-out mice have been shown to exhibit increased morbidity in a model of concanavalin A-mediated T cell-dependent autoimmune hepatitis, as well as increased susceptibility to myelin oligodendrocyte glycoprotein (MOG) peptide-induced experimental autoimmune encephalitis [10,11]. Interestingly, the BTLA knock-out mice have a somewhat similar phenotype to the HVEM knock-out mice in that T cells from the mice exhibited enhanced proliferative responses to in vitro anti-CD3ε stimulation, but not to concanavalin A [1,12]. The BTLA knock-out mice also exhibited increased specific antibody responses and increased susceptibility to MOG peptide-induced experimental autoimmune encephalitis [1].
Several in vivo studies have been performed with HVEM-Ig that demonstrate its beneficial effect in mouse models of transplantation rejection and uveitis [13–16]. However, these studies all predate the identification of the HVEM : BTLA axis, and it is not clear whether these in vivo effects are due to the neutralization of signalling through HVEM by LIGHT and lymphotoxin-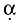 or the actions of the soluble HVEM-Ig through BTLA. No in vivo disease models or mechanism-based studies with a uniquely BTLA specific reagent have been described in the literature.
or the actions of the soluble HVEM-Ig through BTLA. No in vivo disease models or mechanism-based studies with a uniquely BTLA specific reagent have been described in the literature.
Interestingly, Cheung et al. identified the UL144 (Unique Long 144) protein from the human cytomegalovirus (HuCMV) as being capable of binding hBTLA, but not LIGHT, and inhibiting in vitro lymphocyte proliferation [17–19]. HuCMV infection is a serious disease in immunosuppressed patients and the UL144 is one of many open reading frames present in clinical isolates but not in commonly used laboratory strains [20–25]. UL144 is homologous to the N terminal, putative BTLA binding region of hHVEM. There is no known murine equivalent. This suggests that that the virus may have evolved the ability to target the BTLA pathway in an effort to induce immunosuppression in its human host. This raises the intriguing possibility that targeting BTLA may be an attractive pharmacological approach for the treatment of human inflammatory diseases. This hypothesis is supported further by associations of BTLA polymorphisms with clinical rheumatoid arthritis and inflammatory bowel disease and the demonstrated crucial role for BTLA in models of inflammatory bowel disease (IBD) [26–28].
In this study, we set out to determine the exact requirements for BTLA specific reagents to inhibit T and B lymphocyte proliferation in vitro and to test their ability to ameliorate inflammation in a mechanistically relevant in vivo model.
We found that HVEM and a panel of different monoclonal antibodies bound murine BTLA specifically on both B and T cells and that some antibodies inhibited anti-CD3ε-induced T cell proliferation in vitro, but only when constrained appropriately with a putatively cross-linking reagent. They did not affect in vitro T cell proliferation induced in a mixed lymphocyte reaction or antigen-induced proliferation of DO11.10 transgenic T cells. None of these antibodies, nor the HVEM-Fc molecule, had any significant effect on in vitro B cell proliferation. We elucidated further the requirements for inhibition of in vitro T cell proliferation using a beads-based system to demonstrate that the antibodies that inhibited T cell proliferation in vitro were required to be presented to the T cells in a cis, and not trans, format relative to the anti-CD3ε stimulus. We also found that the antibodies that inhibited T cell proliferation in vitro had no significant effect on the antibody-captured interleukin (IL)-2 associated with the in vivo activation of DO11.10 T cells transferred to syngeneic recipient BALB/c mice. These data suggest that there may be specific structural requirements for the BTLA molecule to exert its effect on lymphocyte activation and proliferation.
Materials and methods
Anti-BTLA reagents
Antibodies specific for BTLA (and fluorescently labelled antibodies) were obtained from e-BioSciences (San Diego, CA, USA). Murine BTLA (extracellular domain), murine HVEM (CRD1-4) and mCTLA-4 were made as mouse or human IgG1 Fc fusion proteins as indicated and expressed in a CHO adherent cell line. Single cell clones were isolated and conditioned medium was harvested over 7 days of production. The proteins were purified with a monoclonal antibody (mAb) select column in the Department of Protein Sciences at Amgen Thousand Oaks. mAb 20A9 was used as an irrelevant mouse IgG1 isotype control antibody specific for the CXCL10 chemokine [29].
In vitro murine T cell proliferation assays: plate- and bead-based
Mouse CD4+ T cells were purified from C57BL/6 mouse splenocytes by AutoMACS-negative selection (Miltenyi Biotec, Auburn, CA, USA). In a U-bottomed 96-well plate, 100 000 T cells were activated in vitro by 0·1 µg per plate of hamster anti-mouse CD3ε clone 145-2C11 for 72 h and [3H]-labelled tritium was added to the cell culture medium for the last 18 h; the test reagent was co-immobilized with the activating stimulus at the indicated amounts. In the cross-linked plate, 1 µg per well of a polyclonal goat anti-mFc reagent (Sigma Biochemicals, St Louis, MO, USA) was added at the same time as the activating stimulus and the test reagents were added for the last 18 h at the indicated amounts. Cells were harvested onto a filter after 72 h of stimulation and radioactivity was assessed as a measure of cell proliferation. Analysis of secreted cytokines was by multi-analyte profiling using a kit from LincoPlex (St Charles, MO, USA), as per the manufacturer's instructions. For the bead-based assays, 100 000 T cells in a U-bottomed 96-well plate were activated in vitro by bead-absorbed anti-mouse CD3ε coated at 0·1 µg per 106 cells on tosyl-activated 4·5 µM beads (Dynal Biotech, ASA Corporation/Invitrogen, Oslo, Norway/Carlsbad, CA, USA: catalogue no. 140-13) for 72 h and [3H]-labelled thymidine was added to the cell culture medium for the last 18 h; the test reagent was either co-immobilized with the activating stimulus (cis format) or on a separate bead (trans format) at the indicated amounts. Cells were harvested and proliferation and secreted cytokines analysed as described previously. Proteins were immobilized on the beads, as per the manufacturer's instructions. Briefly, 0·5 ml of the provided Dynabeads were washed twice with phosphate-buffered saline (PBS), resuspended in 200 µl of PBS per tube, and 20 µg of anti-CD3ε and/or the indicated µg amount of anti-BTLA test antibody (or antibodies) reagent was absorbed passively to the beads, mixed well and incubated at room temperature for 60 min. The tube was vortexed (bench top) every 3 min to ensure mixing. Then 100 µl of a 0·5% bovine serum albumin (BSA) solution in PBS was added to each tube and the volume adjusted to 500 µl with PBS to block any unoccupied bead surface. The beads were incubated at 4°C for 3 days with shaking and then washed three times with 0·1% BSA in PBS buffer. They were finally resuspended in 500 µl of 0·1% BSA in PBS to yield a final bead concentration of 4 × 108/ml and the final bead : cell ratio in the well was adjusted to 1:1.
For the mixed lymphocyte reaction (MLR) in vitro assay, T cells were isolated from the spleens of C57BL/6 mice with a pan T cell-negative selection isolation kit (Miltenyi Biotech); antigen-presenting cells (APC) were selected negatively from the spleens of BALB/c mice (Miltenyi Biotech). The APC were incubated with mitomycin C (Sigma) at 25 µg/ml for 30 min at 37°C and then washed three times. T cells were cultured with mitomycin C-treated APC at a 1:1 ratio, with 2 × 105 cells per well in 200 µl volume for 5 days. For the last 16 h, 1 µCi of [3H]-thymidine (MP Biomedicals, Inc., Irvine, CA, USA) was added to each well. The cells were then harvested and [3H]-incorporation measured using a 1450 Microbeta Liquid Scintillation and Luminescence Counter (Perkin Elmer, Sherton, CT, USA).
For the ovalbumin (OVA) antigen-specific T cell proliferation in vitro assay, CD4 T cells were isolated from the spleens of DO11.10 mice by CD4 T cell-negative selection (Miltenyi Biotec) and APCs were isolated from same mice with an AutoMACS T cell depletion kit (Miltenyi Biotec). The APCs were incubated with mitomycin C at 25 µg/ml for 30 min at 37°C and then washed three times. The T cells were stimulated by 0·1 µg/ml OVA peptide in the presence of mitomycin C-treated APC at a 1:1 ratio, with 2 × 105 cells per well in a 200 µl volume. Cell proliferation was measured at day 3 as described above.
In vitro murine B cell proliferation assays
Mouse B cells were purified from C57BL/6 mouse splenocytes by AutoMACS-negative selection (Miltenyi Biotec) and 100 000 cells were incubated in duplicate in 96-well flat-bottomed plates in RPMI-1640 (Invitrogen, Inc.) with 10% heat-inactivated fetal bovine serum (FBS) (54°C for 45 min), 1 mM HEPES and 55 µM β-mercaptoethanol (all from Gibco). Cells were stimulated with 2 µg/ml of lipopolysaccaride (List Biological Laboratories, Inc.) or 1 µg/ml of anti-CD40 mAb (BD Biosciences, San Diego, CA, USA) plus 2 µg/ml of goat F(ab')2 anti-mouse IgM (Jackson ImmunoResearch, West Grove, PA, USA) in the presence of various concentrations of anti-BTLA reagent or mHVEM-Fc protein. Cross-linking was performed as described previously. Cells were incubated for 72 h at 37°C and 5% CO2 and pulsed with radioactive [3H]-thymidine for the last 18 h to assess proliferation.
BIACore surface plasmon resonance
The BIACore 2000, sensor chip CM5, surfactant P-20, HBS-EP [10 mM HEPES, 0·15 M NaCl, 3·4 mM ethylendiamine tetraacetic acid (EDTA), 0·005% P-20, pH 7·4], amine coupling kit and 10 mM acetate pH 4·5 were from BIACore, Inc. (Piscataway, NJ, USA). Immobilization of antibodies to the sensor chip surface was performed according to the manufacturer's instructions, using a continuous flow of 10 mM HEPES, 0·15 M NaCl, 3·4 mM EDTA and 0·005% P-20, pH 7·4 (HBS-EP buffer). Briefly, carboxyl groups on the sensor chip surfaces were activated by injecting 60 µl of a mixture containing 0·2 M N-ethyl-N′ (dimethylaminopropyl)carbodiimide (EDC) and 0·05 M N-hydroxysuccinimide (NHS). Specific surfaces were obtained by injecting antibody diluted in 10 mM acetate, pH 4·5 at a concentration of 30 µg/ml. Excess reactive groups on the surfaces were deactivated by injecting 60 µl of 1 M ethanolamine. Final immobilized levels were ∼9000–12 000 resonance units (RU) for the antibodies. A blank, mock-coupled reference surface was also prepared on the sensor chip. To perform a competition binding analysis of the anti-mBTLA mAbs by BIACore, each antibody was immobilized to a different flow cell of a CM5 sensor chip. Murine BTLA-mFc was captured on the antibody surfaces and then either the immobilized antibody or a different antibody was injected over the captured mBTLA-mFc.
In vivo DO11.10 T cell proliferation
DO11.10 splenocytes, 20 × 106, were adoptively transferred into BALB/c recipients. The next day mice were treated intraperitoneally with 15 mg/kg of anti-BTLA reagent or control reagent. Three h after protein treatment animals were administered 10 µg of biotin-labelled rat anti-mIL-2 (clone JES6-5 H4) to capture secreted IL-2 as described previously. Mice were then injected in the footpad with 100 µg of OVA protein to activate the monoclonal population of transferred DO11.10 T cells. The mice were rested for 18 h before exsanguination and then serum IL-2 was detected using an enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN, USA). Studies that benchmarked the effect of CTLA4-Fc in this model were performed in a similar manner.
Results
Only cross-linked anti-BTLA reagents inhibit anti-CD3ε induced in vitro T cell proliferation
Figure 1 shows the effect of anti-BTLA reagents on the anti-CD3ε-induced proliferation of murine spleen-derived T cells in vitro. We have shown previously that the mHVEM-mFc ligand and all the putative anti-BTLA mAb-stained T and B cells by fluorescence activated cell sorter analysis (FACS) and that the staining could be reversed specifically with soluble mBTLA-mFc (data not shown). We also performed individual titrations with commercial hamster anti-mCD3ε mAb clone 145-2C11 to induce robust and reproducible T cell proliferation in vitro. When the anti-BTLA reagents were co-immobilized on the plate with the stimulus, no significant effect on T cell proliferation was observed. However, when the anti-BTLA reagents were putatively ‘cross-linked’ by coating the plate with a polyclonal goat anti-mouse Fc reagent and then adding the murine reagents, the mHVEM-mFc ligand and some of the anti-BTLA mAb inhibited T cell proliferation dose-responsively – specifically, clones 6 H6, 8F4 and 3F9.D12. A similar effect was seen on the levels of secreted interferon-γ (data not shown). Further studies with the anti-BTLA reagents in the murine in vitro MLR and the murine in vitro DO11.10 antigen-specific T cell proliferation system have shown similar results to the direct plate immobilization assay system in that the anti-BTLA reagents had no significant effect on in vitro T cell proliferation induced by these methods (see Supporting information, Figs S1 and S2, at the end of the paper and online).
Fig. 1.
Anti-CD3ε T cell proliferation is unaffected by co-immobilized anti-B and T lymphocyte attenuator (BTLA) reagents (a): cross-linked herpesvirus entry mediator (HVEM-Fc) and some anti-mBTLA monoclonal antibodies (mAbs) suppress this proliferation (b). Mouse CD4+ T cells were purified from C57BL/6 mouse splenocytes by AutoMACS-negative selection (Miltenyi Biotec). In schematic (a) 100 000 T cells in a U-bottomed 96-well plate were activated in vitro by plate-bound hamster anti-mouse CD3ε (clone 145-2C11 at 0·1 µg per plate) for 72 h and [3H]-labelled tritium was added to the cell culture medium for the last 18 h; the test reagent was co-immobilized with the activating stimulus at the indicated amounts. In the cross-linked plate in schematic (b), 1 µg per well of a polyclonal goat anti-mFc reagent was added just prior to the activating stimulus and the test reagents were added for the last 18 h at the indicated amounts. Cells were harvested onto a filter after 72 h of stimulation and radioactivity was assessed as a measure of cell proliferation. Analysis of secreted cytokines by multi-analyte profiling showed that secreted levels of interferon-γ correlated well with cell proliferation (data not shown). Epitope grouping experiments using BIACore showed that the anti-mBTLA mAbs that inhibited T cell proliferation grouped to the same epitope on the mBTLA molecule (see Fig. S3).
Competition binding experiments with surface plasmon resonance (BIAcore) showed that the anti-BTLA mAb clones that inhibited in vitro T cell proliferation in the ‘cross-linked’ plate format grouped to a similar epitope on the BTLA molecule and, conversely, the clones that had no effect on T cell proliferation grouped to a different epitope (see Fig. S3).
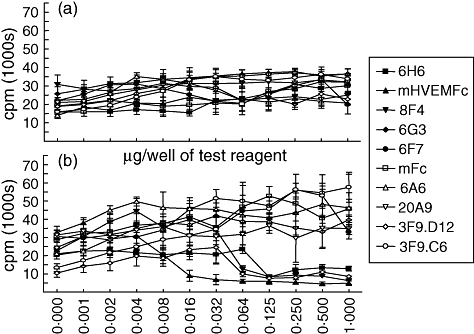
Anti-BTLA reagents have no significant effect on in vitro B cell proliferation
Figure 2 shows the effect of anti-BTLA reagents on the LPS-induced or anti-CD40 plus anti-IgM mAb-induced proliferation of murine spleen derived B cells in vitro. Neither method of induced in vitro B cell proliferation was affected significantly by anti-BTLA antibodies or mHVEM-Fc. No significant inhibition of proliferation was detected with co-immobilized (see Fig. 2) or cross-linked anti-BTLA reagents (data not shown), nor did we see any effect on the lower levels of proliferation induced by an anti-IgM mAb alone (data not shown). Notably, none of the clones that inhibited in vitro T cell proliferation had any significant effect on B cell proliferation induced by any of the above methods.
Fig. 2.
B cell proliferation is not inhibited by co-immobilized anti-B and T lymphocyte attenuator (BTLA) reagents. Mouse B cells were purified from C57BL/6 mouse splenocytes by AutoMACS-negative selection (Miltenyi Biotec). Purified B cells (105) were incubated in duplicate in 96-well flat-bottomed plates in RPMI-1640 (Invitrogen, Inc.) with 10% heat inactivated fetal bovine serum (Gibco). Cells were stimulated with 2 µg/ml of lipopolysaccaride (List Biological Laboratories, Inc.) or 1 µg/ml of anti-CD40 monoclonal antibody (mAb) (BD Biosciences, Inc.) plus 2 µg/ml of goat F(ab')2 anti-mouse immunoglobulin M (IgM) (Jackson ImmunoResearch, West Grove, PA, USA) in the presence of various concentrations of anti-BTLA reagent or murine herpesvirus entry mediator (mHVEM-Fc) protein. Cells were incubated for 72 h at 37°C and 5% CO2 and pulsed with radioactive [3H] thymidine for the last 18 h to assess proliferation.
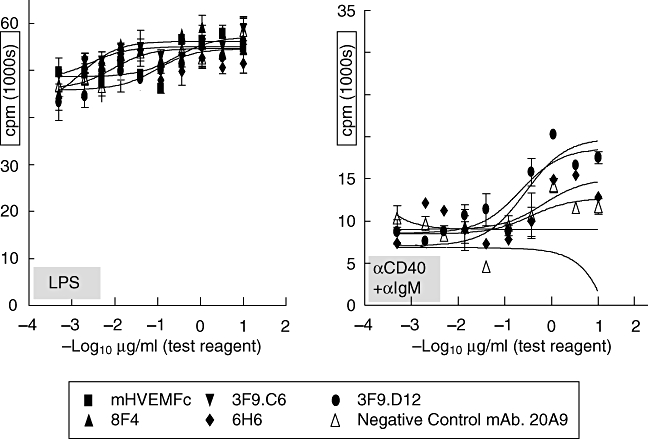
Only anti-BTLA reagents in cis format inhibit in vitro T cell proliferation
In an effort to elucidate further the exact mechanism of how the mHVEM-mFc ligand and some of the anti-BTLA mAbs acted to inhibit T cell proliferation, we used a beads-based approach in addition to direct immobilization on polystyrene plates. Figure 3 shows that, similarly to direct immobilization in the plate, bead-absorbed anti-CD3ε mAb caused T cell proliferation. Some of the anti-BTLA reagents that had been shown previously to inhibit T cell proliferation were tested in this novel format – specifically the mAb 6H6 and the mHVEM-mFc ligand, as well as an isotype control antibody. The test reagents were immobilized on either the same bead as the stimulus (cis format) or a different bead (trans format). Only anti-BTLA reagents in the cis, and not the trans, format relative to the activating stimulus inhibited this T cell proliferation. The control mAb did not have any significant effect on the T cell proliferation whether it was in cis or trans format. Analysis of secreted cytokines by multi-analyte profiling showed that secreted levels of interferon-γ correlated well with cell proliferation and this effect on inhibition of T cell proliferation observed in either the plate-immobilized or beads-based format could be reversed with excess soluble mBTLA-Fc (data not shown).
Fig. 3.
Bead-absorbed αCD3ε causes T cell proliferation: only anti-B and T lymphocyte attenuator (BTLA) reagents in the cis, and not the trans, format relative to the activating stimulus inhibit this proliferation. Mouse CD4+ T cells were purified from C57BL/6 mouse splenocytes by AutoMACS-negative selection (Miltenyi Biotec). In a U-bottomed 96-well plate, 100 000 T cells were activated in vitro by bead-absorbed anti-mouse CD3ε[clone 145-2C11] at 0·1 µg per 106 cells on tosyl activated 4·5 µM beads (Dynal) for 72 h and [3H]-labelled tritium was added to the cell culture medium for the last 18 h: the test reagent was either co-immobilized with the activating stimulus (cis format) or on a separate bead (trans format) at the indicated amounts. Cells were harvested onto a filter after 72 h of stimulation and radioactivity was assessed as a measure of cell proliferation. Analysis of secreted cytokines by multi-analyte profiling showed that secreted levels of interferon-γ correlated well with cell proliferation (data not shown).
Anti-BTLA mAbs do not affect in vivo activation of DO11.10 T cells transferred to syngeneic mice
We were interested to test the effect of the anti-BTLA regents that inhibited in vitro T cell proliferation in a mechanistically relevant in vivo model of inflammation. The most strongly indicated for T cell antagonism was judged to be the DO11.10 T cells syngeneic transfer with in vivo trapping of IL-2 (see later discussion).
Figure 4 shows that a large dynamic range for trapped IL-2 was generated in this model and that this was unaffected by an isotype control antibody and that the IL-2 signal was normalized completely by dosing with recombinant mCTLA4-hFc. None of the anti-BTLA mAbs that had inhibited in vitro T cell proliferation had a significant effect on the levels of trapped IL-2 in this model, even with relatively high dosing of 15 mg/kg. In an effort to determine any additive or synergistic effects of CTLA4-Fc and anti-BTLA reagents in this experimental system, we titrated the effect of CTLA4-Fc and have found that it is extremely effective at a wide range of concentrations, providing almost complete quenching of the signal even at a very low dose of 8 µg per mouse (approximately 0·2 mg/kg) (see Fig. S4). In our experience, this profound suppression of the disease-associated readout leaves an insufficient dynamic range for any additive or synergistic combination studies in this model.
Fig. 4.
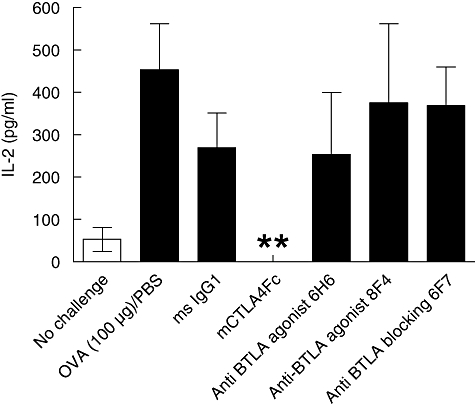
Anti-B and T lymphocyte attenuator (BTLA) monoclonal antibodies (mAbs) do not affect in vivo activation of DO11.10 T cells transferred to syngeneic mice; 20 × 106 DO11.10 splenocytes were adoptively transferred into BALB/c recipients. The next day mice were treated intraperitoneally with 15 mg/kg of anti-BTLA reagent or control reagent (5 mg/kg for mCTLA4-hFc). Three h after protein treatment animals were administered 10 µg of biotin-labelled rat amIL-2 (clone JES6-5H4) to capture secreted interleukin (IL)-2 (Finkelman et al., Int. Immunol., 11, 1999). Mice were then injected in the footpad with 100 µg of ovalbumin protein to activate the monoclonal population of transferred DO11.10 T cells. The mice were rested for 18 h before exsanguination and then serum IL-2 was detected by enzyme-linled immunosorbent assay. A double asterisk indicates no detectable IL-2.
Discussion
In this study we have elucidated further the mechanism of how BTLA acts to affect lymphocyte proliferation. We found that HVEM and a panel of different monoclonal antibodies bound murine BTLA specifically on both B and T cells and that some of the antibodies inhibited anti-CD3ε-induced T cell proliferation in vitro. None of these antibodies, or the HVEM molecule, had any significant effect on in vitro B cell proliferation.
Although some of the anti-BTLA reagents potently inhibited in vitro T cell proliferation, this effect occurred only when the BTLA ligand or the antibodies were in the appropriate format, i.e. putatively cross-linked with a reagent specific for the Fc region of the test agents. Despite the extensive use of this approach in many laboratories, the exact nature of the molecular interaction between the cross-linking reagent, the test agents and the target cells is still unclear. We elucidated further the requirements for inhibition of in vitro T cell proliferation using a beads-based system to immobilize the stimulus and the test agent. This system offers the advantage of either separating or locally clustering these two separate elements that interact with the cell. This system demonstrated clearly that the anti-BTLA reagent could inhibit anti-CD3ε-induced T cell proliferation only when it was in a cis conformation relative to the activating stimulus, and not in a a trans format. This suggests that the anti-BTLA reagent needs to be in close contact with, if not immediately juxtaposed to the stimulus that causes the T cells to proliferate.
Figure 5 shows a schematic illustrating a possible mechanistic explanation for this observation. In Fig. 5a, bead-absorbed anti-CD3ε clusters and activates the TCR and the cell proliferates. Anti-BTLA reagents on the same bead can localize BTLA to synapse, bringing the BTLA molecule in juxtaposition to the TCR. This allows the activation of BTLA to recruit the SHP-2 phosphatase adjacent to the intracellular domain of the TCR, resulting in dephosphorylation of the TCR complex and countering T cell proliferation.
Fig. 5.
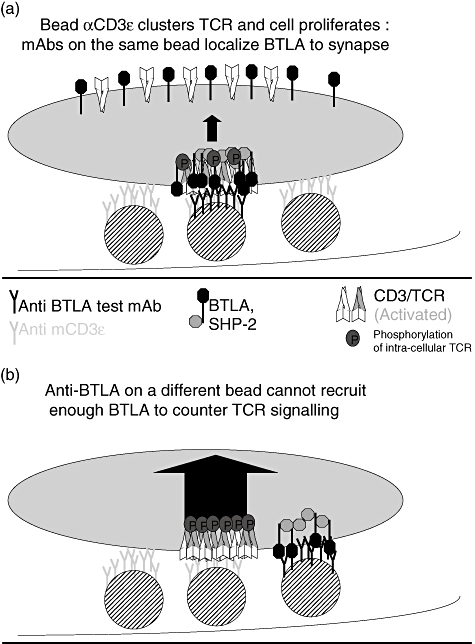
Bead-absorbed αCD3ε causes T cell proliferation: Only anti-B and T lymphocyte attenuator (BTLA) reagents in the cis, and not the trans, format relative to the activating stimulus inhibit this proliferation. Shown above is a hypothetical schematic that models a possible explanation for this observation. (a) Bead-absorbed αCD3ε clusters and activates the T cell receptor (TCR) and the cell proliferates. Anti-BTLA reagents on the same bead can localize BTLA to synapse, bringing the BTLA molecule in juxtaposition to the TCR. This allows the activation of BTLA to recruit the SHP-2 phosphatase adjacent to the intracellular domain of the TCR, resulting in dephosphorylation of the TCR complex and countering T cell proliferation. (b) Bead-absorbed αCD3ε clusters and activates the TCR and the cell proliferates. An anti-BTLA reagent on a different bead is dislocated physically from the immunological synapse and is unable to localize BTLA to the synapse. Hence, the SHP-2 phosphatase cannot be recruited adjacent to the intracellular domain of the TCR and T cell proliferation is unaffected.
In Fig. 5b, bead-absorbed anti-CD3ε clusters and activates the TCR and the cell proliferates. An anti-BTLA reagent on a different bead is dislocated physically from the immunological synapse and is unable to localize BTLA to the synapse. Hence, the SHP-2 phosphatase cannot be recruited adjacent to the intracellular domain of the TCR and T cell proliferation is unaffected.
We propose a model whereby Fig. 5a is analogous to the presence of a cross-linking reagent when the reagents are directly immobilized on the plate. When the cross-linking reagent is used, it brings the stimulus and the anti-BTLA reagent into close physical proximity as they interact and T cell proliferation is inhibited, as shown in Fig. 1b. Without a cross-linking reagent, the stimulus and the anti-BTLA reagent are immobilized directly on the plate and dislocated physically from each other and T cell proliferation is unaffected, as shown in Fig. 1a.
This proposed mechanism of action of an anti-proliferative BTLA-specific reagent is plausible based on the association of BTLA with elements of the TCR signalling complex [1,5,30]. It is also consistent with functional observations described in the literature. Hurchla et al. [2,4] and Sedy et al. [9] demonstrated that HVEM signals through BTLA by co-culturing Chinese hamster ovary (CHO) cells expressing the IAd major histocompatibility complex (MHC) molecule and also expressing either mBTLA or mHVEM with OVA antigen-activated CD4+ DO11.10 cells [2,4,9]. Co-expression of mBTLA had no effect on lymphocyte proliferation and co-expression of mHVEM inhibited lymphocyte proliferation significantly. This HVEM-mediated inhibition of proliferation did not occur if the CD4+ DO11.10 cells were from a BTLA knock-out mouse. In this system, the use of BTLA expressed on the surface of transfected cells is analogous to the use of the beads-based system. It is possible that the anti-BTLA reagent (in this case the HVEM ligand) needs to be juxtaposed similarly to the stimulus causing target cell proliferation (in this case the IAd MHC molecule presenting the OVA antigen).
In a more reduced in vitro proliferation system, Gonzalez et al. demonstrated that full-length mHVEM-Fc inhibited the proliferative responses of in vitro anti-CD3ε-stimulated CD4+ T cells, but only when it was co-immobilized on the plate via anti-mouse Fc (and presumptively cross-linked) and not soluble [31]. They generated similar data with in vitro anti-CD3ε-stimulated primary human CD4+ T cells where co-immobilized hHVEM-Fc (via anti-human Fc) inhibited lymphocyte proliferation significantly but soluble hHVEM-Fc did not. This effect could be blocked with a monoclonal antibody to hBTLA that had otherwise been shown to block the interaction between hBTLA and hHVEM [3]. Again, this is consistent with our observations using cross-linking reagents. Similarly, the Fiala strain of Hu CMV protein in the form of UL144-Fc was shown to inhibit dose-responsively anti-CD3ε and anti-CD28-stimulated proliferation of CD4+ human peripheral blood lymphocytes when cross-linked on the plate [17].
Krieg et al. generated a number of monoclonal antibodies specific for mBTLA and characterized further the rat anti-mBTLA (C57BL/B6) clone PK18 that inhibited proliferation of in vitro anti-CD3ε-stimulated CD3+ and CD4+ purified T cells from wild-type C57BL/B6 mice, but not from BTLA knock-outs [7,8]. Functionally, they showed that the mechanism of proliferation inhibition does not involve elimination of cells, the induction of apoptosis or the induction of putative regulatory CD4+ CD25+ T cells. This is the only published study to demonstrate inhibition of lymphocyte proliferation with a soluble, rather than an immobilized/coated or Fc-bound BTLA-specific reagent, although the required 60 µg/ml concentration needed is very high for such an assay and one cannot rule out the possibility of an artefactual effect on lymphocyte proliferation at such concentrations [7,8].
The BTLA system is newly described and the biology underlying it is complex. Although several different published studies have concluded that the signalling in the HVEM : BTLA axis is unidirectional through BTLA, it is noteworthy that all the published studies have concentrated upon the effects of BTLA- specific reagents on purified T cells (either CD3+, CD4+ or CD8+) and not crude mixed cell populations [2,3]. The study by Krieg et al. used BALB.K splenocytes as a source of antigen-presenting cells with the antigen-activated pigeon cytochrome C-specific T cells and the PK18 mAb inhibited proliferation significantly, but the PK18 anti-mBTLA mAb does not cross-react with BALB.K BTLA [7,8]. The study by Gonzalez et al. showed no effect of soluble mHVEM-mFc on the proliferation of concanavalin A-stimulated BALB/c crude splenocytes, nor was there any effect of soluble hHVEM-Fc on the phytohaemaglutinin-induced proliferation of human peripheral blood mononuclear cells. However, it is unclear if this is because the HVEM-Fc was soluble, as was the case for the purified CD4+ murine T cells, or because the cell population was not purified [3]. It is also possible that any one cell can express both BTLA and HVEM, complicating further the potential molecular interactions. We attempted to define the exact requirements for signalling through BTLA to exert an effect on lymphocyte proliferation. We found that neither the HVEM-Fc ligand nor any of the anti-BTLA mAbs had any significant effect on B cell proliferation in vitro. We found that the ligand and some of the antibodies inhibited T cell proliferation, but only when they were cross-linked with an anti-Fc reagent; this was consistent with several different published studies. We used the beads-based system to separate the stimulus and the test agent physically and found that T cell proliferation could be inhibited only when the test agent was juxtaposed immediately to the stimulus, and we have proposed a model for how this might occur. Other evidence in support of this hypothesis is shown by studies that demonstrate localization of BTLA to the immunological synapse during T cell activation [32]. None of the anti-BTLA reagents tested had any significant effect on the observed in vitro T cell proliferation in other commonly used experimental systems such as the MLR or the OVA antigen-induced system. In our opinion, this observation is a reflection of our hypothesis that an anti-BTLA reagent can act to inhibit T cell proliferation only when it is juxtaposed immediately to the activating stimulus. In the MLR and DO11.10 in vitro systems, the activating stimulus to the T cell is either a polymorphic MHC molecule on another cell or the OVA peptide presented by an MHC molecule on another cell, respectively. Hence, the anti-BTLA test agent is physically unable to interdict the signalling complex that drives TCR signalling and the subsequent T cell activation and proliferation. Indeed, as the stimulus is inherent to the cell–cell interaction, it would not be possible to mitigate the target T cell activation successfully with an exogenous anti-BTLA reagent in this experimental system. Based on our current understanding of BTLA biology, in the frame of our current hypothesis, one would have to engineer genetically the cell that presents the stimulus to the target cell with an appropriate anti-BTLA reagent, such as the HVEM ligand, in order to interdict successfully the target T cell activation. Indeed, this was described by Sedyet al. [9], whereby the presentation was made by CHO cells transfected with the MHC IAd molecule that presented the OVA antigen to the target DO11.10 T cell, causing T cell activation. This was mitigated by transfection of the same CHO cell with the HVEM molecule, i.e. the BTLA ligand.
We extended these studies to look at the effect of a BTLA-specific reagent in vivo. Of the various options for in vivo models, and bearing in mind the lack of any in vivo exposure data for any of these reagents, the most strongly indicated for T cell antagonism was judged to be the DO11.10 T cells syngeneic transfer with in vivo trapping of IL-2. This has the advantages of being short-term, robust and well-validated. This system involves the transfer of ex vivo-activated syngeneic CD4+ T cells with a measure of in vivo proliferation and IL-2 production and hence has a wide dynamic range that is related directly to T cell proliferation [33]. This model was also used by Sedy et al., and proliferation was inhibited by CHO/mHVEM-expressing cells [9]. Furthermore, several T cell function antagonists have been validated in this model [33]. We found that antibodies that inhibited T cell proliferation in vitro had no significant effect on the antibody-captured IL-2 associated with the in vivo activation of DO11.10 T cells transferred to syngeneic recipient BALB/c mice. We propose that this may be because an exogenously administered, soluble BTLA-specific reagent is unable to interdict the immunological synapse that has formed between an antigen-presenting cell and a T cell in vivo. There are few studies that describe the effects of anti-specific anti-BTLA reagents in vivo (as opposed to soluble HVEM-Fc which can bind to other molecules). The study by Truong et al. is a novel and interesting study that describes a synergistic improvement in allograft maintenance when the anti-BTLA mAb clone 6F7 is combined with CTLA4-Fc [34]. Specifically, at day 100 post-transplant approximately 40% of the mice treated with CTLA4-Fc alone have survived and approximately 70% of the mice treated with CTLA4-Fc and the mAb 6F7 have survived. This probably represents a statistically significant improvement, but the dynamic range between the two separate treatment groups is moderate. Furthermore, it is unclear if there is a significant improvement in the in vivo phenotypical behaviour and proliferation (i.e. lymphocyte precursor frequency) of the mice treated with CTLA4-Fc plus mAb 6F7, relative to treatment with CTLA4-Fc alone, and these reagents reportedly do not induce in vitro allospecific unresponsiveness as measured by MLR and CTL assays. In our hands, the anti-BTLA mAb 6F7 does not inhibit T cell proliferation in vitro and it groups to a different epitope on mBTLA relative to the reagents that inhibit T cell proliferation and activation. Hence, we cannot account readily for the reported synergistic improvement in transplant tolerance with the mAb 6F7 that is described in this study. However, differences between different animal facilities and detailed experimental protocols between different laboratories, as well as different preparations of test reagents with varying potencies and pharmacokinetic properties, may provide a partial explanation. It must also be borne in mind that the DO11.10 OVA antigen-activated experimental system is very different mechanistically to the islet allograft in vivo model, as the duration is very short (2–3 days relative to >3 months) and the population of in vivo-activated T cells is monoclonal and specific for a defined antigen, compared to the heterogeneous and variable population of cells and their associated MHC molecules in an islet allograft. However, we believe it is mechanistically relevant to the BTLA pathway, as Sedy et al. described an ex vivo analysis of these cells using a co-culture system with CHO cells presenting the OVA antigen ± the BTLA ligand HVEM, and demonstrated inhibition of DO11.10 T cell proliferation when the HVEM molecule was presented appropriately to the T cells [9]. Taken together, the in vitro and in vivo data set we have generated suggests that there may be specific structural requirements for the BTLA molecule to exert its effect on lymphocyte activation and proliferation.
Disclosure
All authors were employees of Amgen Inc. at the time this work was conducted and the manuscript written.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Anti-B and T lymphocyte attenuator (BTLA) monoclonal antibodies (mAbs) do not inhibit mouse T cell proliferation in the mixed lymphocyte reaction (MLR) in vitro. Mouse T cells and mitomycin C-treated antigen-presenting cells were cultured at a 1:1 ratio in the presence of plate coated anti-BTLA antibodies clone 6G3, 6H6 and mouse immunoglobulin isotype control (10 μg/ml in phosphatebuffered saline, 100 μl per well). Mouse CTLA4-Fc was added to the indicated wells as a positive control inhibitor of T cell proliferation. The cells were cultured for 5 days and [3H]-thymidine was pulsed for the last 18 h. T cell proliferation was measured by scintillation counting as described in the Materials and methods on day 5.
Fig. S2. Anti-B and T lymphocyte attenuator (BTLA) monoclonal antibodies (mAbs) do not inhibit antigen-induced mouse DO11.10 T cell proliferation in vitro. DO11.10 mice CD4+ T cells and mitomycin C-treated antigen-presenting cellswere cultured at a 1:1 ratio in the presence of plate-coated anti-BTLA antibodies clone 6G3, 6H6 and mouse immunoglobulin isotype control (10 μg/ml in PBS, 100 μl per well). Mouse CTLA4 Fc was added to the indicated wells as positive control inhibitor of T cell proliferation. The cells were stimulated with ovalbumin peptide at 0·05 μg/ml for 3 days and [3H]-thymidine was pulsed for the last 18 h. T cell proliferation was measured by scintillation counting on day 5.
Fig. S3. Inhibitory anti-B and T lymphocyte attenuator (BTLA) monoclonal antibodies (mAbs) bind to a different epitope on muBTLA than do non-inhibitory anti-BTLA mAbs. Anti-BTLA mAb 6F7, which does not inhibit in vitro T cell proliferation, was immobilized on a CM5 sensor chip, and mBTLA-mFc was captured on the antibody surface, followed by injection of inhibitory anti-BTLA antibody. If the immobilized antibody and the injected antibody bind to the same epitope, a second binding event will not be observed; if they bind to distinct epitopes, a second binding event will be seen. Events during the experiment are represented by letters, with ‘A’ corresponding to injection of mBTLA-mFc, ‘B’ corresponding to the end of the mBTLA-mFc injection, ‘C’ corresponding to injection of the second mAb, and ‘D’ corresponding to the end of the second mAb injection and start of the buffer wash.
Fig. S4. mCTLA4-Fc inhibits interleukin (IL)-2 production of DO11.10 T cells transferred to syngeneic mice; 20 × 106 DO11.10 splenocytes were transferred adoptively into BALB/c recipients. The next day mice were treated intraperitoneally with mCTLA-hFc reagent at 10, 2 and 0·4 mg/kg, respectively. One control group was treated with cyclosporin A(100 mg/kg) and the protein control group was treatedwith 10 mg/kg of a non-specific Fc protein.Three h after treatment animals were administered 10 μg of biotin-labelled rat amIL-2 (Clone JES6-5 H4) to capture secreted IL-2 (Finkelman et al., Int Immunol, 11, 1999). Mice were then injected in the footpad with 100 μg of ovalbumin protein in 1% alum to activate the monoclonal population of transferred DO11.10 T cells. The mice were rested for 18 h before exsanguination and then serum IL-2 was detected by enzyme-linked immunosorbent assay; n = 5 (standard error of the mean).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Gavrieli M, Watanabe N, Loftin SK, Murphy TL, Murphy KM. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of B and T lymphocyte attenuator required for association with protein tyrosine phosphatases SHP-1 and SHP-2. Biochem Biophys Res Commun. 2003;312:1236–43. doi: 10.1016/j.bbrc.2003.11.070. [DOI] [PubMed] [Google Scholar]
- 2.Hurchla MA, Sedy JR, Drake CG, Murphy KM. B and T lymphocytle attenuator (BTLA) exhibits structural and expression polymorphisms and is highly Induced in anergic CD4+ T cells. J Immunol. 2005;174:3377–85. doi: 10.4049/jimmunol.174.6.3377. [DOI] [PubMed] [Google Scholar]
- 3.Compaan DM, Gonzalez LC, Tom I, Loyet KM, Eaton D, Hymowitz SG. Attenuating lymphocyte activity – the crystal structure of the BTLA–HVEM complex. J Biol Chem. 2005;280:39553–61. doi: 10.1074/jbc.M507629200. [DOI] [PubMed] [Google Scholar]
- 4.Hurchla MA, Sedy JR, Murphy KM. Unexpected role of B and T lymphocyte attenuator in sustaining cell survival during chronic allostimulation. J Immunol. 2007;178:6073–82. doi: 10.4049/jimmunol.178.10.6073. [DOI] [PubMed] [Google Scholar]
- 5.Chemnitz JM, Lanfranco AR, Braunstein I, Riley JL. B and T lymphocyte attenuator-mediated signal transduction provides a potent inhibitory signal to primary human CD4 T cells that can be initiated by multiple phosphotyrosine motifs. J Immunol. 2006;176:6603–14. doi: 10.4049/jimmunol.176.11.6603. [DOI] [PubMed] [Google Scholar]
- 6.Cai GF, Anumanthan A, Brown JA, Greenfield EA, Zhu B, Freeman GJ. CD160 inhibits activation of human CD4(+) T cells through interaction with herpesvirus entry mediator. Nat Immunol. 2008;9:176–85. doi: 10.1038/ni1554. [DOI] [PubMed] [Google Scholar]
- 7.Krieg C, Boyman O, Fu Y-X, Kaye J. B and T lymphocyte attenuator regulates CD8(+) T cell-intrinsic homeostasis and memory cell generation. Nat Immunol. 2007;8:162–71. doi: 10.1038/ni1418. [DOI] [PubMed] [Google Scholar]
- 8.Krieg C, Han P, Stone R, Goularte OD, Kaye J. Functional analysis of B and T lymphocyte attenuator engagement on CD4(+) and CD8(+) T cells. J Immunol. 2005;175:6420–7. doi: 10.4049/jimmunol.175.10.6420. [DOI] [PubMed] [Google Scholar]
- 9.Sedy JR, Gavrieli M, Potter KG, et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol. 2005;6:90–8. doi: 10.1038/ni1144. [DOI] [PubMed] [Google Scholar]
- 10.Tao R, Wang L, Han R, et al. Differential effects of B and T lymphocyte attenutator and programmed death-1 on acceptance of partially versus fully MHC-mismatched cardiac allografts. J Immunol. 2005;175:5774–82. doi: 10.4049/jimmunol.175.9.5774. [DOI] [PubMed] [Google Scholar]
- 11.Tao R, Wang L, Murphy KM, Fraser CC, Hancock WW. Regulatory T cell expression of herpesvirus entry mediator suppresses the function of B and T lymphocyte attenuator-positive effector T cells. J Immunol. 2008;180:6649–55. doi: 10.4049/jimmunol.180.10.6649. [DOI] [PubMed] [Google Scholar]
- 12.An M-M, Fan K-X, Zhang J-D, et al. Lymphtoxin beta receptor-Ig ameliorates TNBS-induced colitis via blocking LIGHT/HVEM signaling. Pharmacol Res. 2005;52:234–44. doi: 10.1016/j.phrs.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 13.Brown GR, Lee EL, El-Hayek J, Kintner K, Luck C. IL-12-independent LIGHT signaling enhances MHC class II disparate CD4+ T cell alloproliferation, IFN-gamma response, and intestinal graft-versus-host disease. J Immunol. 2005;174:4688–95. doi: 10.4049/jimmunol.174.8.4688. [DOI] [PubMed] [Google Scholar]
- 14.Tamada K, Ni J, Zhu G, et al. Cutting edge: selective impairment of CD8+ T cell function in mice lacking the TNF superfamily member LIGHT. J Immunol. 2002;168:4832–5. doi: 10.4049/jimmunol.168.10.4832. [DOI] [PubMed] [Google Scholar]
- 15.Tamada K, Shimozaki K, Chapoval AI, et al. LIGHT, a TNF-like molecule, costimulates T cell proliferation and is required for dendritic cell-mediated allogeneic T cell response. J Immunol. 2000;164:4105–10. doi: 10.4049/jimmunol.164.8.4105. [DOI] [PubMed] [Google Scholar]
- 16.Ye Q, Fraser CC, Gao W, et al. Modulation of LIGHT-HVEM costimulation prolongs cardiac allograft survival. J Exp Med. 2002;195:795–800. doi: 10.1084/jem.20012088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheung TC, Humphreys IR, Potter KG, et al. Evolutionarily divergent herpesviruses modulate T cell activation by targeting the herpesvirus entry mediator cosignaling pathway. Proc Natl Acad Sci USA. 2005;102:13218–23. doi: 10.1073/pnas.0506172102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheung TC, Macauley MG, Oborne LM, Ware CF. Dimerization of HVEM and BTLA in cellular membranes. Cytokine. 2008;43:294. [Google Scholar]
- 19.Cheung TC, Steinberg MW, Oborne LM, et al. Unconventional ligand activation of herpesvirus entry mediator signals cell survival. Proc Natl Acad Sci USA. 2009;106:6244–9. doi: 10.1073/pnas.0902115106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arav-Boger R, Foster CB, Zong J-C, Pass RF. Human cytomegalovirus-encoded alpha-chemokines exhibit high sequence variability in congenitally infected newborns. J Infect Dis. 2006;193:788–91. doi: 10.1086/500508. [DOI] [PubMed] [Google Scholar]
- 21.Arav-Boger R, Willoughby RE, Pass RF, et al. Polymorphisms of the cytomegalovirus (CMV)-encoded tumor necrosis factor-alpha and beta-chemokine receptors in congenital CMV disease. J Infect Dis. 2002;186:1057–64. doi: 10.1086/344238. [DOI] [PubMed] [Google Scholar]
- 22.Bale JF, Petheram SJ, Robertson M, Murph JR, Demmler G. Human cytomegalovirus alpha sequence and UL144 variability in strains from infected children. J Med Virol. 2001;65:90–6. [PubMed] [Google Scholar]
- 23.Picone O, Costa JM, Chaix ML, Ville Y, Rouzioux C, Leruez-Ville M. Human cytomegalovirus UL144 gene polymorphisms in congenital infections. J Clin Microbiol. 2005;43:25–9. doi: 10.1128/JCM.43.1.25-29.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prichard MN, Penfold MET, Duke GM, Spaete RR, Kemble GW. A review of genetic differences between limited and extensively passaged human cytomegalovirus strains. Rev Med Virol. 2001;11:191–200. doi: 10.1002/rmv.315. [DOI] [PubMed] [Google Scholar]
- 25.Stranska R, Schuurman R, Toet M, Verboon-Maciolek M, de Vries LS, van Loon AA. Application of UL144 molecular typing to determine epidemiology of cytomegalovirus infections in preterm infants. J Clin Microbiol. 2006;44:1108–10. doi: 10.1128/JCM.44.3.1108-1110.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mei L, Taylor KD, Landers CJ, McGovern DP, Targan SR, Rotter JI. Genetic variation in BTLA is strongly associated with Crohn's disease. Gastroenterology. 2008;1:A42. [Google Scholar]
- 27.Lin SC, Kuo CC, Chan CH. Association of a BTLA gene polymorphism with the risk of rheumatoid arthritis. J Biomed Sci. 2006;13:853–60. doi: 10.1007/s11373-006-9113-7. [DOI] [PubMed] [Google Scholar]
- 28.Wang J, Anders RA, Wang Y, et al. The critical role of LIGHT in promoting intestinal inflammation and Crohn's disease. J Immunol. 2005;174:8173–82. doi: 10.4049/jimmunol.174.12.8173. [DOI] [PubMed] [Google Scholar]
- 29.Byrne FR, Winters A, Brankow D, et al. An antibody to IP-10 is a potent antagonist of cell migration in vitro and in vivo and does not affect disease in several animal models of inflammation. Autoimmunity. 2009;42:171–82. doi: 10.1080/08916930802629547. [DOI] [PubMed] [Google Scholar]
- 30.Wu T-H, Zhen Y, Zeng C, Yi H-F, Zhao Y. B and T lymphocyte attenuator interacts with CD3zeta and inhibits tyrosine phosphorylation of TCRzeta complex during T-cell activation. Immunol Cell Biol. 2007;85:590–5. doi: 10.1038/sj.icb.7100087. [DOI] [PubMed] [Google Scholar]
- 31.Gonzalez LC, Loyet KM, Calemine-Fenaux J, et al. A coreceptor interaction between the CD28 and TNF receptor family members B and T lymphocyte attenuator and herpesvirus entry mediator. Proc Natl Acad Sci USA. 2005;102:1116–21. doi: 10.1073/pnas.0409071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Owada T, Watanabe N, Oki M, et al. Activation-induced accumulation of B and T lymphocyte attenuator to the immunological synapse in CD4+ T cells. J Leukoc Biol. 2010;87:425–32. doi: 10.1189/jlb.0309138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finkelman FD, Morris SC. Development of an assay to measure in vivo cytokine production in the mouse. Int Immunol. 1999;11:1811–18. doi: 10.1093/intimm/11.11.1811. [DOI] [PubMed] [Google Scholar]
- 34.Truong W, Plester JC, Hancock WW, et al. Combined coinhibitory and costimulatory modulation with anti-BTLA and CTLA4Ig facilitates tolerance in murine islet allografts. Am J Transpl. 2007;7:2663–74. doi: 10.1111/j.1600-6143.2007.01996.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.