

Abstract
The considerable clinical heterogeneity of patients with common variable immunodeficiency disorders (CVID) shares some similarity with bone-marrow failure disorders such as Diamond–Blackfan anaemia (DBA) and Shwachman–Diamond syndrome (SDS), now recognized as defects in ribosome biogenesis or ribosomopathies. The recognition of a patient with DBA who subsequently developed CVID lends support to our previous finding of a heterozygous mutation in the SBDS gene of SBDS in another CVID patient, suggesting that ribosome biogenesis defects are responsible for a subset of CVID. Genetic defects in the ribosomal translational machinery responsible for various bone marrow failure syndromes are recognized readily when they manifest in children, but diagnosing these in adults presenting with complex phenotypes and hypogammaglobulinaemia can be a challenge. In this perspective paper, we discuss our clinical experience in CVID patients with ribosomopathies, and review the immunological abnormalities in other conditions associated with ribosomal dysfunction. With genetic testing available for various bone marrow failure syndromes, our hypothesis that ribosomal abnormalities may be present in patients with CVID could be proved in future studies by testing for mutations in specific ribosomal genes. New knowledge might then be translated into novel therapeutic strategies for patients in this group of immunodeficiency disorders.
Keywords: bone marrow failure, common variable immunodeficiency, Diamond–Blackfan anaemia, ribosome, ribosomopathy
Introduction
Common variable immunodeficiency disorders (CVID) comprise a range of hypogammaglobulinaemias, for which a small number of genetic defects have been identified [1–3]. However, these account for only a small proportion of cases of CVID, and the majority of patients have no identified genetic cause. A number of bone marrow failure syndromes are now recognized to be due to defects in ribosome biogenesis with mutations in genes coding for ribosomal proteins. Various immunological abnormalities are evident in these syndromes and provide proof that failure of optimal ribosome function, ‘ribosomopathies’, can also affect cells of the immune system.
These syndromes are heterogeneous in their clinical presentations: for example, patients with Shwachman–Diamond syndrome (SDS) with confirmed mutations in the SBDS gene (Chr7q11) may not have all the characteristic features of neutropenia, skeletal defects and pancreatic insufficiency [4]. There is emerging evidence that loss of Shwachman–Bodian–Diamond syndrome (SBDS) protein affects haematopoeisis and numbers of circulating B lymphocytes [5]. Craniofacial malformation syndromes such as Treacher–Collins syndrome, caused by haploinsufficiency of the treacle protein, also affect the cells of the immune system [6], and a broader immunological defect has been described in the congenital anaemia of Diamond–Blackfan syndrome (Diamond–Blackfan anaemia: DBA) [7]. The 5q- syndrome, a somatically acquired deletion of chromosome 5q and a subtype of myelodysplastic syndrome, leads to haploinsufficiency of a ribosomal protein that is also implicated in DBA.
The active eukaryotic ribosome, the site of protein synthesis, is composed of 40S and 60S subunits. Formation of the active complex requires synthesis and assembly of core ribosomal proteins, ribosomal RNAs, small nucleolar RNAs and several other associated proteins (see Fig. 1). This process begins in the nucleolus and the preribosomal units are exported into the cytoplasm for final steps in the maturation of ribosomes [8]. The exact functions of many of these proteins remain unknown. Some ribosomal proteins are now known to have extraribosomal functions; for example, the SBDS protein has a role in stabilizing the mitotic spindle. Immunological abnormalities in ribosomopathies may therefore provide clues as to how ribosomal proteins can shape the immune system.
Fig. 1.
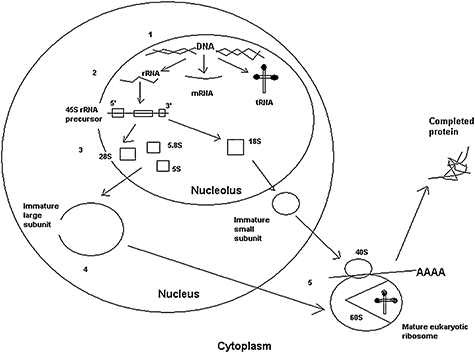
An overview of the biogenesis of the eukaryotic ribosome. Synthesis of ribosomal proteins and assembly of the mature eukaryotic ribosome has several step: (1) DNA transcription and RNA processing; (2) translation of ribosomal RNA (rRNA); (3) modification and processing by small nucleolar RNPs (snoRNPs) and Rnase; (4) formation of immature large and small ribosomal subunits and exit from nucleolus; and (5) formation of mature eukaryotic ribosome[8]. The 60S subunit has a cleft for tRNA and amino acids from the endoplasmic reticulum (ER) are translated into protein. rRNA, ribosomal RNA; mRNA, messenger RNA; tRNA, transfer RNA; AAA, amino acids.
According to internationally accepted criteria, the diagnosis of CVID remains one of exclusion. The currently identified four genetic mutations (ICOS, CD19, TACI, BAFFR) account for fewer than a fifth of cases, with no consensus on which genetic testing should be undertaken in most cases [1]. The current European Society of Immunodeficiency (ESID)/Pan-American Group for Immunodeficiency (PAGID) criteria for CVID include: ‘probable’ CVID in those aged > 2 years with low immunoglobulin (Ig)G and another low isotype level (IgA or IgM) with absent vaccine responses; and ‘possible’ CVID in those with low immunoglobulin of any isotype with absent vaccine responses where other causes of hypogammaglobulinaemia have been excluded [2]. Additional similarities with ribosomopathies and CVID patients include heterogeneous presentations with T cell defects, cytopenias and malignancies [1–3].
The initial description of DBA was of a congenital erythroblastopenia characterized by an early arrest of pre-erythroblast differentiation. The first report of loss-of-function mutations in a gene coding for a ribosomal protein in this disease (non-sense, missense, frameshift, splice-site, complete deletion of one RPS19 allele) generated enormous interest in the clinical effects of disordered ribosome biosynthesis [8,9]. Mutations in the RPS19 gene prevent assembly of the 40S ribosomal subunit, but account for only 25% of DBA patients [9]. However, to our knowledge, there have been no reports of failure of antibody production in DBA. We present our clinical experience with the report of the first case of DBA who subsequently developed antibody deficiency, consistent with a new diagnosis of CVID, with complications of bronchiectasis and managed on immunoglobulin therapy. The previous case of CVID with mutation in the SBDS gene of SDS has been discussed briefly with additional data, as a detailed report was published in a previous issue of this Journal [10]. In the final part of this perspective paper, we review the immunological abnormalities beginning to emerge in ribosomopathy syndromes.
Clinical experience of ribosomopathies and hypogammaglobulinaemia
DBA and CVID
Clinical synopsis including investigations
A 22-year-old female presented with bronchiectasis and hypogammaglobulinemia. DBA had been diagnosed at 1 year of age and required treatment with corticosteroids and blood transfusions until the age of 6 years. There were no associated skeletal, cardiac or congenital defects. Over the next 3 years she suffered from recurrent sinusitis, otitis media, chest infections (sputum cultures positive for Moraxella catarrhalis and Haemophilus species) and viral warts.
She has a sister with features of DBA – low haemoglobin at 10·4 g/dl, raised mean corpuscular volume (MCV), lymphopenia, elevated fetal haemoglobin (HbF) (3%), high erythrocyte adenosine deaminase (eADA) levels, mildly reduced T cell numbers and slight reduction in proliferative responses to standard mitogens. The sister's immunoglobulin levels, including functional antibody levels, are normal and she has not required any specific therapy for her anaemia.
Investigations in infancy showed a normocytic anaemia, normal serum immunoglobulins [IgG 7·3 g/l (normal range 3·0–10·5), IgA 0·28 g/l (0·1–1·2), IgM 1·07 g/l (0·3–1·5)] and good vaccine responses to conjugated Haemophilus influenzae type b and unconjugated pneumococcal polysaccharide vaccines. By the age of 9, serum IgG levels had dropped to 4·94 g/l (normal range 6·0–13·0). Lymphocyte proliferation responses to phytohaemagglutinin, pokeweed mitogen, OKT3, tetanus, varicella and herpes antigens were reduced. Intravenous immunoglobulin (IVIG) replacement therapy was commenced, and stopped after 8 years for reassessment of immune function. Four years later, she had persistent anaemia (Hb 10·0 g/dl, MCV 95·6fl) and low IgG (3·37 g/l), IgA (0·96 g/l) and IgM (0·79 g/l). Bone marrow cytogenetic studies were normal, excluding microdeletions in 19q13 and 5q- syndrome. Specific antibody tests showed absent antibodies against measles and reduced tetanus and pneumococcal antibody levels. She was diagnosed to have common variable immunodeficiency as no other causes of low IgG and low levels of specific antibodies were identified. High resolution CT scan chest showed evidence of right middle lobe bronchiectasis and bilateral lower lobe bronchiectasis worse on the left. Intravenous immunoglobulin therapy was recommenced at this stage.
Lymphocyte subset analysis showed lymphopenia at 833 × 106/µl (normal range 1500–3500), CD3+ T cells 536 (800–2700), helper CD4+ T cells 291 (400–1700), cytotoxic CD8+ T cells 191 (300–1200), CD19+ B cells 158 (100–600) and CD16+CD56+ natural killer cells 32 (90–600). B cell studies showed a reduced class-switched memory B cell subset at 2·5%. Lymphocyte proliferation responses to OKT3, phytohaemagglutinin and pokeweed mitogen remained reduced (see Table 1). Peripheral blood eADA level performed recently was high at 594 (normal range 40–100 u/l), consistent with the diagnosis of DBA. She has remained well on home therapy with weekly subcutaneous immunoglobulin infusions over the last 3 years.
Table 1.
Lymphocyte proliferation results (Diamond–Blackfan syndrome and common variable immunodeficiency disorders case).
| Patient | Control | |
|---|---|---|
| CD3 (OKT3) | ||
| 200 ng/ml | 4326 | 10 304 |
| 100 ng/ml | 4153 | 9 843 |
| 50 ng/ml | 3860 | 8 328 |
| 25 ng/ml | 3129 | 9 025 |
| PHA | ||
| 50 µg/ml | 560 | 36 570 |
| 20 µg/ml | 1150 | 62 300 |
| 5 µg/ml | 920 | 45 350 |
Lymphocyte proliferation was determined using [3H]-thymidine uptake following the manufacturer's recommendations. Numbers denote disintegrations per minute (dpm). PHA: phytohaemagglutinin.
Genetic analyses
Polymerase chain reaction (PCR)-based methods for mutation detection
Genomic DNA was extracted from the patient's leucocytes with a commercial DNA purification kit, as per the manufacturer's instructions. All RPS19, RPS24, RPS17, RPL5, RPL11 and RPL35a coding, promoter and 3′UTR regions (including the boundaries) were amplified by PCR according to previously described methods, with minor changes [11–13]. Direct sequencing of all fragments was carried out in an automatic sequencer. All sequence variations identified were verified on the complementary strand using an independent PCR product.
Multiplex ligand-dependent probe amplification (MLPA) technique for mutations in the RPS19 gene
The MLPA technique, which is used for the detection of complete or partial gene deletions or duplications, was carried out [13,14]. This technique is based on the simultaneous hybridization and ligation of several probes matched to single exons using a single reaction tube, which is followed by PCR and analysis by capillary electrophoresis. Reduced peaks suggest deletions (even on only one exon of a single allele) and enhanced peaks suggest duplication [14]. Informed consent for genetic testing was obtained from the patient and the study was approved by the Trust's Research and Development Department.
Results of genetic analyses
No loss-of-function mutations were identified in RPS19, RPS24, RPS17, RPS5, RPL11 and RPL35a genes that is in keeping with approximately 50% of cases of DBA where no mutations are found in these genes (RPS: ribosomal protein small subunit; RPL: ribosomal protein large subunit). However, heterozygous polymorphisms were identified in RPS24 and RPS17 genes: RPS24 IVSI +26 (c > t); RPS17 IVS2 −73 (g > c), IVS2 −30 (c > t) and nt159 T > C; and homozygous polymorphisms were identified in RPL11 gene: RPL11 −17 (c > g) and IVS5 +39 (a > g) (Fig. 2). The MLPA technique did not reveal any deletion (complete or partial) or duplication in the RPS19 gene (Fig. 3).
Fig. 2.
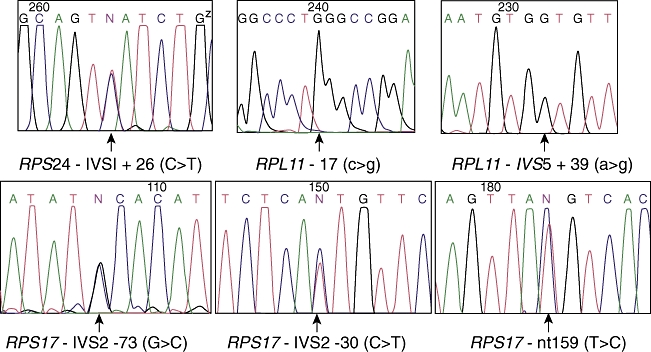
Sequencing results showing the polymorphisms found in the RPS24, RPL11 and RPS17 genes.
Fig. 3.
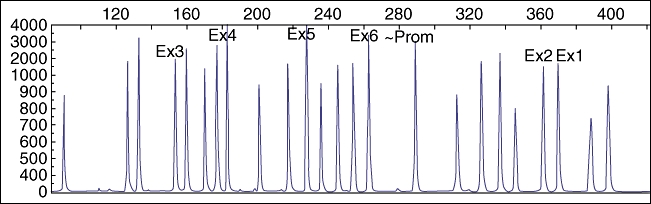
Multiplex ligand-dependent probe amplification (MLPA) results of the RPS19 gene.
Implications
This illustrates a ribosomopathy in a patient with DBA (anaemia, raised adenosine deaminase levels) who subsequently developed CVID. She was dependent on corticosteroids and blood transfusions but went into remission at the age of 6 years. The current definition of ‘remission’ is stable, physiologically acceptable haemoglobin maintained for a minimum of 6 months without corticosteroids, transfusions or other therapy [15]. T cell responses to mitogens were suboptimum, as in a previous case of DBA, which also showed failure of T cell proliferation to human recombinant interleukin (rIL)-2 [16]. Our patient therefore resembles approximately half of DBA patients who do not have mutations in the currently described six ribosomal genes (RPS19, RPS17, RPS24, RPL5, RPL11 and RPL35a), but the laboratory abnormalities (anaemia, raised eADA levels) suggest that other genes affecting ribosomal functions may be involved. A recent paper has described mutations in other genes, RPS7, RPS27A, RPL36 and RPS15, evident in DBA, but we have not looked for mutations in these genes [8]. Hypogammaglobulinaemia, reduced pneumococcal antibodies, defective T cell proliferation responses and reduced class-switched memory B cells, all features consistent with CVID, are not assessed typically in DBA, so many cases may be missed.
Persistent low IgG levels in some cases of DBA may be secondary to corticosteroids used for refractory anaemia, or transient after rituximab therapy [17]. Three reports of use of IVIG in DBA were an attempt to treat the refractory anaemia, and not for treatment of hypogammaglobulinaemia [16,18,19]. The present consensus opinion is that IVIG therapy is ineffective for treatment of refractory anaemia in DBA [15]. However, there are rare DBA patients who have recurrent infections with antibody deficiency (low IgG levels) requiring monthly IVIG infusions (Adrianna Vlachos, personal communication, data not published).
Mutations in the SBDS gene of SDS and hypogammaglobulinaemia
We previously reported a patient with typical features of CVID and complications of bronchiectasis, arthritis, intestinal lymphoid hyperplasia and malabsorption who had a heterozygous mutation in the SBDS gene of SDS [10]. Following publication of the report, the patient was admitted with life-threatening arrhythmias with significant electrolyte imbalances secondary to malabsorption and required percutaneous endoscopic gastrostomy (PEG) insertion. Adjusted Ca2+ levels were 1·86 mmol/l (normal range, 2·2–2·6), vitamin A levels were 0·55 µmol/l (normal range, 0·84–3·6) and 25-hydroxy vitamin D levels were 27 nmol/l (should be > 50 at all times with some seasonal variations). He was continued on pancreatic supplements (pancreozyme), calcium and magnesium supplements and immunoglobulin replacement therapy.
In 2005 lymphocyte subsets showed absolute B cell count at 0·110 × 109/L; B cell subsets (locally derived normal percentages in brackets) – naive (IgD+CD27-) B cells 82% (60–71%), unswitched (IgD+CD27+) memory B cells 16·4% (10–18%) and switched (IgD-CD27+) memory B cells 0·4% (5–15%). By 2009, there was a significant reduction in B cell numbers: 0·046 × 109/l. He had a further prolonged course of admission in the intensive care with pneumonia due to drug-resistant Pseudomonas aeruginosa that proved fatal.
One might consider this late-onset SDS rather than CVID, which is rare, as most SDS patients present quite early and the heterozygous mutation in this case could account for residual functional protein and the ‘late’ presentation. However, he had developed features of CVID long before the SDS phenotype was apparent. Malabsorption, progressive weight loss, bi-cytopenias (anaemia, thrombocytopenia) and recurrent chest infections in spite of adequate trough IgG levels would suggest progressive disease that strengthens the hypothesis that the single ca. 258 + 2T > C mutation resulted in defective ribosomal function.
Some of the interesting features of this case included pelvic kidney, eosinophilia, absence of classical presentation of chronic neutropenia and identification of only one mutation (ca. 258 + 2T > C frameshift mutation) in the SBDS gene. This was the second report of a heterozygous mutation that provided evidence that a single mutation may also result in the clinical phenotype, after a Japanese report of a patient with typical SDS phenotype and with only ca. 258 + 2T > C mutation [20]. Recently, there has been another report of a novel heterozygous mutation in the SBDS gene (exon 1, 98 A > C) in a 4-year-old girl with virtual absence of B cells but normal immunoglobulin levels [21].
Following our finding of the SBDS mutation in one patient, we subsequently checked for SBDS mutation in two other patients.
One patient was a 77-year-old woman with CVID, chronic anaemia due possibly to underlying myelodysplasia (proved on bone marrow biopsy) and thrombocytopenia. The other patient was in his early 40s, with CVID and on IVIG for 8 years with a 2-year history of enteropathy (chronic diarrhoea, ongoing weight loss, coeliac-like disease with no response to gluten-free diet). No mutations in the SBDS gene were found in either of these patients.
SDS and CVID share common features, such as recurrent infections, malabsorption, cytopenias (neutropenia, thrombocytopenia, anaemia), low immunoglobulins ± absent vaccine responses in some cases [10], abnormal liver function tests, autoimmunity and malignancy [myelodysplastic syndrome (MDS), leukaemia], and testing for mutations in the SBDS gene in CVID patients with most of the above features would be worthwhile.
More importantly, testing for SBDS mutations would be important in children with persistent neutropenia, recurrent infections, growth and skeletal abnormalities where the immunodeficiency disorder may have been described as CVID. A scoring system may prove useful in the future when more patients are described.
Review of immunological abnormalities in ribosomopathy syndromes
Ribosomopathies and bone marrow failure syndromes have variable and overlapping clinical presentations, yet most have subtle immune defects and a strong tendency to develop leukaemic transformation. The role of p53 in ribosomal dysfunction is beginning to be understood, such as up-regulation of p53 in haploinsufficiency of certain ribosomal proteins and consequent apoptosis and cell-cycle arrest, offer interesting mechanisms of cellular effects in ribosomopathies [8]. Deciphering subtle defects in the immune system in these patients may help to unravel the complex interaction of ribosomal proteins in the development of specific parts of the immune system. Table 2 lists the syndromes with known mutations in ribosomal genes and the immunological abnormalities.
Table 2.
Immunological abnormalities in ribosomopathy syndromes.
| Syndrome | Ribosomal gene and chromosomal location (% patients with mutation) | Clinical and laboratory features | Immunological abnormalities |
|---|---|---|---|
| Diamond–Blackfan anaemia (DBA) OMIM: #105650 | RPS 19 19q13·2 (25%) [10]RPS 24 10q22-q23 (2%) [11]RPS 17 15q[12]RPL35A 3q29-qter [22] (3%) RPL 5 and 11 (11%) [23]RPS10 and 26[24] | Pallor, growth retardation, congenital anomalies of the head, neck, upper limbs and urogenital system (40%), macrocytic anaemia, raised HbF, EPO and eADA levels, leukaemia [25]RPL5 and RPL11 associated with craniofacial (cleft palate) and abnormal thumbs, respectively [23]Parvovirus B19 seropositivity reaches 50% by age 15 years [26] | Transient low T cells (in three patients), reduced T cell responses to hrIL-2; low B cell number with normal sIg+, absent ConA-induced CD8+ T cell production [7], CVID with reduced T cell mitogen responses (index case) |
| Shwachman–Diamond syndrome (SDS) OMIM: #260400 | SBDS 7q11·21 (90%) ?EFL1[8,27,28]SBDS mutations affect development of 60S subunit | Recurrent infections (85%), pancreatic exocrine insufficiency, metaphyseal chondrodysplasia, short stature, malabsorption, increased risk of malignancy (AML, MDS), neutropenia (95%), thrombocytopenia, aplastic anaemia | Neutrophil chemotaxis defect, defect in B and T cell function and survival, low immunoglobulins, low B cells [10,21], low specific antibodies [10,21], increased apoptosis of CD34+ marrow cells [29–31]; CVID [10] |
| Dyskeratosis congenita •X-linked (OMIM: #305000)•Autosomal dominant •Autosomal recessive | DKC1 at Chr. Xq28 encodes dyskerin TERC (Chr. 3q26); TERT (5p15·53); TINF2 (14q12) NOP10 (NOLA3) Chr. 15q14–15 60% lack an identifiable mutation [32–35]Mutations affect rRNA pseudo-uridylation | Reticulated skin pigmentation, nail dystrophy, mucosal leukoplakia, cerebellar hypoplasia (Hoyeraal–Hreidarsson syndrome), serious infections, osteoporosis, liver and lung fibrosis, aplastic anaemia, early ageing due to reduced telomere length, tumour susceptibility [25] | Low IgM, severe B lymphopenia (X-linked DC); T + B − NK − SCID with DKC1 mutation reported in Hoyeraal–Hreidarsson syndrome [36] |
| Cartilage hair hypoplasia (CHH) OMIM: #250250 | RNase mitochondrial RNA processing (RMRP) RNA gene Chr. 9p21–p12 70A > G (founder mutation, 92% Finnish and 48% non-Finnish) [37]Certain mutations correlate with severe immunogical and haematological abnormalities [38]RMRP mutations affect pre5·8S (part of 60S subunit) | Short stature, hypoplastic hair, metaphyseal chondrodysplasia (±cone-shaped epiphysis), some excess risk of malignancy (lymphoma, liver, duodenal, skin), anaemia (6% dependent on blood transfusions), neutropenia, lymphopenia, infections 56% (Pneumocystis, CMV) [39–41] | Hypogammaglobulinaemia, low IgA and IgG subclass [39], decreased T cell count and proliferation (low CD4+ with increased apoptosis) and CD8 lymphopenia [40,42–44], low B cells [44], defective B cell and fibroblast proliferation [44], severe combined immunodeficiency (Omenn syndrome) [43,45] |
| Treacher–Collins syndrome OMIM: #154500 | Treacle gene TCOF1 5q32–q33·1 (93%) TCOF1 mutations cause 18S rRNA methylation defect that leads to insufficient nucleolar phosphoprotein treacle, and defects in craniofacial development [46]18S is part of 40S ribosomal subunit | Anti-mongoloid slant of eyes, cataracts, coloboma of lid/uveal tract, micrognathia, ear deformities, hypoplastic zygomatic arches, macrostomia, conductive hearing loss, cleft palate, craniosynostosis, anaemia [47] | T cell abnormalities, impaired mitogen responses, decreased B cells with low immunoglobulins (in some craniofacial syndromes) [6] |
| Turner's syndrome | Human sex-linked genes RPS4X and RPS4Y encode 2 isoforms of ribosomal protein S4; RPS4 is a component of 40S subunit; haploinsufficiency of RPS4X hypothesized to lead to Turner's syndrome [48,49] | Girls with 45 XO karyotype, ring X chromosome with mental retardation and kyphoscoliosis, short stature, webbed neck, gonadal failure, cardiovascular and renal malformations, hearing loss, diabetes | CVID [50,51], hypogammaglobulinaemia, T cell immunodeficiency, selective IgA deficiency, coeliac disease [50–56] |
AML: acute myeloid leukaemia; CMV: cytomegalovirus; ConA: concanavalin A; CVID: common variable immunodeficiency disorder; eADA: erythrocyte adenosine deaminase; EPO: erythropoietin; HbF: fetal haemoglobin; hrIL: human recombinant interleukin; Ig: immunoglobulin; MDS: myelodysplastic syndrome; NK: natural killer; rIL: recombinant interleukin; SCID: severe combined immunodeficiency.
Future studies will determine whether our observations of polymorphisms in specific ribosomal genes associated with DBA and the association of symptomatic or asymptomatic hypogammaglobulinaemia. With expanding knowledge and detection of newer ribosomal proteins, sequencing of specific ribosomal genes and/or use of ‘functional’ assays that provide evidence of aberrant pre-ribosomal RNA precursor accumulation would provide more tools to detect newer ribosomopathies that currently do not have a genetic basis [8,57]. Optimal expression of ribosomal proteins appears to be critical for functioning of organ systems (as complete absence of SBDS or RPS19 are incompatible with life [8]) and certain tissues may therefore be more susceptible to decreased production of specific ribosomal proteins when they are produced at limited rates [8,58].
Low IgM levels with B cell lymphopenia have been reported in X-linked dyskeratosis congenita (X-linked DC), with severe combined immunodeficiency (T + B − NK − SCID) reported in the most severe variant of dyskeratosis congenita (Hoyeraal–Hreidarsson syndrome) [36]. Premature ageing is also a feature of this disease [32–34] and TINF2 gene mutation (a component of the telomere protection complex) [35] leading to short telomeres has been described in X-linked DC. It is not clear whether the immune abnormalities are due to the defective tRNA pseudouridylation or the short telomere length.
Turner's syndrome (45,X0) is postulated to have a ribosomal defect due to haploinsufficiency of ribosomal protein RPS4X [48,49]. Variable degrees of antibody deficiency (panhypogammaglobulinaemia [48], low IgM [50,52]) including decreased T and B cell numbers [50,54] and coeliac disease with IgA deficiency have been recognized in this syndrome [53,55]. Some of these patients with Turner's syndrome have clinical syndromes of recurrent sinopulmonary infections and other features overlapping with CVID [50,51].
Conclusions
We have looked at the evidence for ribosomal defects being associated with and possibly causative of immune abnormalities with features of CVID. We describe two such patients with different ribosomal defects who subsequently developed a presentation consistent with CVID. A review of the literature indicates that patients with ribosomal defects may share abnormalities of T or B cell development with many features of CVID, and which may not be recognized as such by non-immunologists. Given that the four established genetic defects account for fewer than a fifth of cases of CVID, this hypothesis could be tested in the future by more detailed studies of ribosome genetics and/or function in CVID.
Disclosure
This work was supported by the Centre for Immunoglobulin Therapy and Department of Immunology, Hull Royal Infirmary. WACS is Director of Centre for Immunoglobulin Therapy, which has received unrestricted educational grants from Octapharma, Baxter, Grifols, CSL-Behring. The rest of the authors have no financial interests to disclose.
References
- 1.Park MA, Li JT, Hagan JB, Maddox DE, Abraham RS. Common variable immunodeficiency: a new look at an old disease. Lancet. 2008;372:489–502. doi: 10.1016/S0140-6736(08)61199-X. [DOI] [PubMed] [Google Scholar]
- 2.Conley M, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies: representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies) Clin Immunol. 1999;93:190–7. doi: 10.1006/clim.1999.4799. [DOI] [PubMed] [Google Scholar]
- 3.Giovannetti A, Pierdominici M, Mazzetta F, et al. Unravelling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178:3932–43. doi: 10.4049/jimmunol.178.6.3932. [DOI] [PubMed] [Google Scholar]
- 4.Hall GW, Dale P, Dodge JA. Shwachman–Diamond syndrome: UK perspective. Arch Dis Child. 2006;91:521–4. doi: 10.1136/adc.2003.046151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rawls AS, Gregory AD, Woloszynek JR, Liu F, Link DC. Lentiviral-mediated RNAi inhibition of SBDS in murine hematopoietic progenitors impairs their hematopoietic potential. Blood. 2007;110:2414–22. doi: 10.1182/blood-2006-03-007112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheuerle AE, Good RA, Habal MB. Involvement of the thymus and cellular immune system in craniofacial malformation syndromes. J Craniofac Surg. 1990;1:88–90. doi: 10.1097/00001665-199001020-00003. [DOI] [PubMed] [Google Scholar]
- 7.Finlay JL, Shahidi NT, Horowitz S, Borcherding W, Hong R. Lymphocyte dysfunction in congenital hypoplastic anemia. J Clin Invest. 1982;70:619–26. doi: 10.1172/JCI110655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010;115:3196–205. doi: 10.1182/blood-2009-10-178129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Draptchinskaia N, Gustavsson P, Andersson B, et al. The gene encoding ribosomal protein S19 is mutated in Diamond–Blackfan anaemia. Nat Genet. 1999;21:169–75. doi: 10.1038/5951. [DOI] [PubMed] [Google Scholar]
- 10.Khan S, Hinks J, Shorto J, Schwarz MJ, Sewell WA. Some cases of common variable immunodeficiency may be due to a mutation in the SBDS gene of Shwachman–Diamond syndrome. Clin Exp Immunol. 2008;151:448–54. doi: 10.1111/j.1365-2249.2007.03556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gazda HT, Grabowska A, Merida-Long LB, et al. Ribosomal protein S24 gene is mutated in Diamond–Blackfan anemia. Am J Hum Genet. 2006;79:1110–8. doi: 10.1086/510020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cmejla R, Cmejlova J, Handrkova H, Petrak J, Pospisilova D. Ribosomal protein S17 gene (RPS17) is mutated in Diamond–Blackfan anemia. Hum Mutat. 2007;28:1178–82. doi: 10.1002/humu.20608. [DOI] [PubMed] [Google Scholar]
- 13.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quarello P, Garelli E, Brusco A, et al. Multiplex ligation-dependent probe amplification (MLPA) enhances molecular diagnosis of Diamond–Blackfan anemia due to RPS19 deficiency. Haematologica. 2008;93:1748–50. doi: 10.3324/haematol.13423. [DOI] [PubMed] [Google Scholar]
- 15.Vlachos A, Ball S, Dahl N, et al. on behalf of the participants of the Sixth Annual Daniella Maria Arturi International Consensus Conference. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008;142:859–76. doi: 10.1111/j.1365-2141.2008.07269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miceli Sopo S, Pesaresi MA, Pastore M, Stabile A. Intravenous immunoglobulin in Diamond–Blackfan anaemia. Eur J Pediatr. 1990;149:779–80. doi: 10.1007/BF01957279. [DOI] [PubMed] [Google Scholar]
- 17.Morimoto A, Kuriyama K, Tsuji K, et al. Use of rituximab to treat refractory Diamond–Blackfan anemia. Eur J Haematol. 2005;74:442–4. doi: 10.1111/j.1600-0609.2004.00394.x. [DOI] [PubMed] [Google Scholar]
- 18.Sumimoto S, Kawai M, Kasajima Y, Hamamoto T. Intravenous gamma-globulin therapy in Diamond–Blackfan anemia. Acta Paediatr Jpn. 1992;34:179–80. doi: 10.1111/j.1442-200x.1992.tb00947.x. [DOI] [PubMed] [Google Scholar]
- 19.Bejaoui M, Fitouri Z, Sfar MT, Lakhoua R. Failure of immunosuppressive therapy and high-dose intravenous immunoglobulins in four transfusion-dependent, steroid-unresponsive Blackfan–Diamond anemia patients. Haematologica. 1993;78:38–9. [PubMed] [Google Scholar]
- 20.Kawakami T, Mitsui T, Kanai M, et al. Genetic analysis of Shwachman–Diamond syndrome: phenotypic heterogeneity in patients carrying identical SBDS mutations. Tohoku J Exp Med. 2005;206:253–9. doi: 10.1620/tjem.206.253. [DOI] [PubMed] [Google Scholar]
- 21.Shah SS, Bacino CA, Sheehan AM, Shearer WT. Diagnosis of primary immunodeficiency: let your eyes do the talking. J Allergy Clin Immunol. 2009;124:1363–4. doi: 10.1016/j.jaci.2009.10.049. [DOI] [PubMed] [Google Scholar]
- 22.Farrar JE, Nater M, Caywood E, et al. Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond–Blackfan anemia. Blood. 2008;112:1582–92. doi: 10.1182/blood-2008-02-140012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gazda HT, Sheen MR, Vlachos A, et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond–Blackfan anemia patients. Am J Hum Genet. 2008;83:769–80. doi: 10.1016/j.ajhg.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doherty L, Sheen MR, Vlachos A, et al. Ribosomal protein genes RPS10 and RPS26 are commonly mutated in Diamond–Blackfan anemia. Am J Hum Genet. 2010;86:222–8. doi: 10.1016/j.ajhg.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Amsterdam A, Sadler KC, Lai K, et al. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2004;2:E139. doi: 10.1371/journal.pbio.0020139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parekh S, Perez A, Yang XY, Billett H. Chronic parvovirus infection and G6PD deficiency masquerading as Diamond–Blackfan anemia. Am J Hematol. 2005;79:54–7. doi: 10.1002/ajh.20313. [DOI] [PubMed] [Google Scholar]
- 27.Yang DQ, Halaby MJ, Zhang Y. The identification of an internal ribosomal entry site in the 5′-untranslated region of p53 mRNA provides a novel mechanism for the regulation of its translation following DNA damage. Oncogene. 2006;25:4613–9. doi: 10.1038/sj.onc.1209483. [DOI] [PubMed] [Google Scholar]
- 28.Ceci M, Gaviraghi C, Gorrini C, et al. Release of eIF6 (p27BBP) from the 60S subunit allows 80S ribosome assembly. Nature. 2003;426:579–84. doi: 10.1038/nature02160. [DOI] [PubMed] [Google Scholar]
- 29.Dror Y, Ginzberg H, Dalal I, et al. Immune function in patients with Shwachman–Diamond syndrome. Br J Haematol. 2001;114:712–7. doi: 10.1046/j.1365-2141.2001.02996.x. [DOI] [PubMed] [Google Scholar]
- 30.Dror Y, Freedman MH. Shwachman–Diamond syndrome marrow cells show abnormally increased apoptosis mediated through the Fas pathway. Blood. 2001;97:3011–6. doi: 10.1182/blood.v97.10.3011. [DOI] [PubMed] [Google Scholar]
- 31.Calado RT, Graf SA, Wilkerson KL, et al. Mutations in the SBDS gene in acquired aplastic anemia. Blood. 2007;110:1141–6. doi: 10.1182/blood-2007-03-080044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knudson M, Kulkarni S, Ballas ZK, Bessler M, Goldman F. Association of immune abnormalities with telomere shortening in autosomal-dominant dyskeratosis congenita. Blood. 2005;105:682–8. doi: 10.1182/blood-2004-04-1673. [DOI] [PubMed] [Google Scholar]
- 33.Vulliamy TJ, Knight SW, Mason PJ, Dokal I. Very short telomeres in the peripheral blood of patients with X-linked and autosomal dyskeratosis congenita. Blood Cells Mol Dis. 2001;27:353–7. doi: 10.1006/bcmd.2001.0389. [DOI] [PubMed] [Google Scholar]
- 34.Walne AJ, Vulliamy T, Marrone A, et al. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum Mol Genet. 2007;16:1619–29. doi: 10.1093/hmg/ddm111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walne AJ, Vulliamy TJ, Beswick R, Kirwan M, Dokal I. TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood. 2008;112:3594–600. doi: 10.1182/blood-2008-05-153445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cossu F, Vulliamy TJ, Marrone A, Badiali M, Cao A, Dokal I. A novel DKC1 mutation, severe combined immunodeficiency (T + B − NK − SCID) and bone marrow transplantation in an infant with Hoyeraal–Hreidarsson syndrome. Br J Haematol. 2002;119:765–8. doi: 10.1046/j.1365-2141.2002.03822.x. [DOI] [PubMed] [Google Scholar]
- 37.Ridanpää M, Jain P, McKusick VA, Francomano CA, Kaitila I. The major mutation in the RMRP gene causing CHH among the Amish is the same as that found in most Finnish cases. Am J Med Genet C Semin Med Genet. 2003;121C:81–3. doi: 10.1002/ajmg.c.20006. [DOI] [PubMed] [Google Scholar]
- 38.Thiel CT, Mortier G, Kaitila I, Reis A, Rauch A. Type and level of RMRP functional impairment predicts phenotype in the cartilage hair hypoplasia–anauxetic dysplasia spectrum. Am J Hum Genet. 2007;81:519–29. doi: 10.1086/521034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mäkitie O, Kaitila I, Savilahti E. Deficiency of humoral immunity in cartilage-hair hypoplasia. J Pediatr. 2000;137:487–92. doi: 10.1067/mpd.2000.108102. [DOI] [PubMed] [Google Scholar]
- 40.Mäkitie O, Kaitila I, Savilahti E. Susceptibility to infections and in vitro immune functions in cartilage-hair hypoplasia. Eur J Pediatr. 1998;157:816–20. doi: 10.1007/s004310050943. [DOI] [PubMed] [Google Scholar]
- 41.Mäkitie O, Kaitila I. Cartilage-hair hypoplasia – clinical manifestations in 108 Finnish patients. Eur J Pediatr. 1993;152:211–7. doi: 10.1007/BF01956147. [DOI] [PubMed] [Google Scholar]
- 42.Kooijman R, van der Burgt CJ, Weemaes CM, Haraldsson A, Scholtens EJ, Zegers BJ. T cell subsets and T cell function in cartilage-hair hypoplasia. Scand J Immunol. 1997;46:209–15. doi: 10.1046/j.1365-3083.1997.d01-112.x. [DOI] [PubMed] [Google Scholar]
- 43.Kavadas FD, Giliani S, Gu Y, et al. Variability of clinical and laboratory features among patients with ribonuclease mitochondrial RNA processing endoribonuclease gene mutations. J Allergy Clin Immunol. 2008;122:1178–84. doi: 10.1016/j.jaci.2008.07.036. [DOI] [PubMed] [Google Scholar]
- 44.Pierce GF, Polmar SH. Lymphocyte dysfunction in cartilage-hair hypoplasia: evidence for an intrinsic defect in cellular proliferation. J Immunol. 1982;129:570–5. [PubMed] [Google Scholar]
- 45.Roifman CM, Gu Y, Cohen A. Mutations in the RNA component of RNase mitochondrial RNA processing might cause Omenn syndrome. J Allergy Clin Immunol. 2006;117:897–903. doi: 10.1016/j.jaci.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 46.Gonzales B, Henning D, So RB, Dixon J, Dixon MJ, Valdez BC. The Treacher Collins syndrome (TCOF1) gene product is involved in pre-rRNA methylation. Hum Mol Genet. 2005;14:2035–43. doi: 10.1093/hmg/ddi208. [DOI] [PubMed] [Google Scholar]
- 47.Hasan R, Inoue S. Diamond–Blackfan anemia associated with Treacher–Collins syndrome. Pediatr Hematol Oncol. 1993;10:261–5. doi: 10.3109/08880019309029494. [DOI] [PubMed] [Google Scholar]
- 48.Fisher EM, Beer-Romero P, Brown LG, et al. Homologous ribosomal protein genes on the human X and Y chromosomes: escape from X inactivation and possible implications for Turner syndrome. Cell. 1990;63:1205–18. doi: 10.1016/0092-8674(90)90416-c. [DOI] [PubMed] [Google Scholar]
- 49.Watanabe M, Zinn AR, Page DC, Nishimoto T. Functional equivalence of human X- and Y-encoded isoforms of ribosomal protein S4 consistent with a role in Turner syndrome. Nat Genet. 1993;4:268–71. doi: 10.1038/ng0793-268. [DOI] [PubMed] [Google Scholar]
- 50.al-Attas RA, Rahi AH, Ahmed el-FE. Common variable immunodeficiency with CD4+ T lymphocytopenia and overproduction of soluble IL-2 receptor associated with Turner's syndrome and dorsal kyphoscoliosis. J Clin Pathol. 1997;50:876–9. doi: 10.1136/jcp.50.10.876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robson SC, Potter PC. Common variable immunodeficiency in association with Turner's syndrome. J Clin Lab Immunol. 1990;32:143–6. [PubMed] [Google Scholar]
- 52.Cacciari E, Masi M, Fantini MP, et al. Serum immunoglobulins and lymphocyte subpopulations derangement in Turner's syndrome. J Immunogenet. 1981;8:337–44. doi: 10.1111/j.1744-313x.1981.tb00938.x. [DOI] [PubMed] [Google Scholar]
- 53.Wood CB, Martin W, Adinolfi M, Polani PE. Immunoglobulins and the X-chromosome. BMJ. 1969;4:110. doi: 10.1136/bmj.4.5675.110-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mock, Markert UR, Vogelsang H, Jäger L. Selective T-cell deficiency in Turner's syndrome. J Investig Allergol Clin Immunol. 2000;10:312–3. [PubMed] [Google Scholar]
- 55.Ferrer Calvete J, Tomás M, Galmes J, Prieto F. Chronic diarrhea with selective IgA deficit associated with Turner's syndrome. An Esp Pediatr. 1982;16:459–63. [PubMed] [Google Scholar]
- 56.Arslan D, Kuyucu T, Kendirci M, Kurtoglu S. Celiac disease and Turner's syndrome: patient report. J Pediatr Endocrinol Metab. 2000;13:1629–31. doi: 10.1515/jpem.2000.13.9.1629. [DOI] [PubMed] [Google Scholar]
- 57.Ding Q, Markesbery WR, Chen Q, Li F, Keller JN. Ribosome dysfunction is an early event in Alzheimer's disease. J Neurosci. 2005;25:9171–5. doi: 10.1523/JNEUROSCI.3040-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ellis SR, Massey AT. Diamond Blackfan anemia: a paradigm for a ribosome-based disease. Med Hypotheses. 2006;66:643–8. doi: 10.1016/j.mehy.2005.09.010. [DOI] [PubMed] [Google Scholar]