

Abstract
Background
Leukotriene D4 (LTD4) belongs to the bioactive lipid group known as eicosanoids and has implications in pathological processes such as inflammation and cancer. Leukotriene D4 exerts its effects mainly through two different G-protein-coupled receptors, CysLT1 and CysLT2. The high affinity LTD4 receptor CysLT1R exhibits tumor-promoting properties by triggering cell proliferation, survival, and migration in intestinal epithelial cells. In addition, increased expression and nuclear localization of CysLT1R correlates with a poorer prognosis for patients with colon cancer.
Methodology/Principal Findings
Using a proximity ligation assay and immunoprecipitation, this study showed that endogenous CysLT1R formed heterodimers with its counter-receptor CysLT2R under basal conditions and that LTD4 triggers reduced dimerization of CysLTRs in intestinal epithelial cells. This effect was dependent upon a parallel LTD4-induced increase in CysLT1R tyrosine phosphorylation. Leukotriene D4 also led to elevated internalization of CysLT1Rs from the plasma membrane and a simultaneous increase at the nucleus. Using sucrose, a clathrin endocytic inhibitor, dominant-negative constructs, and siRNA against arrestin-3, we suggest that a clathrin-, arrestin-3, and Rab-5-dependent process mediated the internalization of CysLT1R. Altering the CysLT1R internalization process at either the clathrin or the arrestin-3 stage led to disruption of LTD4-induced Erk1/2 activation and up-regulation of COX-2 mRNA levels.
Conclusions/Significance
Our data suggests that upon ligand activation, CysLT1R is tyrosine-phosphorylated and released from heterodimers with CysLT2R and, subsequently, internalizes from the plasma membrane to the nuclear membrane in a clathrin-, arrestin-3-, and Rab-5-dependent manner, thus, enabling Erk1/2 signaling and downstream transcription of the COX-2 gene.
Introduction
Patients with prolonged inflammatory conditions such as inflammatory bowel disease (IBD) exhibit increased levels of inflammatory mediators, such as cysteinyl leukotrienes (CysLT; LTC4, LTD4, and LTE4) [1]. The fact that IBD patients have a 30–50% increased risk of developing colorectal cancer [2] implies a possible role of cysteinyl leukotrienes in the coupling between chronic inflammation and the development of colorectal cancer.
Leukotrienes exert their effects through G-protein-coupled receptors (GPCRs). The CysLT1R [3] is a high affinity GPCR for the pro-inflammatory mediator LTD4 that is implicated in many inflammatory conditions [4], [5]. We have shown that LTD4 up-regulates several proteins related to carcinogenesis, such as COX-2, β-catenin, and Bcl-2, via the CysLT1R in intestinal epithelial cells [6], [7]. We have also shown that LTD4 mediates survival [8], [9], proliferation [10], and migration [11] in epithelial cells through the CysLT1R. Up-regulation of the receptors at the plasma membrane and the nuclear membrane was shown in a colon cancer tissue microarray [12]. This up-regulation of the CysLT1R in colon cancer correlates with a poorer prognosis [12], [13], [14]. In contrast to this, increased levels of the CysLT2R, which is also located in the plasma and nuclear membrane, correlates with a better prognosis for patients with colon cancer [14], [15]. Furthermore, LTC4-induced activation of CysLT2R has been shown to promote differentiation of colon cancer cells [15], which suggests a potentially opposite role for the CysLT2R compared to the CysLT1R in the development or progression of colon cancer.
A key regulatory mechanism of GPCR signaling is internalization and trafficking. There are a limited number of publications studying the trafficking of the CysLT1R [16], [17], [18]. Naik et al. demonstrated that in HEK-293 cells over expressing the CysLT1R, the internalization of the receptor is Protein Kinase C (PKC)-dependent [16]. Furthermore, our group has demonstrated that the nuclear localization sequence (NLS) domain, which contains the PKC sites, is required for internalization and Erk1/2 signaling via the CysLT1R [12]. Capra et al. showed that, unlike the homologous desensitization induced by LTD4, the heterologous desensitization of the CysLT1R via the P2YR is PKC-dependent [16], [17], suggesting that CysLT1R regulation can be cell specific. Previous results from our laboratory suggest that, upon stimulation with LTD4, the CysLT1R translocates from the plasma membrane to the outer nuclear membrane of Int 407 cells [12]. The internalization and trafficking of GPCRs are often implicated in GPCR-related pathologies, such as in the case of retinitis pigmentosa, which is reported to be a result of improper intracellular trafficking and localization of rhodopsin receptors [19], [20].
An important aspect of GPCR regulation is the ability to dimerize. GPCRs can induce signals as hetero-, homo-dimers or oligomers [21]. Moreover, GPCR dimerization has been shown to be needed for their proper expression, stronger ligand binding, phosphorylation, and internalization [21]. Dimerized GPCRs may have signaling properties distinct from those of monomeric receptors [22], [23]. Receptor-mediated endocytosis is a mechanism by which the cell regulates the magnitude and duration of external stimuli [24], [25]. There have been extensive investigations into endocytosis via clathrin-coated pits, resulting in it being the best-characterized mechanism for GPCR internalization [26], [27]. Clathrin-coated pits are membrane invaginations coated with clathrin. Upon ligand binding, G-protein-coupled receptor kinases (GRKs) or protein kinases, such as PKC, phosphorylate GPCRs. This phosphorylation leads to the recruitment of arrestin, which, in turn, targets the GPCR to the clathrin-coated pits. However, certain GPCRs, such as the leukotriene B4 receptor 1 (BLT1R), when transfected into Cos-7 and HEK-293 cells, may internalize independently of arrestins [28]. Different Rab proteins are involved in vesicle trafficking and regulate their directionality. Rab-5, -11 and -21, in particular, are involved in the trafficking of early endosomes [29], [30], [31]. Once internalized, the receptor is either recycled through early endosomes, sent for degradation to the lysosomes [32], or transported to the nucleus [33], [34], [35]. A less studied internalization pathway is the one through caveolae. Caveolae are membrane invaginations, rich in caveolin proteins and cholesterol. Certain GPCRs, such as the M1 receptor and the glucagon peptide 1 receptor, are internalized and have been shown to be internalized via this pathway [36], [37]. However, the mechanism of how these GPCRs are targeted into the caveolae is still unknown. Other GPCRs, like the β-adregenic receptors β1AR, and β2AR, are enriched in the caveolae, but they are not internalized through this pathway [38], [39].
The aim of this study was to investigate the underlying regulatory mechanisms leading to the internalization of CysLT1R. We demonstrate that LTD4 induces tyrosine phosphorylation and internalizes the CysLT1R. Furthermore, we suggest that the LTD4-induced CysLT1R translocation to the nucleius, or disruption of this internalization at various stages, could affect its overall signaling process.
Materials and Methods
Chemicals
Antibodies against heavy-chain clathrin were from BD Transduction Laboratories (Erembodegem, Belgium). The LTD4 was from Cayman Chemical Company (Ann Arbor, MI), and N-terminal CysLT1R was from Innovagen (Lund, Sweden). The ZM198615 was a gift from AstraZeneca (R&D, Lund, Sweden), and ECL Western blot detection reagents and Hyperfilm were from Amersham International (Buckinghamshire, UK). The source for Protein A sepharose was GE Healthcare (Uppsala, Sweden). The phospho-Erk1/2 antibody was from New England BioLabs Inc, (Beverly, MA). Antibodies against arrestin-3 were purchased from Cell Signaling (Boston, MA) and Santa Cruz (Santa Cruz, CA). The arrestin-3 and scrambled siRNA were from Santa Cruz (Santa Cruz, CA). Peroxidase-linked goat anti-rabbit antibodies and fluorescence mounting medium were from Dako A/S (Copenhagen, Denmark). Lipofectamine 2000 and all cell culture media were from Invitrogen (Carlsbad, CA) and Alexa 488 and Alexa 546 were from Molecular Probes Inc. (Leiden, Netherlands). The RNeasy MinElute Spin Column was from Qiagen (Hilden, Germany). Genistein was from Calbiochem (San Diego, CA). The Flag M2 antibodies, light-chain clathrin antibodies, and all other chemicals were of analytical grade and were purchased from Sigma Chemical Company (St. Louis, MO).
Cell Culture
Non-transformed human intestinal epithelial cells, Int 407 cells exhibiting typical epithelial growth and morphology [40], and the human colorectal adenocarcinoma cell line Caco-2 (ATCC HTB-37) were cultured as described previously [41]. Cells were cultured to approximately 80% confluency and regularly tested to ensure the absence of mycoplasma.
The in situ proximity ligation assay
The in situ proximity ligation assay (PLA), Duolink™, was from Olink Bioscience (Uppsala, Sweden) and performed according to the manufacturer's instructions [42], with slight modifications. Briefly, Int 407 cells were grown in 4-well plates to 50% confluency, serum starved, and stimulated with LTD4 or LTC4 (40 nM) for indicated time points and fixed for 15 minutes in 4% ice-cold PFA/PBS. Blocking in a 3% BSA/PBS for 1 hour followed. Thereafter, the cells were stained with anti-rabbit CysLT1R, anti-goat CysLT2R antibodies, and anti-phospho-tyrosine antibodies (1∶250 in 3% BSA/PBS) overnight at 4°C. This was followed by washing five times in PBS-T and incubation with PLA probes minus and plus (anti-goat DNA minus strand and anti-rabbit DNA-plus strand, diluted 1∶5) in 3% BSA/PBS for 2 hours at 37°C. Alternatively, as a negative control, the CysLT2R antibody, or the DNA-plus probe, was omitted. Furthermore, as a positive control, the Duolink control kit contains two primary antibodies targeting different epitopes of chicken TK1 protein. Thereafter, the cells were washed twice in PBS-T and hybridized at 37°C for 15 minutes, followed by ligation for 15 minutes at 37°C. The cells were washed 1× in PBS-T and treated with polymerase for amplification for 90 minutes at 37°C. The detection of PLA-amplicons (red dots) was carried out using the “563 detection kit” provided by Olink Bioscience. This kit includes the Hoechst 33342 dye for nuclear staining (blue) and the Alexa Fluor 488-phalloidin/actin for cytoplasmic staining (green). The cells were then mounted and examined using a Nikon TE300 microscope (60×1.4 plan apochromat oil immersion objective), integrated into fluorescent microscopy. The red dots were counted using the MATLAB/Blob Finder software from Olink Bioscience (Uppsala, Sweden) [42].
Electron Microscopy
Cells stimulated with or without 40 nM LTD4 or LTC4 for 15 or 30 minutes were used for electron microscopy and prepared as described previously [12]. Briefly, 5×106 cells were pelleted at 4°C immediately after being placed in a fixative (4% paraformaldehyde and 0.1% glutaraldehyde). The pellets were dehydrated in ethanol for 1 hour at room temperature and then embedded in Lowicryl [43]. Ultra thin sections were cut on a microtome and mounted on nickel grids. For immunostaining, the grids were floated on drops of immune reagents placed on a sheet of parafilm. Free aldehyde groups were then blocked with 50 mmol/L glycine, and the grids were subsequently incubated with 5% (v/v) donkey serum in PBS supplemented with 0.2% bovine serum albumin (BSA; pH 7.6) for 15 minutes. Overnight incubation with the primary antibody (dilution 1∶100) at 4°C followed this blocking procedure. The grids were subsequently washed by placing them, successively, on 10 drops of incubation buffer (5 minutes on each drop), after which the sections were incubated with the gold-conjugated secondary antibody by letting them float on drops containing the gold conjugate reagent (diluted 1∶20 in incubation buffer) for 60 minutes at room temperature. After further washing on 10 drops of incubation buffer, the sections were postfixed in 2% glutaraldehyde. Finally, the sections were washed with distilled water, poststained with uranyl acetate and lead citrate, and examined using a Jeol 1200 EX transmission electron microscope operated at 60 kV accelerating voltage, as previously described [12]. The antibody directed against the CysLT1 was labeled with 10-nm colloidal thiocyanate gold and CysLT2 with 5-nm colloidal thiocyanate gold [44] for the LTC4 experiment. But, 10-nm colloidal thiocyanate gold for CysLT2R and 5-nm colloidal thiocyanate gold for CysLT1R in the LTD4 experiment. The images were recorded with a Gatan Multiscan 791 CCD camera. Researchers examined sixty cellular profiles for evaluation.
Immunofluorescent Staining
Cells were grown on cover slips to 50–60% confluency, pre-treated, or not, with ZM198,615 (40 µM, 15 minutes) and then treated with, or without, LTD4 (80 nM, 5 minutes or as indicated). Cells were washed once and kept in 1.5% serum-containing medium for 15 or 20 minutes. Thereafter, the cells were fixed for 15 minutes in 4% PFA/PBS, followed by blocking in a 3% BSA/PBS for 1 hour for anti-CysLT1R, 5% goat serum and 1% TritonX100/PBS for anti-Flag, or 3% milk/PBS for anti-clathrin. Cells were then incubated overnight with anti-CysLT1R (1∶250) in a 3% BSA/PBS or 1% goat serum in PBS-Tween (PBS-T) for Flag (1∶2500), and clathrin (1∶250) in 2% BSA/PBS. Cells were washed five times in PBS and incubated for 1 hour with secondary antibody goat anti-rabbit IgG Alexa 488 or 546 (3% BSA/PBS 1∶250) for CysLT1R and clathrin or 1% goat serum 1∶800 for Flag antibodies. Following five washes with PBS, the cover slips were mounted on glass slides with a fluorescence-mounting medium and examined using a Nikon TE300 microscope (60×1.4 plan-apochromat oil immersion objective), integrated in fluorescent microscopy.
Transfection
Cells were grown on cover slips to 50–60% confluency. Transfection was performed with GFP-DN-Eps-15, GFP-Eps-15, Flag-CysLT1R, or GFP-Rab-5 constructs, using lipofectamine according to the manufacturer's protocol. Briefly, cells were transfected for 6 hours in serum-free medium and left to rest for 48 hours in complete medium before analysis. For siRNA, cells were grown to about 60% confluency in 6-well plates, transfected in serum-antibiotic-free medium, with 80 pmol siRNA against arrestin-3 or scrambled siRNA using lipofectamine 2000. After 6 hours of transfection, 1 mL serum-free medium was added to each well and cells were left overnight. The medium was then changed to normal growth medium and cells were left to rest for an additional 48 hours before being lysed or used for FACS analysis.
Cell Lysates, Immunoprecipitation and Fractionation
Cells were left in serum-free medium for 2 hours, pre-treated with Filipin (5 µg/mL, 1 hour), sucrose (0.4 M, 1 hour), or cycloheximide (100 µg/mL, 1 hour) and then treated with, or without, LTD4 (80 nM) for indicated time points. The stimulations were terminated by the addition of ice-cold lysis buffer A (20 mM sodium Hepes pH 8.0, 2 mM MgCl2, 1 mM EDTA, 5 mM sodium orthovandate, 60 µg/mL phenylmethylsulfonyl fluoride (PMSF), and 4 µg/mL leupeptin) and the cells were placed on ice. The cells were then scraped from the flasks. The supernatant was collected from the cell lysate preparation after a centrifugation at 200×g for 10 minutes at 4°C and after a centrifugation at 10,000×g for 15 minutes at 4°C. The samples were compensated to equal protein content and pre-cleared with 1 µg of rabbit IgG and 15 µl of protein A-sepharose for overnight at 4°C. The samples were immunoprecipitated with 5 µg of CysLT2R antibody for 2 hours at 4°C. Thereafter, 20 µg of protein A-sepharose beads were added, and the samples were rotated for an additional 1 hour at 4°C. The precipitates were washed multiple times with lysis buffer A. For fractionation, the cells were subjected to N2-decompression at 1,000 psi for 10 minutes, using a cell disruption bomb (Parr Instrument Company, Moline). The intact nuclei were collected by centrifugation at 200×g and washed twice in buffer A. The supernatant was centrifuged at 10,000×g for 10 minutes, and the resulting supernatant was fractioned into cytosol and plasma membrane fractions by centrifugation at 200,000 g for 1 hour.
Gel Electrophoresis and Immunoblotting
Cell lysates were solubilized by boiling in sample buffer (62 mM Tris pH 6.8, 1.0% SDS, 10% glycerol, 15 mg/mL dithiothreitol, and 0.05% bromphenol blue), loaded, and subjected to electrophoresis on 10% homogeneous polyacrylamide gels. The separated proteins were electrophoretically transferred to PVDF membranes. The CysLT1R membranes were incubated overnight at 4°C with anti-CysLT1R and CysLT2R (diluted 1∶250 in 3% BSA/PBS) and for 1 hour at room temperature for anti-actin (1∶2000 in 2% BSA/PBS). After washing three times, the membranes were incubated for 1 hour at room temperature with HRP-conjugated secondary antibody (1∶5000 in 1% BSA/PBS for CysLT1R and CysLT2R or 1∶3000 in 1% BSA/PBS for actin), and then the membranes were washed three to six times. Thereafter, the membranes were incubated with ECL Western blot detection reagents and exposed to Hyperfilm-ECL to visualize immunoreactive proteins.
DNA Isolation and Sequencing
Cells were grown to 80% confluency, scraped, and resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0, 15 mM EDTA, 200 mM NaCl, and 0.5% SDS). The mixture was incubated overnight at 45°C with Proteinase K (Fermentas, Vilnius, Lithuania). Phenol was added and mixed for 10 minutes. The mixture was centrifuged at 600 g for 10 minutes at 10°C. The upper clear aqueous layer was carefully transferred to a new tube. An equal volume of phenol∶chloroform∶isoamyl alcohol (24∶24∶1) was added and mixed, by gentle inversion, for about 10 minutes and centrifuged at 500 g for 10 minutes at 10°C. The upper clear aqueous layer was transferred to a new tube. An equal volume of chloroform∶isoamyl alcohol (24∶1) was added, mixed for 10 minutes, and centrifuged at 500 g for 10 minutes at 10°C. The upper clear aqueous layer was transferred to a new tube. One-tenth of the volume of 3 M sodium acetate, pH 5.2, and double volumes of 100% isopropanol were added and allowed to stand for 1 hour at −20°C. The samples were centrifuged at 11,000 g for 10 minutes at 4°C thereafter. The supernatant was discarded and the pellet washed with 70% ethanol. The resultant pellet was dried and dissolved in TE-buffer (10 mM Tris, pH 8.0, 1 mM EDTA). The Department of Clinical Chemistry at Skåne University Hospital (SUS), Malmö, Sweden, performed the sequencing.
RT-PCR
Cells were scraped on ice in PBS, homogenized 10 times with a 20 G needle, and then centrifuged for 2 minutes at 10,000 g. The pellet was resuspended in 1 ml TRIzol and immediately frozen at −80°C. The RNA was isolated using the phenol-chloroform extraction method. The RNA was dissolved in RNase-free H2O and purified on RNeasy MinElute Spin Columns. The cDNA synthesis was performed using Superscript™ II reverse transcriptase. Next, 2 µg of cDNA was mixed with 0.9 µM TaqMan primers and master mix and amplified at 60°C in a Mx3005P (Stratagene qPCR system). The following primer set was used: COX-2: Hs01573475_g1 and GAPDH: Hs00266705_g1. The samples were analyzed and normalized against a housekeeping gene (GAPDH) using the MX-Pro software (Stratagene).
Flow Cytometry
Int 407 or Caco-2 cells were cultured as described previously; thereafter, they were serum-starved before they were either transfected with siRNA against arrestin-3 or scrambled siRNA and then stimulated with or without, 80 nM LTD4 for 5 or 30 minutes. The cells were detached by the addition of versen or trypsin-EDTA, respectively. The collected cells were first washed with cold PBS supplemented with 0.2 mM EDTA and then with PBS containing 0.5% bovine serum albumin. The cells (1×106) were first fixed using the IC-Fixation buffer (cat # 00-8222; eBioscience, San Diego, Ca) before doing cell surface staining for CysLT1R or further permabilized for intracellular staining of arrestin-3 using the Permabilization Buffer (cat # 00-8333; eBioscience, San Diego, Ca). Following the recommendation given by the manufacturer and supplemented with additional washing steps, the cells were stained with the anti-CysLT1R primary antibody (5 µg/mL) or the anti human arrestin-3 antibody (5 µg/mL) followed by incubations with either goat anti-Rabbit IgG or goat anti-mouse IgG secondary antibody both conjugated respectively with ALEXA-488 (1∶100 in 0.5% BSA/PBS). An equivalent amount of non-specific rabbit or mouse IgG was used as controls. A single-color, immunofluorescence, flow cytometry analysis was performed on a FACSCalibur (Becton Dickinson) and data were analyzed using software (CellQuest; Becton Dickinson). Each measurement was based on the analysis of 10,000 cells.
Statistical Analysis
Results are expressed as mean ± SEM. Differences between experimental groups were assessed by a Student's t test and one way ANOVA. P values of <0.05 were considered significant. *P<0.05 and ** P<0.01 and ***P<0.001.
Results
Hetrodimerization of the CysLTRs and Tyrosine Phosphorylation of the CysLT1R
Because of the overlapping localization of the CysLT1R and CysLT2R at the plasma membrane and nuclear membrane, we investigated a potential heterodimerization of the receptors. Previous studies have demonstrated that the CysLT1R and CysLT2R might dimerize in mast cells [45]. Heterodimerization of the CysLT1R and CysLT2R was examined in Int 407 cells using the in situ proximity ligation assay (PLA) (Fig. 1). With this assay, protein-protein interactions in situ can be detected and visualized; when the secondary antibody is in close proximity, a fluorescent labeling of the DNA product is produced (red dots) [42]. Image analysis is based on counting the number of red dots/cell. The negative control without the CysLT2R antibody does not produce any red dots (Fig. 1A). Cells stained with both the CysLT1 and CysLT2 receptor antibodies showed that the receptors were heterodimerized under basal conditions (Fig. 1). The heterodimers (red dots) were concentrated to the plasma membrane and the nuclear region (Fig. 1A). Stimulation with LTC4 (40 nM) for 5 minutes caused a slight increase in the number of heterodimerized receptors, however this effect was not statistically significant (Fig. 1B). These results suggest that CysLT1R and CysLT2R are dimerized already, under basal conditions, and remain dimerized, even after LTC4 stimulation.
Figure 1. Receptor heterodimerization detected by a in situ proximity ligation assay (PLA).

(A) Briefly, Int 407 cells were grown to 50% confluency, stimulated with or without LTD4 (40 nM), LTC4 (40 nM), or pre-incubation with genistein (50 µg/ml) for 30 minutes. The receptor interactions were studied employing PLA, treated according to the manufacturer's instructions using the CysLT1R antibody (1∶250) and the CysLT2R antibody (1∶250) and mounted on glass slides with a fluorescence-mounting medium. Alternatively, the CysLT2R antibody was omitted as a negative control. The mounted slides were examined using a Nikon TE300 microscope (60×1.4 plan apochromat oil immersion objective), integrated in fluorescent microscopy. The detection of PLA-amplicons (red dots) was carried out using the “563 detection kit”. This kit includes the Hoechst 33342 dye for nuclear staining (blue) and the Alexa Fluor 488-phalloidin/actin for cytoplasmic staining (green). The red dots indicate close proximity between cellular bound antibodies, and they were counted using the MATLAB/Blobfinder software. (B) The data are given as percent of control and represent means ± S.E.M. of at least three separate experiments. The statistical analysis was performed with a Student's t test. *P<0.05 and ** P<0.01. The scale bar represents 10 µm.
We found a statistically significant decrease in heterodimerization of the receptors 5 minutes after LTD4 stimulation (an average of less than 4 dots/cell compared to an average of 60 dots/cell in the control; Fig. 1B). However, after 60 minutes of LTD4 stimulation, a slight increase in heterodimerization (an average of 13 dots/cell) compared to the 5 minutes value was observed. Interestingly, the heterodimers (red dots) were mainly localized to the nuclear region. To further confirm the association between the receptors, we preformed immunoprecipitation with the CysLT2R antibody. With this approach we could confirm an association between the CysLT2R and the CysLT1R that was reduced (P<0.05) after LTD4 stimulation (Figure S1).
Both receptors contained several tyrosine phosphorylation sites, which might be important for activation and internalization. We therefore, investigated if tyrosine phosphorylation was involved in the decreased heterodimerization seen after LTD4 stimulation. Indeed, we found that the effect of LTD4 was abolished in cells pretreated with genistein, a broad phosphotyrosine inhibitor (Fig. 1B).
As demonstrated, LTD4 (40 nM) induced tyrosine phosphorylation of the CysLT1R after 5 and 60 minutes of stimulation (an average of 10 dots/cell as compared to 1 dot/cell when not stimulated), whilst LTC4 (40 nM) did not induce any detectable increase in tyrosine phosphorylation of the CysLT1R (Fig. 2B). Genistein significantly reduced the LTD4-induced tyrosine phosphorylation of CysLT1R (an average of 3 dots/cell; Fig. 2B). Neither LTC4 nor LTD4 induced any detectable tyrosine phosphorylation of the CysLT2R (data not shown). We also investigated threonine phosphorylation of the receptors with the PLA technique. For this experiment, we used an antibody for anti-phospho-threonine (Abnova Taiwan Corp), but we were not able to detect any threonine phosphorylation upon LTD4 or LTC4 stimulation of either of these receptors during the time points tested (data not shown).
Figure 2. The CysLT1R tyrosine phosphorylation detected by in situ proximity ligation assay (PLA).

Briefly, Int 407 cells were grown to 50% confluency, stimulated with or without LTD4 (40 nM), LTC4 (40 nM), or pre-incubation with genistein (50 µg/ml) for 30 minutes. The receptor tyrosine phosphorylation was studied employing PLA, treated according to the manufacturer's instructions using (A) the CysLT1R antibody (1∶250) and the phosphor-Tyr antibody (1∶250) and mounted on glass slides with a fluorescence-mounting medium. The mounted slides were examined using a Nikon TE300 microscope (60×1.4 plan apochromat oil immersion objective), integrated in fluorescent microscopy. The detection of PLA-amplicons (red dots) was carried out using the “563 detection kit”. This kit includes the Hoechst 33342 dye for nuclear staining (blue) and the Alexa Fluor 488-phalloidin/actin for cytoplasmic staining (green). The red dots indicate close proximity between cellular bound antibodies, and they were counted using the MATLAB/Blobfinder software. (B) The data are given as percent of control and represent means ± S.E.M. of at least three separate experiments. The statistical analysis was performed with a Student's t test. *P<0.05 and ** P<0.01. The scale bar represents 10 µm.
Internalization of the CysLT1R and the CysLT2R
We next examined the regulation of low affinity CysLT2R in conjunction with the CysLT1R. Int 407 cells were primarily stimulated with 40 nM LTD4 and receptor internalization and dimerization were visualized using electron microscopy (Fig. 3A). The CysLT1R was labeled with 10-nm colloidal thiocyanate gold particles and CysLT2R was labeled with 5-nm colloidal thiocyanate gold particles; as a result, both heterodimers and homodimers could be seen. Interestingly, upon LTD4 stimulation, it was mainly the CysLT1R that was internalized and localized to the nucleus, both after 15 minutes (53%) and 30 minutes (63%), compared to the CysLT2R (38% and 40%, respectively) (Fig. 3A). We also found a reduction of the CysLT1R at the plasma membrane in cells stimulated with LTD4 for 15 and 30 minutes (13 and 19%, respectively, as compared to 36% in the control). However, the majority of the CysLT2R did not internalize during this period. We then investigated the effect of LTC4 on receptor internalization (Fig. 3B). In these experiments, the CysLT1R was labeled with 5-nm colloidal thiocyanate gold particles and CysLT2R was labeled with 10-nm colloidal thiocyanate gold particles. Interestingly, both receptors internalized upon 15 minutes of 40 nM LTC4 stimulation, and this could be explained by the decrease from the plasma membrane and a small increase in the cystosol of both CysLT1R and CysLT2R (Fig. 3B). The receptor levels were restored to the plasma membrane again after 30 minutes of LTC4 stimulation (Fig. 3B). This was further confirmed by Western blot analysis of the plasma membrane fractions, showing a significant LTC4-induced decrease in the CysLT1R and CysLT2R expression in the plasma membrane after 5-15 minutes of stimulation, effects that are reversed after 30 minutes (Figure S2).
Figure 3. Electron microscopy images of CysLT1R and CysLT2R.

Electron microscopy of Int 407 cells treated without or with (A) LTD4 (40 nM, 15 or 30 minutes) or (B) LTC4 (40 nM, 15 or 30 minutes). Samples of intact cells used for electron microscopy were prepared by pelleting 5×106 cells immediately after adding a fixative (4% paraformaldehyde and 0.1% glutaraldehyde). Ultra thin sections were cut on a microtome and mounted on nickel grids, followed by overnight incubation with the primary antibody against CysLT1R and CysLT2R. (A) The antibody directed against the CysLT1R was labeled with 10-nm colloidal thiocyanate gold (black arrow) and CysLT2R with 5-nm colloidal thiocyanate gold (white arrow). The scale bar represents 0.2 µm. (B) The antibody directed against the CysLT1R was labeled with 5-nm colloidal thiocyanate gold (white arrow) and CysLT2R with 10-nm colloidal thiocyanate gold (black arrow). The scale bar represents 0.1 µm. The specimens were examined using a Jeol JEM 1230 electron microscope operated at 60 kV accelerating voltage, and images were recorded with a Gatan Multiscan 791 CCD camera.
Internalization and Recycling of the CysLT1R
Previous results from our lab have shown that the CysLT1R is localized to the plasma and nuclear membranes of intestinal epithelial and colon cancer cells [12]. In this study, we investigated how the CysLT1R is internalized and increased at the nuclear membrane. In agreement with our previous results, we demonstrated that the endogenous CysLT1R is localized to both the plasma membrane and nuclear region of unstimulated Int 407 cells using fluorescent microscopy (Fig. 4A). We also showed that, upon 5 minutes of stimulation with 80 nM LTD4, the CysLT1 receptor is rapidly internalized (Fig. 4A). The internalization is seen as intracellular punctuated dots (Fig. 4A), which can be blocked by pre-treatment with the CysLT1R antagonist ZM198,615 (40 µM, 15 minutes; Fig. 4A) or PKC inhibitor GF109203X (2 µM, 15 minutes; data not shown). The internalization was also confirmed by Western blot (Fig. 4B). We next transfected Int 407 cells with a Flag-tagged CysLT1R construct, stained with a Flag antibody. The distribution of Flag-tagged CysLT1R is more uniformly distributed than the endogenous receptor, most likely due to the Flag construct. However, the Flag-tagged CysLT1R was also localized to both the plasma membrane and nuclear region, similar to endogenous CysLT1R staining, as it was internalized after 5 minutes of LTD4 stimulation and could be significantly blocked by the specific CysLT1R antagonist, ZM198,615 (40 µM, 15 minutes; Fig. 4C). In summary, the endogenous and the over expressed Flag-tagged CysLT1R was localized to both the plasma membrane and the nuclear region of the cell, and was internalized after 5 minutes of LTD4 stimulation. Receptor recycling was a key mechanism regulating many different receptors [25]; therefore, the recycling of the CysLT1R was investigated. After 5 minutes of stimulation with LTD4, the ligand was removed by changing the medium to a LTD4-free growth medium for an additional 15–20 minutes before they were fixed and stained. The Flag-tagged CysLT1R recycled back to the plasma membrane 15–20 minutes after stimulation (Fig. 4C). We also investigated the endogenous receptor localization in Caco-2 cells, which showed a similar pattern, but no internalization could be detected after 5 minutes of stimulation with 80 nM LTD4 (Fig. 4D). We, therefore, performed Western blot analyses of the plasma membrane fractions of the Caco-2 cells. No significant decrease of the endogenous receptor after 5–60 minutes of LTD4 stimulation could be detected in Caco-2 cells (Fig. 4E). However, in cells transfected with the Flag-tagged CysLT1R, the receptor internalization could be detected after 20 minutes of LTD4 stimulation (Fig. 4F). This internalization was sensitive to pre-treatment with the receptor antagonist ZM198,615 (Fig. 4F). We investigated if the undetectable internalization of endogenous CysLT1R in Caco-2 cells was due to a mutation in the endogenous receptor of colon cancer cells; the CysLT1R from three colon-cancer cell lines (Caco-2, SW-480, and HCT-116) were sequenced. However, no mutation in the CysLT1R sequence was detected (data not shown).
Figure 4. The localization and internalization of the CysLT1R in epithelial cells.

Immunofluorescent staining of endogenous CysLT1R (A, D) and Flag-tagged CysLT1R (C, F) in Int 407 (A, C) and Caco-2 cells (D, F). Cells were grown on cover slips to 50–60% confluency pre-treated, or not, with ZM198,615 (40 µM, 15 minutes) and treated with or without 80 nM LTD4 for 5 minutes or as indicated. The cells were fixed and stained with CysLT1R antibody (1∶250) or Flag antibody (1∶2500) and mounted as described in Methods. The mounted slides were examined using a Nikon TE300 microscope (100×1.4 plan-apochromat oil immersion objective). The scale bar represents 10 µm. Int 407 cells (B) and Caco-2 cells (E) were grown to 80% confluency, serum-starved for 2 hours and treated with or without 80 nM LTD4 for indicated time points. Plasma membrane fractions were prepared as described in Methods and samples were subjected to SDS-polyacrylamide gel electrophoresis and Western blot analysis. The PDVF membranes were stained with the CysLT1R (1∶1000) and re-probed with actin (1∶2000) antibodies. The data are given as percent of control and represent means ± S.E.M. of at least three separate experiments. The statistical analysis was performed with a Student's t test. *P<0.05 and ** P<0.01. (G) A representative FACS analysis histogram overlay displaying the relative fluorescence intensity of CysLT1R surface expression is shown for Caco-2 cells.
We next performed FACS analysis of the endogenous CysLT1R in the colon cancer cell line, Caco-2, to confirm the CysLT1R transfection data with the endogenous receptor, using a more sensitive method. Figure 4G shows the overlay for FACS histograms of Caco-2 cells, where the CysLT1R internalization could be detected after 5 minutes of LTD4 stimulation. Other than the shift in histogram peaks, as shown in Figure 4G, we also evaluated the change in both median and mean fluorescent intensity of CysLT1R expression (data not shown). Based on the findings obtained from all the different approaches, our results confirmed that the CysLT1R is internalized in both colon cancer cells and non-transformed intestinal epithelial cells.
Co-localization of Clathrin with the CysLT1R
We next investigated the internalization pathway of the CysLT1R in these cell lines. GPCRs mainly internalizes via clathrin-coated pits. Next we, therefore, investigated if the internalization of the CysLT1R was clathrin-dependent. Int 407 and Caco-2 cells were transiently transfected with the Flag-tagged CysLT1R and co-transfected with Flag and clathrin antibodies. Under basal conditions, co-localization of the CysLT1R with clathrin was detected at both the plasma membrane and nuclear regions (Fig. 5A). As demonstrated in both cell lines, the internalized receptor co-localized with clathrin upon LTD4 stimulation (Fig. 5A). In order to further confirm if the receptor was internalized via the clathrin pathway, we next used GFP-dominant negative Eps-15 (DN-Eps-15) constructs, as Eps-15 is a protein involved exclusively in the formation of clathrin-coated pits and the lack of Eps-15 activity prevents the formation of clathrin-coated vesicles [46].
Figure 5. Expression and co-localization of clathrin or Eps-15 with the CysLT1R in Int 407 and Caco-2 cells.

(A) Representative fluorescent microscope images show cells that were fixed, permeabilized, and stained with primary antibodies against either Flag and clathrin (1∶250) using either Alexa-488 or -546 conjugated secondary antibodies; (B) GFP-DN-Eps-15 or (C) GFP-Eps-15 and Flag-CysLT1R transfected cells, stimulated, or not, with 80 nM LTD4 and stained with Flag antibody. The mounted slides were examined using a Nikon TE300 microscope (60× or 100×1.4 plan-apochromat oil immersion objective). The scale bar represents 10 µm.
Int 407 cells and Caco-2 cells were transfected with GFP-DN-Eps-15 and Flag-tagged CysLT1R, and stimulated, or not, with LTD4. The Flag-tagged CysLT1R was not internalized upon stimulation in cells over expressing the DN-Eps-15 construct (Fig. 5B). Conversely, in cells co-transfected with the GFP-Eps-15 construct and the Flag-CysLT1R receptor, internalization of the receptor upon LTD4 stimulation was observed (Fig. 5C). This suggests that the CysLT1R is internalized in a clathrin-dependent manner in both cell lines.
CysLT1R Internalizes in a Rab-5- and Arrestin-3 -dependent Manner
The initial step after receptor internalization is the transfer of the receptors into early endosomes. Trafficking of early endosomes and clathrin-coated vesicles is often regulated by the GTPase Rab-5. Therefore, we next examined the role of Rab-5 in the CysLT1R internalization. Cells were co-transfected with GFP-Rab-5 and the Flag-CysLT1R. In unstimulated cells, the CysLT1R was localized at the plasma membrane (Fig. 6A, B). Upon stimulation with LTD4, Rab-5 positive vesicles were formed in Int 407 and Caco-2 cells and the CysLT1R co-localized in these vesicles (Fig. 6A, B).
Figure 6. Co-localization of CysLT1R and Rab-5 protein in Int 407 and Caco-2 cells and arrestin-3-dependent internalization of the CysLT1R.

Fluorescent microscope images showing cells that were fixed, permeabilized, and stained with primary antibodies against Flag (1∶2500) using Alexa-546 conjugated secondary antibodies, Flag-CysLT1R, and GFP-Rab-5 in Int 407 cells (A) and Caco-2 cells (B). Cells were grown on cover slips to 50-60% confluency, transfected with Flag-CysLT1R and GFP-Rab-5, left to rest for 48 hours, and treated with or without 80 nM LTD4. The mounted slides were examined using a Nikon TE300 microscope (60× or 100×1.4 plan-apochromat oil immersion objective). (C, D) Cells were transfected, or not, with siRNA against arrestin-3 or scrambled siRNA, serum-starved, and stimulated, or not, with LTD4 (80 nM, 5 minutes). For FACS analysis, Int 407 cells (1×106 cells) were either first fixed and permabilized before intracellular staining for arrestin-3 or used directly for CysLT1R cell surface staining. Moreover, whole lysates or plasma membrane fractions were made and subjected to SDS-polyacrylamide gel electrophoresis and analyzed for arrestin-3 or CysLT1R protein expression using Western blot analysis. All membranes were re-probed for actin to ensure equal loading. The blots are representative of three separate experiments. The scale bar represents 10 µm.
GPCR internalization and desensitization is either arrestin-dependent or independent. We, therefore, proceeded to down-regulate arrestin-3 using siRNA. Treatment with siRNA resulted in an approximate 50% reduction of arrestin-3 protein expression, as demonstrated by FACS and Western blot (Fig. 6C). This reduction of arrestin-3 protein expression significantly impaired the LTD4-induced internalization of the CysLT1R (Fig. 6D). This data suggests that the CysLT1R is internalized in an arrestin-3-dependent manner in intestinal epithelial cells.
CysLT1R Increases at the Nuclear Membrane upon LTD4 Stimulation
As previously mentioned, it has been shown that the CysLT1R is also localized at the nuclear membrane [12] and that the nuclear localization is increased in colorectal adenocarcinomas and facilitates survival and proliferation [12], [47]. We, therefore, investigated if the receptor at the nuclear membrane was affected by ligand stimulation. Western blot analysis of nuclear fractions of Int 407 and Caco-2 cells demonstrated that the CysLT1R was already significantly up-regulated after 15 minutes of LTD4 stimulation in Int 407 cells (Fig. 7A), after 10 minutes in Caco-2 cells (Fig. 8A), and continues to increase up to 1 hour after stimulation in both cell lines (Figs. 7A, 8A). In order to investigate if this accumulation is due to translocation of CysLT1R from the plasma membrane, cells were pre-treated with an inhibitor of clathrin-coated pit formation, sucrose, and as a control, we also used the caveolae inhibitor Filipin. The cells were, thereafter, fractioned into plasma and nuclear membranes, and stained with the CysLT1R antibody. As shown in Figure 7B, receptor internalization in Int 407 cells was blocked by the clathrin inhibitor, sucrose, but not by the caveolae inhibitor Filipin. Similarly, the increase in the nuclear fraction from the same experiment was blocked by sucrose, but not by Filipin (Fig. 7C). In Caco-2 cells, inhibiting clathrin with sucrose and stimulating the cells with LTD4 led to an increase of the CysLT1R at the plasma membrane (Fig. 8B) and, as a consequence, also inhibited the LTD4-induced increase at the nuclear membrane (Fig. 8C). The increase at the plasma membrane of Caco-2 cells suggests that LTD4 stimulation signals the recruitment of the CysLT1R to the plasma membrane and, when receptor internalization is blocked, it leads to a net increase of the receptor at the plasma membrane. In Int 407 cells, however, inhibiting clathrin with sucrose led to blocking receptor internalization and nuclear increase, but did not lead to an increase at the plasma membrane.
Figure 7. Regulation and function of CysLT1R at the plasma and nuclear membrane in Int 407 cells.

Int 407 cells were grown to 80% confluency, serum-starved for 2 hours, stimulated, or not, with 80 nM LTD4, lysed, fractioned into plasma and nuclear membranes, subjected to SDS-polyacrylamide gel electrophoresis, and stained for the CysLT1R by Western blot. (B–D) Cells were pre-treated with or without sucrose, Filipin, or cycloheximide, stimulated, or not, with 80 nM LTD4 for 5 minutes or as indicated, lysed, fractioned into the plasma membrane (B) and nucleus (A, C), or whole cell lysate (D) and subjected to gel electrophoresis. The PDVF membranes were then stained with the CysLT1R antibody (1∶1000) and re-probed for actin (1∶2000) or lamin B (1∶1000) to ensure equal loading. The data are given as percent of control and represent means ± S.E.M. of at least three separate experiments. The statistical analysis was performed with a Student's t test. *P<0.05 and ** P<0.01.
Figure 8. Regulation of CysLT1R at the plasma and nuclear membrane in Caco-2 cells.

Caco-2 cells were grown to 80% confluency, serum-starved for 2 hours, stimulated, or not, with 80 nM LTD4, lysed, fractioned into plasma and nuclear membranes, subjected to SDS-polyacrylamide gel electrophoresis, and stained for the CysLT1R by Western blot. Cells were pre-treated with or without sucrose (B, C), Filipin (C), or cyclohexamide (1 hour) (C, D), stimulated, or not, with 80 nM LTD4 for 5 minutes or as indicated, lysed, fractioned into the plasma membrane (B) and nucleus (A, C, D), and subjected to gel electrophoresis. The PDVF membranes were then stained with the CysLT1R antibody (1∶1000) and re-probed for actin (1∶2000) or lamin B (1∶1000) to ensure equal loading. The data are given as percent of control and represent means ± S.E.M. of at least three separate experiments. The statistical analysis was performed with a Student's t test. *P<0.05 and ** P<0.01.
Our data demonstrates that both the internalization from the plasma membrane and the accumulation of the CysLT1R at the nucleus are clathrin-dependent, thus indicating that the receptor is translocating. We also explored other possibilities leading to the nuclear accumulation of the CysLT1R. We, therefore, investigated if the increase of the CysLT1R at the nuclear membrane could be due to de novo synthesis. However, cycloheximide (an inhibitor of protein synthesis) does not affect the increase at the nuclear membrane, suggesting that the accumulation of the CysLT1R is not due to new synthesis of the receptors (Figs. 7C, 8C). Furthermore, stimulation with LTD4 up to 1 hour did not increase CysLT1R expression levels in whole cell lysates (data not shown), further supporting the idea that the nuclear accumulation of CysLT1R is not due to new synthesis.
We have previously identified that increased expression of the CysLT1R in colon cancer patients correlates with a poorer prognosis [13]. Increased levels of CysLT1R in colon cancer cells can originate from a slower degradation of the receptor in cancer cells compared to non-transformed cells. Therefore, we next investigated the degradation of the CysLT1R. Cells were pre-incubated with cycloheximide and stimulated with LTD4 for various time points. We found a slight decrease of the CysLT1R after 6 hours of stimulation with LTD4 in Int 407 cells (Fig. 7D) and after 9 hours of stimulation in Caco-2 cells (Fig. 8D). We found it unrealistic that this small difference in receptor level could explain the increased expression level seen in colon cancer cells.
We have previously demonstrated that LTD4 via the CysLT1R induces Erk1/2 phosphorylation [9]. We have now shown that blocking the internalization of the CysLT1R does not reduce this phosphorylation but, instead, a slight increase of the signal is detected (Fig. 9A). Arrestins are also involved in Erk1/2 signaling downstream of GPCRs [48]. We, therefore, investigated the potential functional effect of arrestin-3 knockdown on CysLT1R signaling. Here we demonstrated that down-regulating arrestin-3 decreases LTD4-induced Erk1/2 phosphorylation (Fig. 9B). We next investigated the potential functional effect of clathrin inhibition by sucrose on CysLT1R signaling. We stimulated cells with LTD4 with, or without, sucrose and investigated the effect on one of the target genes for CysLT1R, COX-2. We found that sucrose decreases the LTD4-induced mRNA level of COX-2 (Fig. 9C); this data suggests that in contrast to Erk1/2 phosphorylation, the internalization of the receptor is important for activation of the COX-2 gene.
Figure 9. Effect of sucrose and arrestin-3 on CysLT1R signaling.
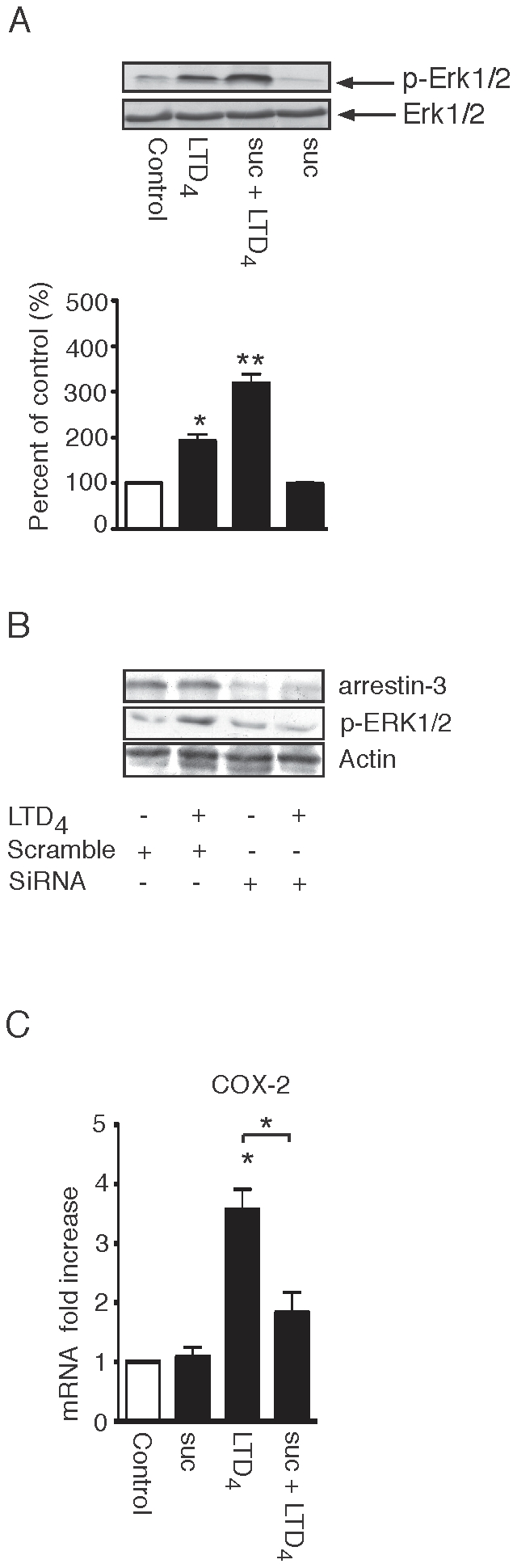
Int 407 cells were grown to 80% confluency, serum-starved for 2 hours, pre-treated, or not, with (A) sucrose for 1 hour or (B) siRNA (arrestin-3 or scrambled) and with or without 40 nM LTD4 stimulation. Cell lysates were prepared as described in Methods and samples were subjected to SDS-polyacrylamide gel electrophoresis and Western blot analysis. The PDVF membranes were stained with the phospho-Erk1/2, total Erk1/2 (1∶1000), or arrestin-3 and actin (1∶2000) antibodies. All membranes were re-probed for actin to ensure equal loading. (C) shows Q-PCR of COX-2 mRNA from Int 407 cells pre-treated, or not, with sucrose and stimulated, or not, with LTD4 (80 nM, 1 hour). The RT-PCR was performed as described in Methods, using primers for COX-2. The data are given as fold increase compared to control and represent means ± S.E.M. The statistical analysis was performed with a Student's t test. *P<0.05 and ** P<0.01.
Discussion
GPCRs have been extremely successful drug targets for a multitude of diseases [49], [50] as with the CysLT1R antagonist, Montelukast, which is currently used as a treatment for asthma [51]. The assembly of GPCRs as homo- and hetero-oligomers and their phosphorylation and association with a vast array of trafficking and signal-modulating proteins are emerging as major mechanisms underlying the functioning of GPCRs. It has become increasingly evident that GPCR signaling, expression, localization, and trafficking often play a role in disease development and progression [52]. One example is retinitis pigmentosa, which results from improper intracellular trafficking and localization of the rhodopsin receptors [19], [20]. Furthermore, previous studies by our group show that increased nuclear expression of the CysLT1R correlates with a poorer prognosis for colon cancer patients [12], [13]. Here, we wanted to investigate the trafficking of the CysLT1R, which is a major regulatory mechanism of GPCR signaling. Previous studies have shown that LTD4 binds the CysLT1R with a higher affinity than LTC4 [3]. In this study, we conclude that LTC4 and LTD4 affect the trafficking of CysLT1R differently, suggesting ligand-specific signaling. We also demonstrate that LTD4 mainly internalizes the CysLT1R, supporting our previous findings that LTD4-induced cell survival, cell proliferation, and cell migration are mediated through the CysLT1R. This is further supported by the fact that LTD4 stimulation decreases the dimerization observed between the CysLT1R and CysLT2R, as demonstrated by the in situ proximity ligation assay and immunoprecipitation data. It is interesting to note that after 60 minutes of stimulation with LTD4, the amount of heterodimers observed is concentrated in the nuclear region, supporting the results of nuclear accumulation of the CysLT1R. The LTC4, on the other hand, does not lead to a nuclear accumulation of either receptor, but induces internalization of both CysLT1 and CysLT2 receptors, which might be due to the preserving of the receptor dimers. Another interesting observation was the effect of the ligand-induced tyrosine phosphorylation of CysLT1R. LTD4 induced tyrosine phosphorylation of the CysLT1R, but not of the CysLT2R, which clearly shows the specificity of the ligand-induced signaling. This correlates well with the fact that CysLT1R is the high affinity receptor and affects cell proliferation, survival, and cell migration, whereas CysLT2R does not [8], [9]. Moreover, these results also support our previous findings that inhibition of the CysLT1R leads to growth inhibition and cell death [47]. The CysLT2R has been shown to be a negative regulator of the mitogenic effect of the CysLT1R upon LTD4 stimulation in mast cells [53].
We found that in Int 407 cells, the internalization of the CysLT1R could be detected after 5 minutes of stimulation, which could be blocked by a specific CysLT1R antagonist. However, in Caco-2 cells, the endogenous internalization was more difficult to detect. This may be due to a high turnover of the receptor at the plasma membrane upon stimulation. This hypothesis is supported by the fact that internalization blocking experiments lead to an accumulation of the receptor at the plasma membrane, which cannot be seen in Int 407 cells. However, using FACS analysis with the Caco-2 cells, we could detect a small but significant internalization of the endogenous receptors after LTD4 stimulation, supporting that there is an internalization of the receptor.
We next investigated how the CysLT1R is internalized. We found that CysLT1 is internalized in a clathrin/Rab-5-dependent pathway; we also suggest that this internalization is arrestin-3-dependent. Previous publication studying CysLT1R internalization in other cells has demonstrated that this process is arrestin-3 independent [16]. In that study, dominant negative (DN)-arrestin-3 constructs were used in HEK-293 cells. The results demonstrated that when arrestin-3 was over expressed, receptor internalization was increased. However, over expression with a dominant negative construct lead to a non-significant decrease of receptor internalization. These results were further supported by experiments in MEF cells from arrestin-3 deficient mice [16]. To conclude, whether CysLT1R required arrestin to internalize or not, we used siRNA against endogenous arrestin-3 and demonstrated that the loss of the receptor from the plasma membrane is inhibited. Our results show that CysLT1R requires arrestin-3 for internalization in epithelial cells. Furthermore, GPCRs can activate Erk1/2 in an arrestin-dependent manner [48]. As shown in our results, down-regulation of arrestin-3 disrupts LTD4-induced Erk1/2 phosphorylation. In summary, these data demonstrate that the CysLT1R is internalized from the plasma membrane in a clathrin-, Rab-5-, and arrestin-3-dependent manner and that inhibition of arrestin-3 also affects the signaling downstream of the CysLT1R.
As previously shown, the CysLT1R is also localized at the nuclear membrane. We demonstrated here that the increase of the receptor at the nuclear membrane coincides with the loss from the plasma membrane. Both the accumulation and the cell surface loss of the CysLT1R are clathrin-dependent. The accumulation of the CysLT1R at the nucleus is not due to new synthesis of the receptor, although there is a possibility of the existence of an internal pool of the receptor that could be responsible for the accumulation. In fact, an internal pool could be one possibility of receptor recruitment to the plasma membrane and a high turnover of CysLT1R upon LTD4 stimulation in Caco-2 cells. In order to investigate the role of the nuclear accumulation of the CysLT1R, we investigated the signaling of the receptor. By inhibiting the formation of clathrin-coated pits and, thereby, inhibiting receptor accumulation at the nuclear membrane, LTD4-induced COX-2 mRNA up-regulation was decreased. This suggests that the CysLT1R accumulation at the nucleus, or its internalization, is required for certain signaling pathways.
Taken together, our results show how the receptor is trafficking from the plasma membrane to the nucleus and demonstrates different regulation of CysLT1R signaling.
Supporting Information
Co-Immunoprecipitation of the CysLTRs in colon cancer cells. Briefly HCT-116 cells were grown to 80% confluency and lysed. Lysates containing 1 mg/ml protein were incubated with rabbit anti-CysLT2R antibody, after which 20 µg of protein A plus agarose was added. The beads then were washed three times mixed with sample buffer, boiled and centrifuged. The proteins were then separated on SDS-polyacrylamide gels. The separated proteins were electrophoretically transferred to a polyvinylidene difluoride (PDVF) membrane incubated with a primary antibody against CysLT1R overnight. Thereafter the membrane was exposed to hyperfilm-ECL to visualize immunoreactive proteins. The membrane was then re-probed with a CysLT2R antibody. The blots shown are representative and the data are given as percent of control and represents means ± S.E.M. of three separate experiments and the statistical analysis were performed with Student's t test. *P<0.05.
(0.36 MB TIF)
The internalization of CysLT1R and CysLT2R after LTC4 stimulation. Int 407 cells were grown to 80% confluency and then treated with or without 40 nM LTC4 for indicated periods of time. Plasma membrane fractions were prepared as described in Materials and Methods and samples were subjected to SDS-polyacrylamide gel electrophoresis and Western blot analysis. The PDVF membranes were stained with CysLT1R, CysLT2R (both 1∶1000) or actin (1∶2000) antibodies. The blots shown are representative and the data are given as percent of control and represents means ± S.E.M. of three separate experiments and the statistical analysis were performed with Student's t test. *P<0.05 and ** P<0.01.
(0.08 MB TIF)
Acknowledgments
The GFP-Rab-5 was a kind gift from Dr J. Ivaska, VTT Biotechnology Centre, Turku, Finland. We thank M. Juhas, L. Olsson and E. Heathcote for expert technical assistance.
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work was supported by grants to AS by the Swedish Cancer Foundation, the Swedish Medical Research Council, the Foundations at Malmö University Hospital, the Ruth and Richard Julin Foundation, Gunnar Nilsson's Cancer Foundation, and the Österlund Foundation and to LP and WS from the Royal Physiographic Society in Lund. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Stenson WF. Role of eicosanoids as mediators of inflammation in inflammatory bowel disease. Scand J Gastroenterol. 1990;(Suppl 172):13–18. doi: 10.3109/00365529009091903. [DOI] [PubMed] [Google Scholar]
- 2.Ekbom A, Helmick C, Zack M, Adami HO. Ulcerative colitis and colorectal cancer. A population-based study. N Engl J Med. 1990;323:1228–1233. doi: 10.1056/NEJM199011013231802. [DOI] [PubMed] [Google Scholar]
- 3.Lynch KR, O'Neill GP, Liu Q, Im DS, Sawyer N, et al. Characterization of the human cysteinyl leukotriene CysLT1 receptor. Nature. 1999;399:789–793. doi: 10.1038/21658. [DOI] [PubMed] [Google Scholar]
- 4.Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 5.Serhan CN, Haeggström JZ, Leslie CC. Lipid mediator networks in cell signaling: update and impact of cytokines. FASEB J. 1996;10:1147–1158. doi: 10.1096/fasebj.10.10.8751717. [DOI] [PubMed] [Google Scholar]
- 6.Mezhybovska M, Wikstrom K, Öhd JF, Sjölander A. The inflammatory mediator leukotriene D4 induces beta-catenin signaling and its association with antiapoptotic Bcl-2 in intestinal epithelial cells. J Biol Chem. 2006;281:6776–6784. doi: 10.1074/jbc.M509999200. [DOI] [PubMed] [Google Scholar]
- 7.Wikström K, Öhd JF, Sjölander A. Regulation of leukotriene-dependent induction of cyclooxygenase-2 and Bcl-2. Biochem Biophys Res Com. 2003;302:330–335. doi: 10.1016/s0006-291x(03)00187-6. [DOI] [PubMed] [Google Scholar]
- 8.Öhd JF, Wikström K, Sjölander A. Leukotrienes induce cell-survival signaling in intestinal epithelial cells. Gastroenterology. 2000;119:1007–1018. doi: 10.1053/gast.2000.18141. [DOI] [PubMed] [Google Scholar]
- 9.Paruchuri S, Hallberg B, Juhas M, Larsson C, Sjölander A. Leukotriene D(4) activates MAPK through a Ras-independent but PKCepsilon-dependent pathway in intestinal epithelial cells. J Cell Sci. 2002;115:1883–1893. doi: 10.1242/jcs.115.9.1883. [DOI] [PubMed] [Google Scholar]
- 10.Paruchuri S, Sjölander A. Leukotriene D4 mediates survival and proliferation via separate but parallel pathways in the human intestinal Epithelial cell line Int 407. J Biol Chem. 2003;278:45577–45585. doi: 10.1074/jbc.M302881200. [DOI] [PubMed] [Google Scholar]
- 11.Paruchuri S, Broom O, Dib K, Sjölander A. The pro-inflammatory mediator leukotriene D4 induces phosphatidylinositol 3-kinase and Rac-dependent migration of intestinal epithelial cells. J Biol Chem. 2005;280:13538–13544. doi: 10.1074/jbc.M409811200. [DOI] [PubMed] [Google Scholar]
- 12.Nielsen Kamp C, Campbell J, Öhd JF, Mörgelin M, Riesbeck K, et al. A novel localization of the G-protein-coupled CysLT1 receptor in the nucleus of colorectal adenocarcinoma cells. Cancer Res. 2005;65:732–742. [PubMed] [Google Scholar]
- 13.Öhd JF, Nielsen CK, Campbell J, Landberg G, Löfberg H, et al. Expression of the leukotriene D4 receptor CysLT1, COX-2, and other cell survival factors in colorectal adenocarcinomas. Gastroenterology. 2003;124:57–70. doi: 10.1053/gast.2003.50011. [DOI] [PubMed] [Google Scholar]
- 14.Magnusson C, Mezhybovska M, Lorinc E, Fernebro E, Nilbert M, et al. Low expression of CysLT1R and high expression of CysLT2R mediate good prognosis in colorectal cancer. Eur J Cancer. 2010;46:826–835. doi: 10.1016/j.ejca.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 15.Magnusson C, Ehrnström R, Olsen J, Sjölander A. An increased expression of cysteinyl leukotriene 2 receptor in colorectal adenocarcinomas correlates with high differentiation. Cancer Res. 2007;67:9190–9198. doi: 10.1158/0008-5472.CAN-07-0771. [DOI] [PubMed] [Google Scholar]
- 16.Naik S, Billington CK, Pascual RM, Deshpande DA, Stefano FP, et al. Regulation of cysteinyl leukotriene type 1 receptor internalization and signaling. J Biol Chem. 2005;280:8722–8732. doi: 10.1074/jbc.M413014200. [DOI] [PubMed] [Google Scholar]
- 17.Capra V, Ravasi S, Accomazzo MR, Citro S, Grimoldi M, et al. CysLT1 receptor is a target for extracellular nucleotide-induced heterologous desensitization: a possible feedback mechanism in inflammation. J Cell Sci. 2005;118:5625–5636. doi: 10.1242/jcs.02668. [DOI] [PubMed] [Google Scholar]
- 18.Deshpande DA, Pascual RM, Wang SW, Eckman DM, Riemer EC, et al. PKC-dependent regulation of the receptor locus dominates functional consequences of cysteinyl leukotriene type 1 receptor activation. FASEB J. 2007;21:2335–2342. doi: 10.1096/fj.06-8060com. [DOI] [PubMed] [Google Scholar]
- 19.Edwards SW, Tan CM, Limbird LE. Localization of G-protein-coupled receptors in health and disease. Trends Pharmacol Sci. 2000;21:304–308. doi: 10.1016/s0165-6147(00)01513-3. [DOI] [PubMed] [Google Scholar]
- 20.Cahill CM, Holdridge SV, Morinville A. Trafficking of delta-opioid receptors and other G-protein-coupled receptors: implications for pain and analgesia. Trends Pharmacol Sci. 2007;28:23–31. doi: 10.1016/j.tips.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Millar RP, Newton CL. The year in G protein-coupled receptor research. Mol Endocrinol. 2009;24:261–274. doi: 10.1210/me.2009-0473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bai M. Dimerization of G-protein-coupled receptors: roles in signal transduction. Cell Signal. 2004;16:175–186. doi: 10.1016/s0898-6568(03)00128-1. [DOI] [PubMed] [Google Scholar]
- 23.Milligan G. G protein-coupled receptor dimerization: function and ligand pharmacology. Mol Pharmacol. 2004;66:1–7. doi: 10.1124/mol.104.000497.. [DOI] [PubMed] [Google Scholar]
- 24.Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- 25.Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- 26.Wolfe BL, Trejo J. Clathrin-dependent mechanisms of G protein-coupled receptor endocytosis. Traffic. 2007;8:462–470. doi: 10.1111/j.1600-0854.2007.00551.x. [DOI] [PubMed] [Google Scholar]
- 27.García Lopez MA AMA, Lamaze C, Martínez-A C, Fischer T. Inhibition of dynamin prevents CCL2-mediated endocytosis of CCR2 and activation of ERK1/2. Cell Signal. 2009;21:1748–1757. doi: 10.1016/j.cellsig.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 28.Chen Z, Gaudreau R, Le Gouill C, Rola-Pleszczynski M, Stankova J. Agonist-induced internalization of leukotriene B(4) receptor 1 requires G-protein-coupled receptor kinase 2 but not arrestins. Mol Pharmacol. 2004;66:377–386. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 29.Seachrist JL, Ferguson SS. Regulation of G protein-coupled receptor endocytosis and trafficking by Rab GTPases. Life Sci. 2003;74:225–235. doi: 10.1016/j.lfs.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 30.Sonnichsen B, De Renzis S, Nielsen E, Rietdorf J, Zerial M. Distinct membrane domains on endosomes in the recycling pathway visualized by multicolor imaging of Rab4, Rab5, and Rab11. J Cell Biol. 2000;149:901–914. doi: 10.1083/jcb.149.4.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwartz SL, Cao C, Pylypenko O, Rak A, Wandinger-Ness A. Rab GTPases at a glance. J Cell Sci. 2007;120:3905–3910. doi: 10.1242/jcs.015909. [DOI] [PubMed] [Google Scholar]
- 32.Marchese A, Paing MM, Temple BR, Trejo J. G protein-coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol. 2008;48:601–629. doi: 10.1146/annurev.pharmtox.48.113006.094646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goetzl EJ. Diverse pathways for nuclear signaling by G protein-coupled receptors and their ligands. FASEB J. 2007;21:638–642. doi: 10.1096/fj.06-6624hyp. [DOI] [PubMed] [Google Scholar]
- 34.Zhu T, Gobeil F, Vazquez-Tello A, Leduc M, Rihakova L, et al. Intracrine signaling through lipid mediators and their cognate nuclear G-protein-coupled receptors: a paradigm based on PGE2, PAF, and LPA1 receptors. Can J Phys Pharmacol. 2006;84:377–391. doi: 10.1139/y05-147. [DOI] [PubMed] [Google Scholar]
- 35.Gobeil F, Jr, Dumont I, Marrache AM, Vazquez-Tello A, Bernier SG, et al. Regulation of eNOS expression in brain endothelial cells by perinuclear EP(3) receptors. Circ Res. 2002;90:682–689. doi: 10.1161/01.res.0000013303.17964.7a. [DOI] [PubMed] [Google Scholar]
- 36.Shmuel M, Nodel-Berner E, Hyman T, Rouvinski A, Altschuler Y. Caveolin 2 regulates endocytosis and trafficking of the M1 muscarinic receptor in MDCK epithelial cells. Mol Biol Cell. 2007;18:1570–1585. doi: 10.1091/mbc.E06-07-0618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Syme CA, Zhang L, Bisello A. Caveolin-1 regulates cellular trafficking and function of the glucagon-like Peptide 1 receptor. Mol Endocrinol. 2006;20:3400–3411. doi: 10.1210/me.2006-0178. [DOI] [PubMed] [Google Scholar]
- 38.Insel PA, Head BP, Ostrom RS, Patel HH, Swaney JS, et al. Caveolae and lipid rafts: G protein-coupled receptor signaling microdomains in cardiac myocytes. Ann N Y Acad Sci. 2005;1047:166–172. doi: 10.1196/annals.1341.015. [DOI] [PubMed] [Google Scholar]
- 39.Ostrom RS, Insel PA. The evolving role of lipid rafts and caveolae in G protein-coupled receptor signaling: implications for molecular pharmacology. Br J Pharmacol. 2004;143:235–245. doi: 10.1038/sj.bjp.0705930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henle G, Deinhardt F. The establishment of strains of human cells in tissue culture. J Immunol. 1957;79:54–59. [PubMed] [Google Scholar]
- 41.Parhamifar L, Jeppsson B, Sjölander A. Activation of cPLA2 is required for leukotriene D4-induced proliferation in colon cancer cells. Carcinogenesis. 2005;26:1988–1998. doi: 10.1093/carcin/bgi159. [DOI] [PubMed] [Google Scholar]
- 42.Söderberg O, Leuchowius KJ, Gullberg M, Jarvius M, Weibrecht I, et al. Characterizing proteins and their interactions in cells and tissues using the in situ proximity ligation assay. Methods. 2008;45:227–232. doi: 10.1016/j.ymeth.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 43.Carlemalm E, Villiger W, Hobot JA, Acetarin JD, Kellenberger E. Low temperature embedding with Lowicryl resins: two new formulations and some applications. J Microsc. 1985;140:55–63. doi: 10.1111/j.1365-2818.1985.tb02660.x. [DOI] [PubMed] [Google Scholar]
- 44.Baschong W, Lucocq JM, Roth J. “Thiocyanate gold”: small (2-3 nm) colloidal gold for affinity cytochemical labeling in electron microscopy. Histochemistry. 1985;83:409–411. doi: 10.1007/BF00509201. [DOI] [PubMed] [Google Scholar]
- 45.Jiang Y, Borrelli LA, Kanaoka Y, Bacskai BJ, Boyce JA. CysLT2 receptors interact with CysLT1 receptors and down-modulate cysteinyl leukotriene dependent mitogenic responses of mast cells. Blood. 2007;110:3263–3270. doi: 10.1182/blood-2007-07-100453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benmerah A, Poupon V, Cerf-Bensussan N, Dautry-Varsat A. Mapping of Eps15 domains involved in its targeting to clathrin-coated pits. J Biol Chem. 2000;275:3288–3295. doi: 10.1074/jbc.275.5.3288. [DOI] [PubMed] [Google Scholar]
- 47.Paruchuri S, Mezhybovska M, Juhas M, Sjölander A. Endogenous production of leukotriene D4 mediates autocrine survival and proliferation via CysLT1 receptor signalling in intestinal epithelial cells. Oncogene. 2006;25:6660–6665. doi: 10.1038/sj.onc.1209666. [DOI] [PubMed] [Google Scholar]
- 48.Tohgo A, Choy EW, Gesty-Palmer D, Pierce KL, Laporte S, et al. The stability of the G protein-coupled receptor-beta-arrestin interaction determines the mechanism and functional consequence of ERK activation. J Biol Chem. 2003;278:6258–6267. doi: 10.1074/jbc.M212231200. [DOI] [PubMed] [Google Scholar]
- 49.Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1:727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- 50.Drews J. Drug discovery: a historical perspective. Science. 2000;287:1960–1964. doi: 10.1126/science.287.5460.1960. [DOI] [PubMed] [Google Scholar]
- 51.Joos GF. Which patients with asthma could benefit from using anti-leukotriene drugs: an evidence based review. Pol Arch Med Wewn. 2008;118:689–690. [PubMed] [Google Scholar]
- 52.Thompson MD, Siminovitch KA, Cole DE. G protein-coupled receptor pharmacogenetics. Methods Mol Biol. 2008;448:139–185. doi: 10.1007/978-1-59745-205-2_8. [DOI] [PubMed] [Google Scholar]
- 53.Paruchuri S, Jiang Y, Feng C, Francis SA, Plutzky J, et al. Leukotriene E4 activates peroxisome proliferator-activated receptor gamma and induces prostaglandin D2 generation by human mast cells. J Biol Chem. 2008;283:16477–16487. doi: 10.1074/jbc.M705822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Co-Immunoprecipitation of the CysLTRs in colon cancer cells. Briefly HCT-116 cells were grown to 80% confluency and lysed. Lysates containing 1 mg/ml protein were incubated with rabbit anti-CysLT2R antibody, after which 20 µg of protein A plus agarose was added. The beads then were washed three times mixed with sample buffer, boiled and centrifuged. The proteins were then separated on SDS-polyacrylamide gels. The separated proteins were electrophoretically transferred to a polyvinylidene difluoride (PDVF) membrane incubated with a primary antibody against CysLT1R overnight. Thereafter the membrane was exposed to hyperfilm-ECL to visualize immunoreactive proteins. The membrane was then re-probed with a CysLT2R antibody. The blots shown are representative and the data are given as percent of control and represents means ± S.E.M. of three separate experiments and the statistical analysis were performed with Student's t test. *P<0.05.
(0.36 MB TIF)
The internalization of CysLT1R and CysLT2R after LTC4 stimulation. Int 407 cells were grown to 80% confluency and then treated with or without 40 nM LTC4 for indicated periods of time. Plasma membrane fractions were prepared as described in Materials and Methods and samples were subjected to SDS-polyacrylamide gel electrophoresis and Western blot analysis. The PDVF membranes were stained with CysLT1R, CysLT2R (both 1∶1000) or actin (1∶2000) antibodies. The blots shown are representative and the data are given as percent of control and represents means ± S.E.M. of three separate experiments and the statistical analysis were performed with Student's t test. *P<0.05 and ** P<0.01.
(0.08 MB TIF)