

In a recent commentary on gene × environment (G×E) interactions in Molecular Psychiatry, Belsky et al (2009) argue that many G×E findings are misinterpreted through adherence to a dominant model of disease vulnerability that assumes an ordinal interaction between specific genetic variation and the presence/absence of environmental adversity (diathesis-stress). They cite numerous studies of people with putative “risk” alleles of certain monoamine-regulating polymorphisms who experience psychiatric outcomes (e.g., depression) more frequently in adverse circumstances, yet less frequently in salutary environments, compared to those with other genotypes. Belsky et al suggest this cross-over (or disordinal) interaction reflects “differential susceptibility” to environmental influences (heightened plasticity) among individuals possessing these alleles (“differential” denoting the potential for both worse and better outcomes), rather than genetic vulnerability to outcomes that are specifically negative and expressed only in adversity. Differential susceptibility has distinct parallels in the “reaction norm,” a concept introduced by Woltereck in 1909 and a staple of experimental research in biology and evolutionary genetics. Here, I suggest that framing differential susceptibility within a reaction norm perspective clarifies the role of phenotypic plasticity in G×E interaction.
A reaction norm (RN) refers to the spectrum of phenotypic variation produced when individuals of the same genotype are exposed to varying environmental conditions. Hypothetical RNs in Figure 1 plot variation in a model phenotype as a function of an unspecified environmental factor. Each line depicts the RN of a single genotype. Genotype “a” is highly plastic, since the gradient of environmental variation maps to a broad range of phenotype values. Genotypes “b” and “c” have shallow RNs, yielding narrow distributions of the phenotype over the same environmental gradient. Because “b” and “c” have identical slopes, the large difference in average phenotype between them reflects a genetic main effect (G). G×E is present instead whenever the phenotypic response to like environmental variation differs between genotypes (i.e., the slopes of corresponding RNs differ). Here, G×E exists in the relation of Genotype “a” with both “b” (disordinal G×E) and “c” (ordinal G×E). If the phenotype were the likelihood of experiencing depression and the environmental factor a gradient of life stress, the interaction of “a” and “c” would comport with a diathesis-stress model and that of “a” and “b” with the differential susceptibility model.
Figure 1.
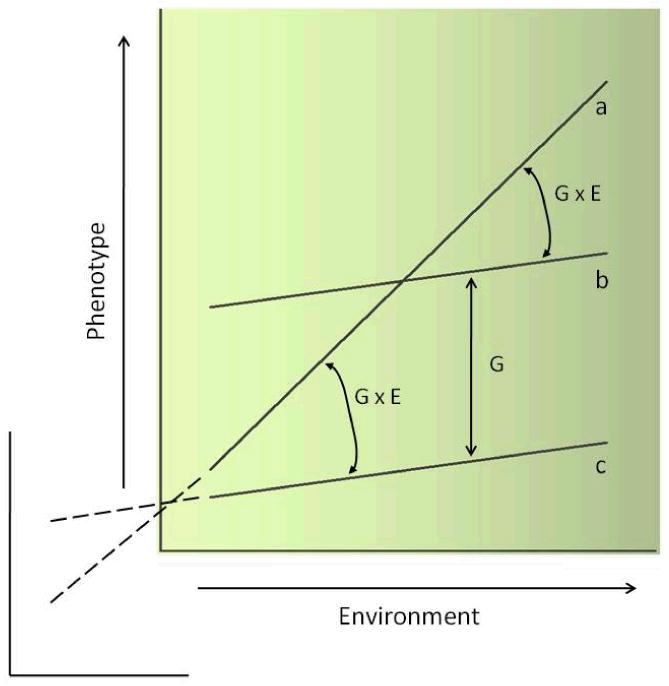
Reaction norms of three hypothetical genotypes: a, b, and c. G = genetic main effect; G×E = gene × environment interaction
How is phenotypic plasticity related to G×E interaction? First, plasticity (great or small) is a property of a genotype and therefore unrelated to the RNs of other genotypes. The plasticity of Genotype “a,” for example, is the same whether in interaction with “b” or “c.” Second, only the intersection of RNs distinguishes disordinal from ordinal G×E (not, for instance, a greater difference in relative plasticity between interacting genotypes). And third, the “for worse and better” argument of Belsky et al – the differential part of differential susceptibility – demands only that different genotypes produce the same phenotype at an intermediate location along an environmental gradient (i.e., disordinal G×E). As the authors anticipate, disordinal G×E may be common but elude detection if the measured phenotype and environment reflect only a portion of their natural ranges. If axes in the figure could be lengthened meaningfully (dashed lines), the RNs of Genotypes “a” and “c” would eventually cross. Thus, the generality of disordinal G×E rests importantly on understanding the natural ranges of environmental variation (e.g., whether positive parenting is coextensive dimensionally with negative parenting) and of behavioral phenotypes (e.g., whether positive and negative affect are ends of a bipolar continuum or independent).
Finally, the RN framework offers perspective on the “for worse and better” property of differential susceptibility and the related suggestion that selection sculpts adaptive variation in plasticity. Biology does not assign value to phenotypes, and selection only recognizes reproductive success. If study phenotypes mirror reproductive fitness, though, a non-trivial consequence of disordinal G×E (crossing RNs) is to preserve genotypic variation, since the relative fitness of competing genotypes differs across environments – sometimes favoring one genotype, sometimes the other. However, this theoretical possibility is plausible here only if the behavioral phenotypes of G×E studies also predict reproductive outcomes. If not, RNs and their varying slopes are more likely by-products of other regulatory processes. For the several genes cited by Belsky et al. a by-product explanation might seem especially likely since monoamine neurotransmitters contribute to the central control of so many biological and behavioral phenotypes (e.g., metabolic, cardiovascular, neuroendocrine, and autononomic activity, as well as sleep, appetite, locomotor and sexual activity, and affective and appetitive motivation). Whatever its implications for evolutionary speculation, though, the RN framework may help clarify ambiguities and frame new questions in G×E research, and perhaps most usefully, create bridges between the “new” genetics of behavior and longstanding, informative streams of biological science.
References
- Belsky J, Jonassaint C, Pluess M, Stanton M, Brummett B, Williams R. Vulnerability genes or plasticity genes? Mol Psychiatry. 2009;14:746–754. doi: 10.1038/mp.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]