

Abstract
The environmental pollutant 2,3,7,8-tetracholorodibenzo-p-dioxin (TCDD) is known to cause a wide variety of toxic effects, including hepatotoxicity, by way of the aryl hydrocarbon receptor (AHR). Although inducible expression of cytochrome P450 (CYP) 1A1 and CYP1A2 is associated with liver injury caused by high-dose TCDD, the specific role of the AHR-CYP1 cascade in hepatotoxicity remains unclear. We investigated the effects of AHR activation under conditions of cholestasis. We administered oral TCDD to mice at a dose that can effectively induce Cyp1 gene expression without overt liver toxicity and then ligated their bile ducts. TCDD pretreatment enhanced bile duct ligation (BDL)-induced increases in liver and plasma bile acids, bilirubin, and aminotransferases. Histology of TCDD-pretreated BDL mice revealed massive hepatic necrosis without any increase in number of apoptotic cells. Whereas induction of AHR-target genes by TCDD was observed similarly in sham-operated as well as in BDL mice, TCDD pretreatment of BDL mice altered the expression of hepatic genes involved in bile acid synthesis and transport. Increased plasma proinflammatory cytokines, tumor necrosis factor and interleukin-1β, in BDL mice were further elevated by TCDD pretreatment. Liver injury by TCDD plus BDL, such as increased plasma bile acids, bilirubin and aminotransferases, liver necrosis, and increased tumor necrosis factor production, was exaggerated in Cyp1a1/1a2(−/−) double knockout mice. These findings indicate that TCDD aggravates cholestatic liver damage and that the presence of CYP1A1 and CYP1A2 plays a protective role in liver damage caused by TCDD and BDL.
Keywords: Aryl hydrocarbon receptor, TCDD, Cholestasis, Hepatotoxicity, Tumor necrosis factor, Bile duct ligation
1. Introduction
The environmental pollutant 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) causes a wide spectrum of toxic effects, such as hepatotoxicity, immunosuppression, wasting, and carcinogenesis (reviewed in Bock and Kohle, 2006). Industrial and military exposures to TCDD have been linked to detrimental health effects. TCDD exposure activates the aryl hydrocarbon receptor (AHR), a transcription factor that contains basic helix-loop-helix (bHLH) and Per-Arnt-Sim (PAS) domains, and AHR requires heterodimerization with another bHLH-PAS protein, AHR nuclear translocator (ARNT), for transcriptional activation. The AHR-ARNT heterodimer induces expression of a group of genes called the [Ah] gene battery, including the cytochrome P450 genes (Cyp1a1, Cyp1a2 and Cyp1b1), which are involved in metabolism of foreign chemicals (reviewed in Nebert et al., 2000; Uno and Makishima, 2009). CYP1 enzymes also metabolize endogenous compounds, including eicosanoids and 6-formylindolo[3,2-b]carbazole-6-carboxylic acid, a tryptophan photoproduct (Nebert and Karp, 2008; Wincent et al., 2009). AHR activation by TCDD modulates expression of genes involved in cell proliferation and differentiation, a mechanism possibly leading to carcinogenesis and teratogenesis (Bock and Kohle, 2006). AHR activation also exerts nongenomic actions, such as activation of mitogen-activated protein kinase cascades and formation of a ubiquitin ligase complex (Fritsche et al., 2007; Ohtake et al., 2007). Studies using Ahr(−/−) knockout mice have demonstrated that AHR is essential for the induction of [Ah] gene battery members and most of the toxicological effects of TCDD (Fernandez-Salguero et al., 1996; Nebert et al., 2000).
Benzo[a]pyrene (BaP) is an AHR-activating polycyclic aromatic hydrocarbon found in industrial incineration products, cigarette smoke, and charcoal-grilled food and is implicated as a causative agent in lung and head-and-neck cancers, immunosuppression, and atherosclerosis by way of AHR-mediated metabolic activation (Uno and Makishima, 2009). BaP is converted to highly mutagenic and carcinogenic metabolites by xenobiotic-metabolizing enzymes, some of which are induced by AHR activation. The metabolic activation of BaP is likely due to the combined effects of CYP1 enzymes and other enzymes (Endo et al., 2008). In contrast to BaP, TCDD is virtually not metabolized in cells (Bock and Kohle, 2006). CYP1A2, an AHR target, is involved in TCDD accumulation in the liver as a TCDD-binding “sink” protein (Diliberto et al., 1997; Dragin et al., 2006). Although CYP1A2 does not play a role in the acute oxidative stress response following TCDD exposure (Slezak et al., 1999), AHR-dependent CYP1A2 and CYP1A1 induction contributes to uroporphyria, which is suggested as a mechanism for TCDD-induced liver toxicity (Smith et al., 2001; Uno et al., 2004b). On the other hand, mice deficient in CYP1A1 and CYP1A2 induction are more sensitive to acute TCDD hepatotoxicity (Nukaya et al., 2009). An understanding of the role of the AHR-CYP1 cascade in TCDD-induced liver toxicity still requires further investigation.
Cholestasis is associated with hepatic and systemic accumulation of toxic biliary compounds, such as bile acids and bilirubin, and subsequent liver damage (Zollner et al., 2006). Decreased secretion of bile acids into the intestine induces proliferation and translocation of intestinal bacteria, which can result in endotoxemia and sepsis (Inagaki et al., 2006). Endotoxin and proinflammatory cytokines, such as tumor necrosis factor (TNF), are implicated in endotoxin-induced cholestasis and exacerbate liver injury. Transcription factors of the nuclear receptor superfamily are known to modulate bile acid metabolism and pathogenesis of cholestasis (Zollner et al., 2006). The bile acid-sensing nuclear receptors, farnesoid X receptor (FXR; NR1H4) (supplementary Table 1 provides a list of all mouse genes and mRNA levels that were included in the present study), pregnane X receptor (PXR; NR1I2) and vitamin D receptor (VDR; NR1I1), have been investigated in the bile-duct ligation (BDL) model of cholestasis. FXR activation by synthetic ligands protects against cholestatic liver damage by decreasing expression of bile acid biosynthetic genes, such as sterol 12α-hydroxylase, and by increased expression of genes involved in bile acid transport, such as those participating in the bile salt export pump ATP-binding cassette transporter B11 (ABCB11; also called BSEP; encoded by the Abcb11 gene). FXR also plays a role in protection of the intestine from bacterial invasion (Inagaki et al., 2006). However, Fxr(−/−) knockout mice exhibit resistance to obstructive cholestasis (Stedman et al., 2006), which seems to contradict the prior finding. PXR agonists enhance bile acid detoxication by inducing an import transporter, the organic anion transporting polypeptide-1a4 (encoded by the Slco1a4 gene), the enzyme CYP3A11 (encoded by the Cyp3a11 gene), and a basolateral export transporter, multidrug resistance-associated protein-3 (encoded by the Abcc3 gene), resulting in decreased serum bile acids and increased urinary bile acid excretion (Wagner et al., 2005). The VDR ligand induces intestinal expression of mouse Cyp3a11 and human CYP3A4 (Matsubara et al., 2008) and enhances the metabolism of bile acids, particularly urinary excretion, by increasing the expression of bile acid transporter genes in mice (Nishida et al., 2009). VDR activation represses proinflammatory cytokine expression in BDL mice, although it does not alter bile acid accumulation (Ogura et al., 2009). Thus, the nuclear receptors modulate pathogenesis of cholestasis. AHR, which is a bHLH-PAS transcription factor and does not belong to the nuclear receptor superfamily, is involved in regulation of CYP gene expression and xenobiotic metabolism. However, the role of the AHR-CYP1 cascade during cholestasis has not been elucidated. In this study, we investigated the in vivo role of AHR activation and CYP1A induction during cholestasis using BDL mice.
2. Materials and methods
2.1. Animals and Treatment
C57BL/6J mice were obtained from Charles River Laboratories Japan (Yokohama, Japan) and were maintained under controlled temperature (23 ± 1 °C) and humidity (45–65 %) with free access to water and chow (Lab. Animal Diet MF, Oriental Yeast, Tokyo). Cyp1a1/1a2(−/−) double-knockout mice has been described (Dragin et al., 2007), and were backcrossed to C57BL/6J mice for 8 generations. Experiments were conducted using 7- to 8-week-old male mice. Five days prior to BDL, mice were orally administered TCDD (Wako Pure Chemicals, Osaka, Japan) dissolved in corn oil at a single dose of 15 μg/kg, while controls were received oral corn oil alone (Figure 1A). For BDL, mice were anesthetized with diethyl ether, and the common bile duct was ligated (Ogura et al., 2009). Sham surgery was performed under the same conditions with the exception of BDL. Plasma and liver were collected 3 days after surgery. Tissues were snap-frozen in liquid nitrogen or dry ice and stored until analysis. Plasma bilirubin, aspartate and alanine aminotransferases, alkaline phosphatase, γ-glutamyl transpeptidase, and plasma and liver total bile acids were quantified with Bilirubin BII-Testwako, Transaminase CII-Testwako, LabAssay ALP (Wako Pure Chemicals), IatroLG γ-GTrate(J)II (Mitsubishi Chemical Medience Corporation, Tokyo, Japan), and Total bile acid-Testwako (Wako Pure Chemicals), respectively (Nishida et al., 2009). Plasma cytokine levels were determined with the Biosource enzyme-linked immunosorbent assay kits (Invitrogen Corporation, Carlsbad, CA). The experimental protocol adhered to the Guidelines for Animal Experiments of the Nihon University School of Medicine and was approved by the Ethics Review Committee for Animal Experimentation of Nihon University School of Medicine.
Figure 1.
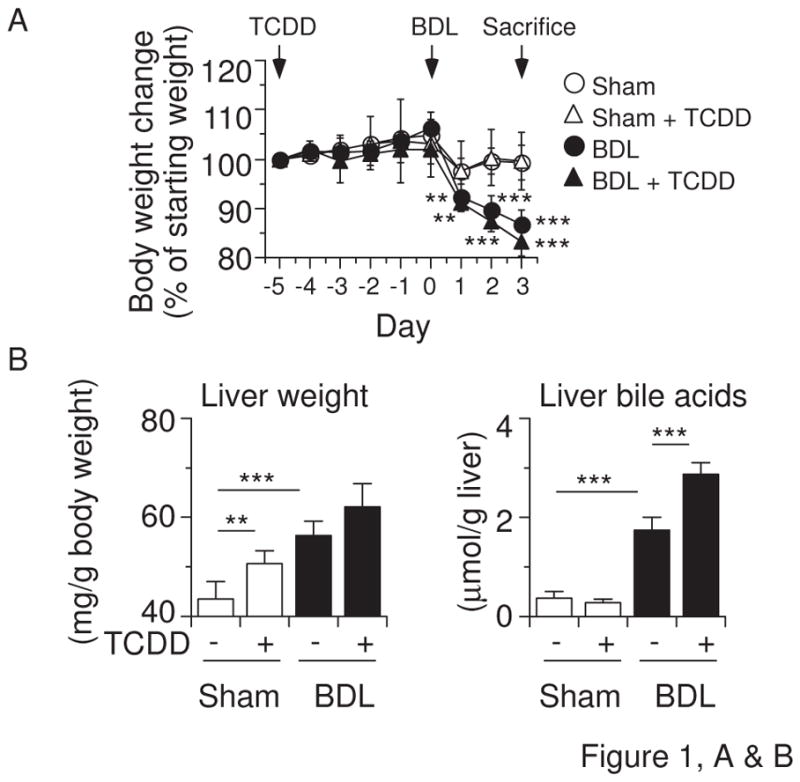
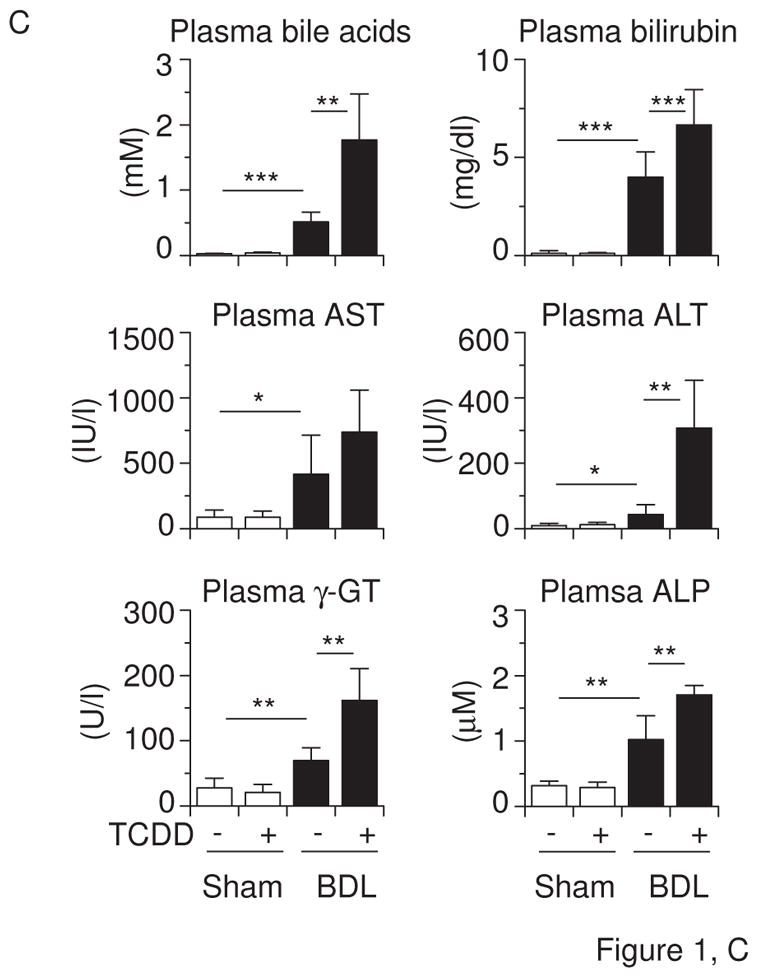
Effects of TCDD pretreatment and BDL on (A) body weight, (B) liver weight and bile acids, (C) plasma bile acids, bilirubin, aspartate aminotransferase (AST), alanine aminotransferase (ALT), γ-glutamyl transpeptidase (γ-GT), and alkaline phosphatase (ALP) in mice. Mice (n = 6 per group) were administered the vehicle corn oil or TCDD (15 μg/kg) via gavage on “Day −5” and subjected to sham operation or BDL on “Day 0”. Liver and blood were collected on “Day +3”. Values represent the means ± S.D. (A) **P < 0.01; ***P < 0.001 vs. vehicle-pretreated sham-operated mice. There was no significant difference between vehicle-pretreated sham-operated mice and TCDD-pretreated BDL mice. (B, C) *P < 0.05; **P < 0.01; ***P < 0.001; between indicated groups.
2.2. Real-time quantitative polymerase chain reaction (qRT-PCR)
Total RNA from liver was prepared by the acid guanidine thiocyanate-phenol/chloroform method (Nishida et al., 2009). The cDNAs were synthesized using the ImProm-II Reverse Transcription system (Promega Corporation, Madison, WI). The qRT-PCR was performed on the ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, CA) using Power SYBR Green PCR Master Mix (Applied Biosystems). Primers for the CYP3A11, AHR, ARNT, TNFSF10, and TNFRSF10B mRNAs are listed in Table 1, and other primers have been reported previously (Dong et al., 2009; Nishida et al., 2009; Ogura et al., 2009; Uno et al., 2009). Relative mRNA levels were calculated by the comparative threshold cycle method using glyceraldehyde-3-phosphate dehydrogenase as the internal control (Nishida et al., 2009).
Table 1.
Primer sequences used for qRT-PCR
| mRNA | Sequence (5′ to 3′) | Amplicon size | GenBank accession number |
|---|---|---|---|
| AHR | f, GTG TGC CCC CAG CAA GA r, CAA ACG TGC CGT TGA TTT G |
55 bp | NM_013464.4 |
| ARNT | f, GGC TCA CGA AGG TCG TTC AT r, ACA GGG TCC ACG GAG CTA GT |
59 bp | NM_009709.3 |
| CYP3A11 | f, GGA TGA GAT CGA TGA GGC TCT G r, CAG GTA TTC CAT CTC CAT CAC AGT |
73 bp | NM_007818 |
| TNFRSF10B | f, AAC ACG GAA CCT GGC AAG A r, TTT CCG TTT ACC GGA ACC A |
61 bp | NM_020275.3 |
| TNFSF10 | f, CAT TTC TCA ACC ACG TGC TCT T r, AGG CCC TCC TGC TCG AT |
57 bp | NM_009425.2 |
f, forward primer; r, reverse primer.
2.3. Western Blotting
Microsomes (S9 fraction) and membrane fractions from liver were prepared with Mem-PER Eukaryotic Membrane Protein Extraction Reagent Kit (Thermo Fisher Scientific Inc., Rockford, IL). They were subjected to SDS-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. Western blot analysis was performed using a polyclonal anti-ABCB11 antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and a polyclonal anti-CYP1A1/1A2 antibody (Daiichi Pure Chemicals, Tokyo, Japan), and detected with an alkaline phosphatase conjugate substrate system (Endo et al., 2008; Uno et al., 2008).
2.4. Histology
Liver samples were fixed in 10% neutral buffered formalin and embedded in paraffin. Tissue sections were stained with hematoxylin and eosin. The terminal deoxynucleotidyltransferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL) assay was performed using the ApopTag apoptosis detection kit (Millipore, Billerica, MA). Immunohistochemistry was performed using a monoclonal anti-Ki-67 antibody (Dako Denmark A/S, Glostrup, Denmark) and a polyclonal anti-cleaved caspase-3 antibody (Cell Signaling Technology, Danvers, MA), visualized with the Simple Stain Mouse MAX-PO (Nichirei Corporation, Tokyo, Japan).
2.5. Statistical Analysis
Data are presented as means ± S.D. The unpaired two-group Student’s t-test was performed to assess significant differences. P-values of < 0.05 were considered statistically significant.
3. Results
3.1. TCDD aggravates BDL-induced liver damage
To examine the effects of AHR-CYP1 induction on cholestasis, we pretreated mice with a dose of TCDD that is known to maximally induce Cyp1 gene expression without overt toxicity (Uno et al., 2004a). We did not observe weight loss in TCDD-treated mice (Figure 1A). Five days after TCDD pretreatment, we performed sham or BDL surgery. BDL decreased the body weight, compared with that following sham surgery, but we saw no difference in body weight between TCDD-pretreated and control vehicle-pretreated mice (Figure 1A). TCDD did not change liver or plasma bile acids, plasma bilirubin, aminotransferases, γ-glutamyl transpeptidase, and alkaline phosphatase in sham-operated mice, although it did increase liver weight (Figures 1B & 1C). TCDD pretreatment in BDL mice further increased the accumulation of liver bile acids (Figure 1B) and plasma bile acids, bilirubin, aminotransferases, γ-glutamyl transpeptidase, and alkaline phosphatase (Figure 1C).
Liver histology showed that TCDD treatment induces local infiltration of inflammatory cells, and a small number of TUNEL-positive hepatocytes were found only in portions of the pericentral and periportal areas, but not in inflamed regions (Figure 2). A limited number of hepatocytes in inflammatory foci were positive for cleaved caspase-3 in mice treated with TCDD. BDL liver showed scattered focal necrosis of hepatocytes, with small populations of TUNEL-positive cells and cleaved caspase-3-positive cells (Figure 2). TCDD pretreatment induced massive necrosis but did not increase the number of TUNEL-positive cells or cleaved caspase-3-positive cells (Figure 2). Cells positive for Ki-67, a proliferation marker, were not observed in any group (data not shown). These findings suggest that TCDD pretreatment induces severe liver damage in BDL mice, which is likely caused by necrosis rather than apoptosis.
Figure 2.
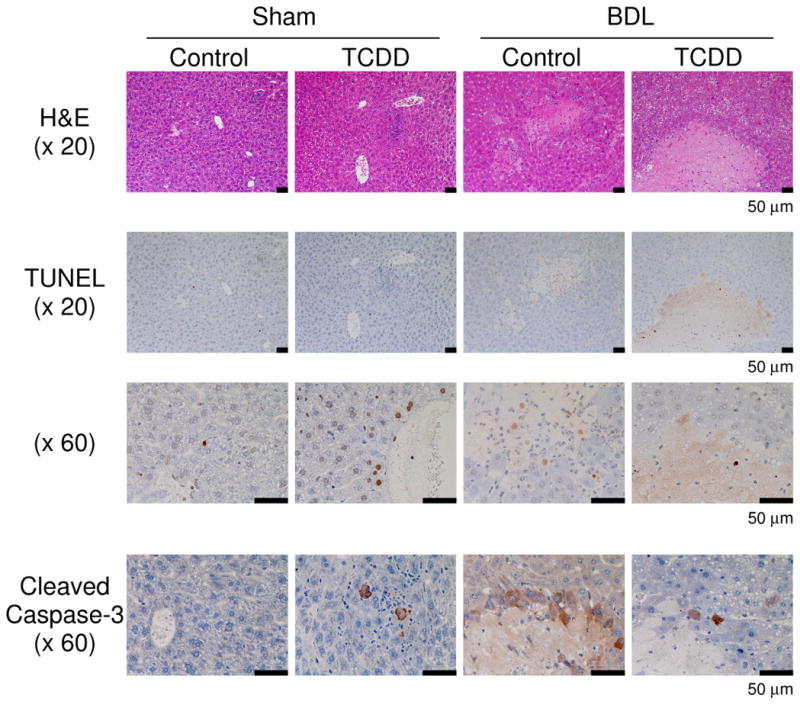
TCDD pretreatment induces massive hepatic necrosis in BDL mice. Hematoxylin and eosin staining (H&E; x 20 magnification), TUNEL assays (x 20 magnification and x 60 magnification), and cleaved caspase-3 staining (x 60 magnification) were performed on liver sections from mice treated as described in Figure 1.
We also examined the cholestatic effect on the expression of AHR-target genes. BDL alone decreased CYP1A2 and increased CYP1B1 mRNA levels in sham-operated mice (Figure 3A, insets). In contrast, TCDD strongly induced hepatic CYP1A1, CYP1A2 and CYP1B1 mRNA levels, and there was no significant difference in these induced levels between sham-operated and BDL mice (Figure 3A). Although TCDD slightly increased AHR and ARNT mRNA expression, BDL did not induce robust changes in these expressions (Figure 3B). TCDD treatment strongly induced hepatic CYP1A1 and CYP1A2 proteins in sham-operated mice as well as in BDL mice (Figure 4). These data indicate that cholestasis does not affect induction of the AHR-CYP1 cascade by TCDD.
Figure 3.
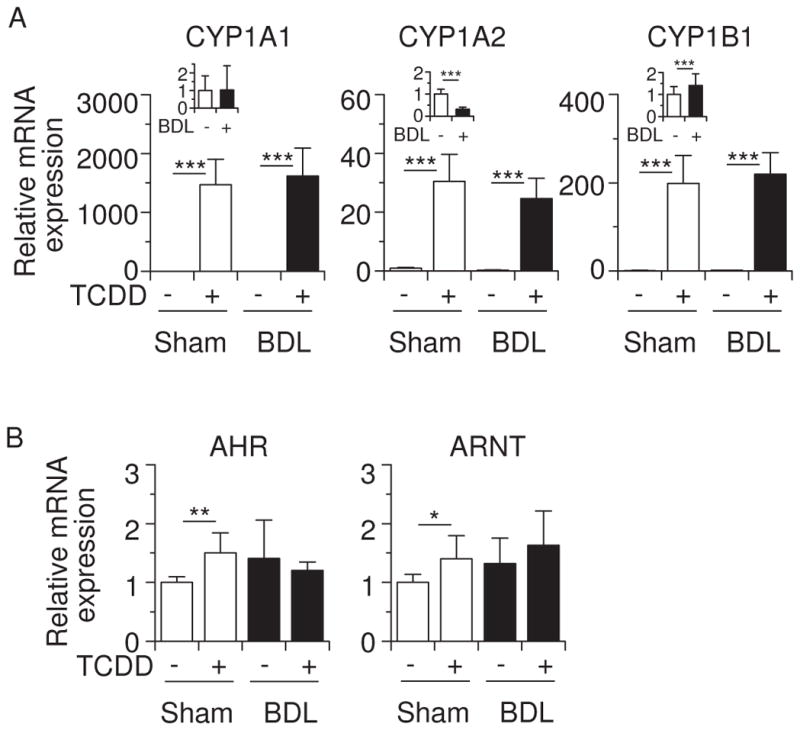
Effects of TCDD and BDL on hepatic mRNA expression of AHR-target genes. (A) CYP1A1, CYP1A2 and CYP1B1 mRNA levels. (B) AHR and ARNT mRNA levels. Total RNA was prepared from the mice shown in Figure 1. In this and subsequent figures, expression of the indicated mRNA levels was measured by qRT-PCR using glyceraldehyde-3-phosphate dehydrogenase as the internal control. Values for normalized mRNA expression are relative to those of corn oil-pretreated sham-operated mice. Effects of BDL on CYP1A1, CYP1A2 and CYP1B1 mRNA expression in corn oil-pretreated mice are shown in insets of (A). Values represent the means ± S.D. *P < 0.05; **P < 0.01; ***P < 0.001.
Figure 4.
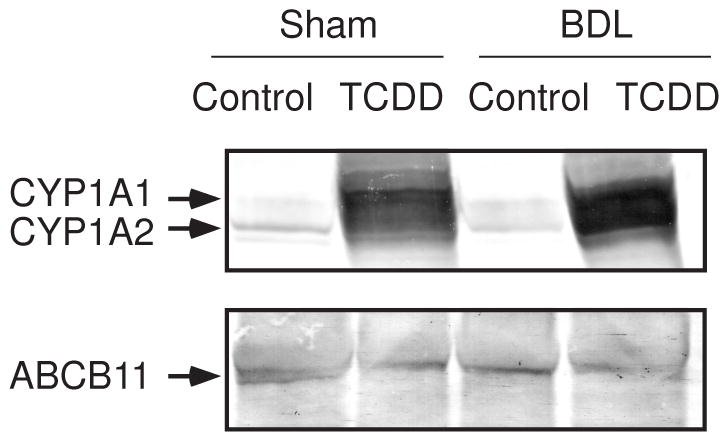
Expression of hepatic CYP1A1, CYP1A2 and ABCB11 proteins. Each lane was loaded with 15 μg of microsomal proteins and 40 μg of membrane proteins for CYP1A1/CYP1A2 and ABCB11, respectively. Microsomes and membrane fractions were prepared from the mice shown in Figure 1. Experiments were repeated with similar results.
3.2. TCDD modifies expression of genes implicated in bile acid metabolism
BDL is known to alter expression of genes that are involved in bile acid metabolism (Zollner et al., 2006). We examined the effect of TCDD pretreatment on the expression of bile acid metabolism-related genes in BDL mice. Cholesterol 7α-hydroxylase (CYP7A1) catalyzes the rate-limiting step of the classical bile acid synthesis pathway, and one or more of the CYP3A enzymes are involved in the metabolic conversion of xenobiotics and endogenous substrates, including bile acids, to more polar derivatives (Xie and Evans, 2001; Makishima, 2005). Figure 5A shows that BDL increased CYP7A1 and CYP3A11 mRNA levels, as had been previously shown (Stedman et al., 2004; Inagaki et al., 2005), and TCDD pretreatment suppressed these induced CYP7A1 and CYP3A11 mRNA levels in BDL mice. Sodium taurocholate-cotransporting polypeptide, and organic anion transporter proteins are involved in bile acid uptake at the basolateral membrane of hepatocytes (Zollner et al., 2006). TCDD pretreatment effectively decreased SLCO1A1 mRNA levels in BDL mice (Figure 5B). The canalicular export pumps ABCB11 and ABCC2 (also called multidrug resistance-associated protein 2; MRP2) excrete bile acids from the canalicular membranes, whereas ABCC3 (MRP3), ABCC4 (MRP4), and OSTB all play a role in the alternative excretion of bile acids from hepatocytes into the systemic circulation (Zollner et al., 2006). Consistent with previous findings (Wagner et al., 2003; Maher et al., 2005; Boyer et al., 2006; Stedman et al., 2006), TCDD increased ABCC2, ABCC3, ABCC4 mRNA, and BDL induced the ABCB11, ABCC3, ABCC4, OSTA and OSTB mRNA levels (Figure 5B). TCDD is well known to induce numerous P450 enzymes, including CYP1, which might influence the regulation of ABCB transporter expression. TCDD pretreatment suppressed ABCB11 mRNA induction by BDL, and TCDD-induced expression of ABCC2 mRNA was not significant in BDL mice (Figure 5B). BDL slightly increased hepatic ABCB11 proteins and TCDD pretreatment decreased its expression (Figure 4). These findings suggest that canalicular excretion of bile acids may be repressed in TCDD-pretreated BDL mice. Further studies are needed to elucidate the functional relevance of ABCB11 expression. ABCC4 mRNA levels were effectively stimulated in TCDD-pretreated BDL mice. However, any combined increases caused by TCDD plus BDL were not seen in ABCC2, ABCC3 or OSTB mRNA levels (Figure 5B).
Figure 5.
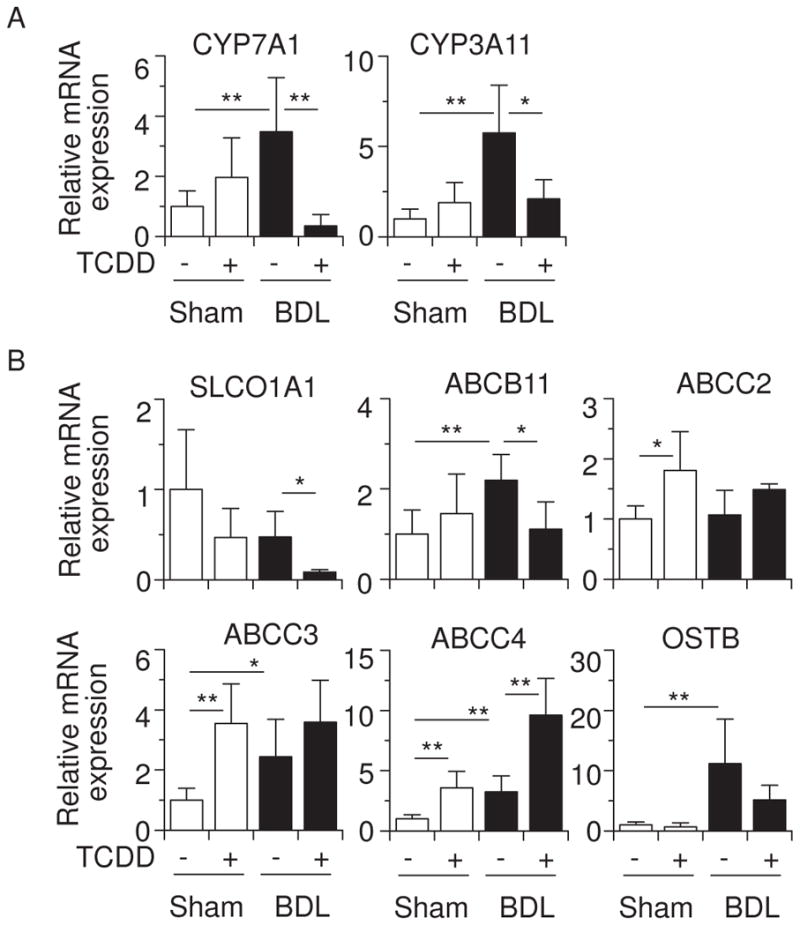
Effects of TCDD and BDL on hepatic mRNA levels of genes involved in bile acid metabolism. (A) CYP7A1 and CYP3A11 mRNA. (B) Expression of SLC01A1, ABCB11, ABCC2, ABCC3, ABCC4 and OSTB mRNA. Values represent the means ± S.D. *P < 0.05; **P < 0.01.
3.3. TCDD enhances cytokine production in BDL mice
TCDD-induced liver toxicity is dependent on inflammatory cytokines such as TNF and interleukin-1β (IL1B) (Pande et al., 2005; Uno and Makishima, 2009). BDL also stimulates inflammatory cytokine production (Ogura et al., 2009). Figure 6A shows that BDL caused an increase in plasma TNF and IL1B mRNA levels, and TCDD pretreatment further enhanced the production of these cytokines. TCDD also raised hepatic TNF and IL1B mRNA levels in sham-operated mice, but these induced levels were not altered in BDL mice (Figure 6B). These data suggest that increased TNF and IL1B in TCDD-pretreated BDL mice are derived from extrahepatic tissues and/or the result of posttranslational modification. The TNF-related apoptosis-inducing ligand TNFSF10 (TRAIL) and its receptor, death receptor-5 TNFRSF10B (DR5; TRAIL receptor 2), have been shown to contribute to liver injury by BDL (Takeda et al., 2008). We therefore examined the Tnfsf10 and Tnfrsf10b expression in these mice. TCDD slightly increased TNFSF10 and TNFRSF10B mRNA levels in sham-operated mice (Figure 6B). BDL increased mRNA expression of TNFRSF10B but not of TNFSF10. In BDL mice, TCDD pretreatment did not alter TNFRSF10B mRNA levels, while further decreasing TNFSF10 mRNA levels. These findings suggest that TNFSF10- and TNFRSF10B-mediated apoptosis is probably not involved in liver injury in TCDD-pretreated BDL mice.
Figure 6.
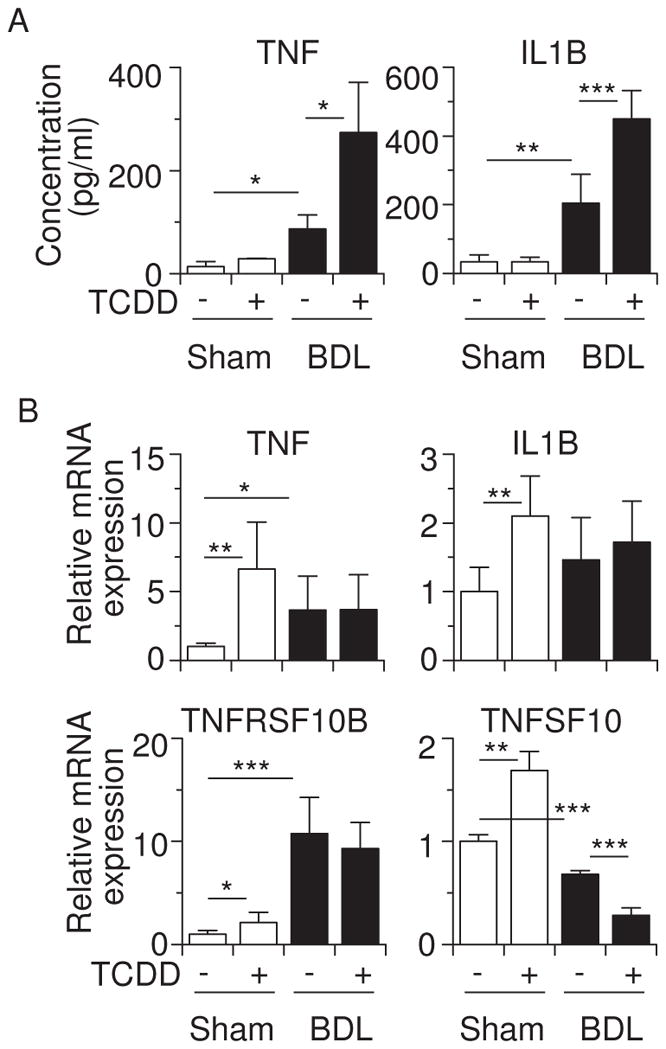
TCDD pretreatment enhances release of TNF and IL1B in BDL mice. (A) Plasma levels of TNF and IL1B proteins. (B) Hepatic TNF, ILIB, TNFRSF10B, and TNFSF10 mRNA levels. Values represent the means ± S.D. *P < 0.05; **P < 0.01; ***P < 0.001.
3.4. TCDD induces disseminated hepatocyte necrosis in cholestatic Cyp1a1/1a2(−/−) mice
Cyp1a2(−/−) knockout mice are partially resistant to high-dose TCDD-induced hepatic toxicity, while being completely protected against TCDD-induced uroporphyria (Smith et al., 2001), and Cyp1a1(−/−) mice show a decreased accumulation of hepatic uroporphyrin levels, compared with that in Cyp1a1(+/+) mice (Uno et al., 2004b). On the other hand, the loss of CYP1A1 and/or CYP1A2 induction in mice leads to increased acute hepatotoxicity induced by TCDD (Nukaya et al., 2009). Hence, we wished to examine the involvement of TCDD-induced CYP1A1 and CYP1A2 in liver toxicity during cholestasis by analyzing Cyp1a1/1a2(−/−) double-knockout mice. TCDD decreased body weight, but increased liver weight, in mice having the absence of both the Cyp1a1 and Cyp1a2 genes (Figure 7A). TCDD pretreatment plus BDL increased plasma bile acids in Cyp1a1/1a2(−/−) mice to levels comparable to that of wild-type mice (Figures 7B & 1B). Interestingly, plasma levels of bilirubin and aminotransferases were drastically elevated in TCDD-pretreated cholestatic Cyp1a1/1a2(−/−) mice (Figure 7B). Plasma levels of γ-glutamyl transpeptidase and alkaline phosphatase increased by TCDD plus BDL in Cyp1a1/1a2(−/−) mice were slightly more than those in wild-type mice (Figures 7B & 1C). Whereas TCDD or BDL alone induced mild histological changes, BDL in combination with TCDD pretreatment resulted in massive diffuse hepatic necrosis (Figure 7C) and strikingly elevated plasma TNF levels (Figure 7D). These findings indicate that the complete absence of CYP1A1 and CYP1A2 results in a far greater deterioration in liver damage and inflammatory response induced by TCDD pretreatment plus BDL. It therefore appears that CYP1A1 and CYP1A2 play a protective role in TCDD-induced hepatotoxicity in cholestatic mice.
Figure 7.
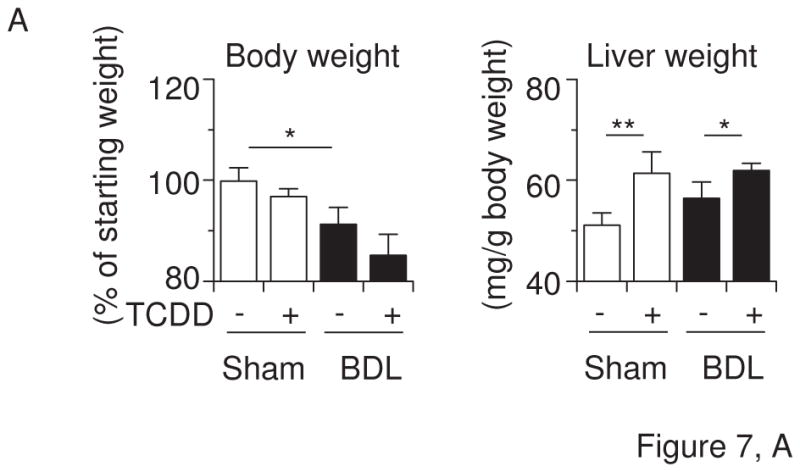
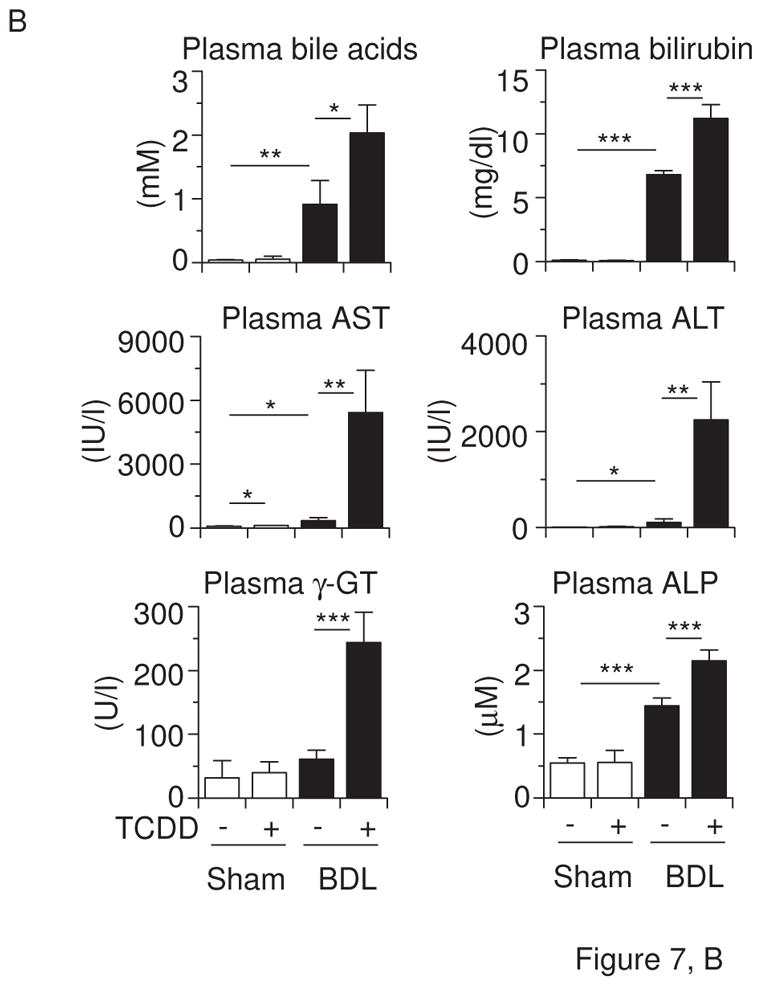
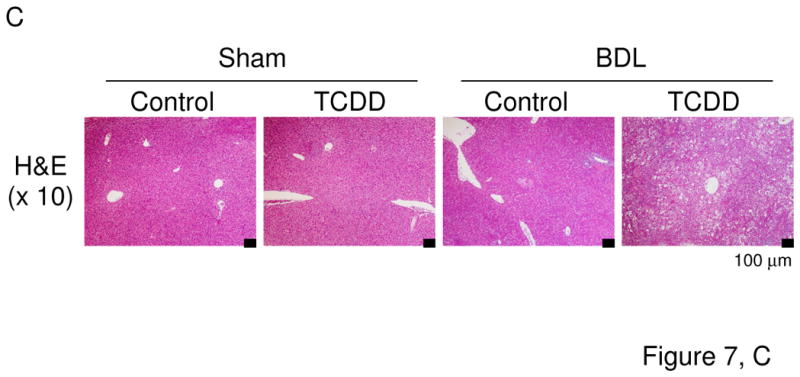

Liver damage by TCDD and BDL is exaggerated in Cyp1a1/1a2(−/−) double-knockout mice. (A) Body weight changes and liver weight. (B) Plasma bile acids, bilirubin, aspartate aminotransferase (AST), alanine aminotransferase (ALT), γ-glutamyl transpeptidase (γ-GT), and alkaline phosphatase (ALP). (C) Hematoxylin and eosin staining (H&E) of liver sections (x10 magnification). (D) Plasma TNF protein concentrations. Cyp1a1/1a2(−/−) mice (n = 4 per group) were administered corn oil alone or TCDD (15 μg/kg) via gavage on “Day −5” and subjected to sham operation or BDL on “Day 0”. Liver and blood were collected on “Day +3” by the same method as shown in Figure 1. Values represent the means ± S.D. *P < 0.05; **P < 0.01; ***P < 0.001.
4. Discussion
In this study, we found that TCDD pretreatment enhances liver damage and inflammatory cytokine production in BDL mice. High-dose TCDD treatment (200 μg/kg) is known to cause liver toxicity, characterized by a marked accumulation of lipids within hepatocytes and inflammatory cell infiltration in an AHR-dependent manner (Fernandez-Salguero et al., 1996). Whereas a single 75-μg/kg dose of TCDD given to wild-type mice causes severe uroporphyria and liver injury 5 weeks later, Cyp1a2(−/−) mice treated with TCDD show no increases in hepatic porphyrin levels or hepatocellular damage (Smith et al., 2001). Cyp1a1(−/−) mice exhibit less accumulation of uroporphyrin in the liver 18 days after a single intraperitoneal 200-μg/kg dose of TCDD (Uno et al., 2004b). These findings suggest that CYP1A2 and CYP1A1 mediate TCDD-induced hepatocellular injury associated with uroporphyria. Thus, TCDD-activated AHR induces expression of CYP1A1 and CYP1A2, and it would appear that these CYP1 enzymes might contribute to the etiology of high-dose TCDD-induced toxicity. On the other hand, mice deficient in CYP1A1 and CYP1A2 induction are more sensitive to acute hepatocellular damage and hepatic inflammation induced by a single intraperitoneal dose of TCDD (32 or 64 μg/kg) than wild-type mice (Nukaya et al., 2009). In the present study (Figures 1, 2 & 7), we showed that a single oral 15-μg/kg dose of TCDD alone to wild-type mice results in no damage and that TCDD produces liver damage under conditions of cholestasis. Moreover, in the cholestatic liver of Cyp1a1/1a2(−/−) double-knockout mice, TCDD caused much more striking damage than that under conditions of cholestasis in wild-type mice. Our findings suggest that AHR-induced CYP1A enzymes play a protective role against low-dose TCDD toxicity in cholestatic liver. Consistent with the previous report (Nukaya et al., 2009), CYP1A1 and CYP1A2 induction is a protective response against acute TCDD-induced hepatotoxicity. CYP1A1 and CYP1A2 are involved in metabolism of xenobiotics as well as endogenous compounds, including bilirubin, eicosanoids and 6-formylindolo[3,2-b]carbazole-6-carboxylic acid (Zaccaro et al., 2001; Nebert and Karp, 2008; Wincent et al., 2009). Cyp1a1/1a2/1b1(−/−) triple-knockout mice show an exaggerated response to zymosan-induced peritonitis, suggesting an anti-inflammatory role of the CYP1 enzymes (Dragin et al., 2008). In the present study, liver damage in TCDD-pretreated BDL Cyp1a1/1a2(−/−) mice was associated with increased plasma levels of bilirubin and TNF (Figure 7). It seems likely that the CYP1 enzymes might be involved in the critical synthesis and/or degradation of specific eicosanoids (Nebert and Karp, 2008) that are responding to toxic endogenous compounds and/or inflammatory signal molecules, which have accumulated in the cholestatic liver. Disruption of Cyp1a1 or Cyp1a2 has been demonstrated to decrease TCDD-induced hepatic injury in subchronic toxicity studies (Smith et al., 2001; Uno et al., 2004b). CYP1 enzymes may play a different role in subchronic or chronic TCDD toxicity.
TCDD pretreatment enhanced the production of inflammatory cytokines TNF and IL1B in BDL mice (Figure 6). TCDD exposure is known to increase lipopolysaccharide-induced toxicity in mice by enhancing serum TNF levels, and treatment of mice with anti-TNF antibody reduces TCDD-mediated mortality (Uno and Makishima, 2009). Tnfrsf1a/Tnfrsf1b/Il1r1(−/−) triple-knockout mice (lacking two receptors for TNF and one receptor for IL1R1) are resistant to TCDD-induced hepatocellular damage (Pande et al., 2005). Increased production of inflammatory cytokines such as TNF may contribute to the exaggerated liver damage that we have seen in TCDD-pretreated BDL mice in the present study. The TNFSF10/TNFRSF10B pathway has been shown to be involved in hepatocyte apoptosis in BDL mice (Takeda et al., 2008). Although BDL increased hepatic TNFRSF10B mRNA expression, TCDD pretreatment did not alter TNFRSF10B mRNA levels, whereas TCDD decreased TNFSF10 mRNA levels in BDL mice (Figure 6). Apoptotic cells were not greater in number in the liver of TCDD-pretreated BDL mice (Figure 2). These data suggest that TNFSF10/TNFRSF10B-mediated apoptosis is not a principal component in causing severe liver damage in TCDD-pretreated BDL mice. Further studies are needed to elucidate the mechanisms of liver injury by TCDD plus BDL.
Hepatic and plasma bile acids were markedly increased in BDL mice pretreated with TCDD (Figure 1). BDL increased CYP7A1 mRNA levels (Figure 5). Fibroblast growth factor-15, which is known to be induced by FXR in the intestine, represses Cyp7a1 transcription (Inagaki et al., 2005). BDL decreases intestinal bile acids, leading to decreased FXR-induced fibroblast growth factor-15 production, thereby derepressing hepatic CYP7A1 expression. Interestingly, TCDD pretreatment repressed Cyp7a1 expression in BDL mice (Figure 5). This strong Cyp7a1 repression may be due to increased inflammatory cytokines (Li et al., 2006). BDL increased CYP3A11, ABCB11, ABCC2, ABCC3 and OSTB mRNA levels and ABCB11 proteins (Figures 5 & 4). Because these genes are also elevated by the bile acid receptors FXR and PXR (Wagner et al., 2003; Wagner et al., 2005; Boyer et al., 2006; Stedman et al., 2006), it is likely that accumulated bile acids may stimulate transcription by way of these receptors. The increases of CYP3A11, ABCB11 and OSTB mRNA levels and ABCB11 proteins in BDL mice were repressed by TCDD pretreatment (Figures 5 & 4). TCDD also decreased SLCO1A1 expression. Proinflammatory cytokines are likely to be involved in the repression of these bile acid metabolism-related genes (Aitken et al., 2006; Klaassen and Aleksunes, 2010). In TCDD-pretreated BDL mice, we found that the bile acid-synthesizing enzyme CYP7A1, as well as the import transporter SLCO1A1, is decreased, whereas the export transporters ABCC2 and ABCC3 are induced. These changes probably reflect a mechanism whereby elevated hepatic bile acids can be lowered. However, we observed that hepatic and plasma bile acids are further enhanced in these mice (Figure 1). The TCDD-mediated decrease in expression of CYP3A11, combined with decreases in ABCB11 and OSTB, might lead to the hepatocytes becoming more vulnerable to toxicity caused by the accumulation of bile acids. Bile acids and TCDD together enhance the production of inflammatory cytokines such as TNF and IL1B in macrophages (Miyake et al., 2000; Kerkvliet, 2009). A vicious cycle of bile acid accumulation, inflammation, and hepatocyte necrosis may therefore be involved in causing the severe liver toxicity that we find in TCDD-pretreated BDL mice. A further understanding of TCDD/cholestasis-induced liver injury should provide insights into the elucidation of pathogenesis seen in chemically induced liver diseases.
Supplementary Material
Acknowledgments
Funding
This work was supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology, Japan [Grant-in-Aid for Scientific Research on Priority Areas (No. 18077995)] and, in part, by NIH grants R01 ES014403 and P30 ES06096.
The authors thank members of the Makishima laboratory for technical assistance and helpful comments, Dr. Masami Hashizume of Hist Science Laboratory Co., Ltd. (Ome, Japan) for help with histology; and Dr. Satoshi Nunomura and Dr. Chisei Ra of Advanced Medical Research Center, Nihon University Graduate School of Medical Science for help in cytokine analysis.
Footnotes
Conflict of interest statement
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aitken AE, Richardson TA, Morgan ET. Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu Rev Pharmacol Toxicol. 2006;46:123–149. doi: 10.1146/annurev.pharmtox.46.120604.141059. [DOI] [PubMed] [Google Scholar]
- Bock KW, Kohle C. Ah receptor: Dioxin-mediated toxic responses as hints to deregulated physiologic functions. Biochem Pharmacol. 2006;72:393–404. doi: 10.1016/j.bcp.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Boyer JL, Trauner M, Mennone A, Soroka CJ, Cai SY, Moustafa T, Zollner G, Lee JY, Ballatori N. Upregulation of a basolateral FXR-dependent bile acid efflux transporter OSTα-OSTβ in cholestasis in humans and rodents. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1124–1130. doi: 10.1152/ajpgi.00539.2005. [DOI] [PubMed] [Google Scholar]
- Diliberto JJ, Burgin D, Birnbaum LS. Role of CYP1A2 in hepatic sequestration of dioxin: studies using CYP1A2 knock-out mice. Biochem Biophys Res Commun. 1997;236:431–433. doi: 10.1006/bbrc.1997.6973. [DOI] [PubMed] [Google Scholar]
- Dong H, Dalton TP, Miller ML, Chen Y, Uno S, Shi Z, Shertzer HG, Bansal S, Avadhani NG, Nebert DW. Knock-in mouse lines expressing either mitochondrial or microsomal CYP1A1: differing responses to dietary benzo[α]pyrene as proof of principle. Mol Pharmacol. 2009;75:555–567. doi: 10.1124/mol.108.051888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragin N, Dalton TP, Miller ML, Shertzer HG, Nebert DW. For dioxin-induced birth defects, mouse or human CYP1A2 in maternal liver protects whereas mouse CYP1A1 and CYP1B1 are inconsequential. J Biol Chem. 2006;281:18591–18600. doi: 10.1074/jbc.M601159200. [DOI] [PubMed] [Google Scholar]
- Dragin N, Shi Z, Madan R, Karp CL, Sartor MA, Chen C, Gonzalez FJ, Nebert DW. Phenotype of the Cyp1a1/1a2/1b1(−/−) triple-knockout mouse. Mol Pharmacol. 2008;73:1844–1856. doi: 10.1124/mol.108.045658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragin N, Uno S, Wang B, Dalton TP, Nebert DW. Generation of ‘humanized’ hCYP1A1_1A2_Cyp1a1/1a2(−/−) mouse line. Biochem Biophys Res Commun. 2007;359:635–642. doi: 10.1016/j.bbrc.2007.05.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo K, Uno S, Seki T, Ariga T, Kusumi Y, Mitsumata M, Yamada S, Makishima M. Inhibition of aryl hydrocarbon receptor transactivation and DNA adduct formation by CYP1 isoform-selective metabolic deactivation of benzo[a]pyrene. Toxicol Appl Pharmacol. 2008;230:135–143. doi: 10.1016/j.taap.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, Gonzalez FJ. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol Appl Pharmacol. 1996;140:173–179. doi: 10.1006/taap.1996.0210. [DOI] [PubMed] [Google Scholar]
- Fritsche E, Schäfer C, Calles C, Bernsmann T, Bernshausen T, Wurm M, Hübenthal U, Cline JE, Hajimiragha H, Schroeder P, Klotz LO, Rannug A, Fürst P, Hanenberg H, Abel J, Krutmann J. Lightening up the UV response by identification of the arylhydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc Natl Acad Sci U S A. 2007;104:8851–8856. doi: 10.1073/pnas.0701764104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, Gerard RD, Repa JJ, Mangelsdorf DJ, Kliewer SA. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005;2:217–225. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson JA, Repa JJ, Mangelsdorf DJ, Kliewer SA. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci U S A. 2006;103:3920–3925. doi: 10.1073/pnas.0509592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerkvliet NI. AHR-mediated immunomodulation: The role of altered gene transcription. Biochem Pharmacol. 2009;77:746–760. doi: 10.1016/j.bcp.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen CD, Aleksunes LM. Xenobiotic, bile acid, and cholesterol transporters: function and regulation. Pharmacol Rev. 2010;62:1–96. doi: 10.1124/pr.109.002014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Jahan A, Chiang JYL. Bile acids and cytokines inhibit the human cholesterol 7α-hydroxylase gene via the JNK/c-jun pathway in human liver cells. Hepatology. 2006;43:1202–1210. doi: 10.1002/hep.21183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher JM, Cheng X, Slitt AL, Dieter MZ, Klaassen CD. Induction of the multidrug resistance-associated protein family of transporters by chemical activators of receptor-mediated pathways in mouse liver. Drug Metab Dispos. 2005;33:956–962. doi: 10.1124/dmd.105.003798. [DOI] [PubMed] [Google Scholar]
- Makishima M. Nuclear receptors as targets for drug development: regulation of cholesterol and bile acid metabolism by nuclear receptors. J Pharmacol Sci. 2005;97:177–183. doi: 10.1254/jphs.fmj04008x4. [DOI] [PubMed] [Google Scholar]
- Matsubara T, Yoshinari K, Aoyama K, Sugawara M, Sekiya Y, Nagata K, Yamazoe Y. Role of vitamin D receptor in the lithocholic acid-mediated CYP3A induction in vitro and in vivo. Drug Metab Dispos. 2008;36:2058–2063. doi: 10.1124/dmd.108.021501. [DOI] [PubMed] [Google Scholar]
- Miyake JH, Wang SL, Davis RA. Bile acid induction of cytokine expression by macrophages correlates with repression of hepatic cholesterol 7α-hydroxylase. J Biol Chem. 2000;275:21805–21808. doi: 10.1074/jbc.C000275200. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Karp CL. Endogenous functions of the aryl hydrocarbon receptor (AHR): intersection of cytochrome P450 1 (CYP1)-metabolized eicosanoids and AHR biology. J Biol Chem. 2008;283:36061–36065. doi: 10.1074/jbc.R800053200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebert DW, Roe AL, Dieter MZ, Solis WA, Yang Y, Dalton TP. Role of the aromatic hydrocarbon receptor and [Ah] gene battery in the oxidative stress response, cell cycle control, and apoptosis. Biochem Pharmacol. 2000;59:65–85. doi: 10.1016/s0006-2952(99)00310-x. [DOI] [PubMed] [Google Scholar]
- Nishida S, Ozeki J, Makishima M. Modulation of bile acid metabolism by 1α-hydroxyvitamin D3 administration in mice. Drug Metab Dispos. 2009;37:2037–2044. doi: 10.1124/dmd.109.027334. [DOI] [PubMed] [Google Scholar]
- Nukaya M, Moran S, Bradfield CA. The role of the dioxin-responsive element cluster between the Cyp1a1 and Cyp1a2 loci in aryl hydrocarbon receptor biology. Proc Natl Acad Sci U S A. 2009;106:4923–4928. doi: 10.1073/pnas.0809613106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura M, Nishida S, Ishizawa M, Sakurai K, Shimizu M, Matsuo S, Amano S, Uno S, Makishima M. Vitamin D3 modulates the expression of bile acid regulatory genes and represses inflammation in bile duct-ligated mice. J Pharmacol Exp Ther. 2009;328:564–570. doi: 10.1124/jpet.108.145987. [DOI] [PubMed] [Google Scholar]
- Ohtake F, Baba A, Takada I, Okada M, Iwasaki K, Miki H, Takahashi S, Kouzmenko A, Nohara K, Chiba T, Fujii-Kuriyama Y, Kato S. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature. 2007;446:562–566. doi: 10.1038/nature05683. [DOI] [PubMed] [Google Scholar]
- Pande K, Moran SM, Bradfield CA. Aspects of dioxin toxicity are mediated by interleukin 1-like cytokines. Mol Pharmacol. 2005;67:1393–1398. doi: 10.1124/mol.105.010983. [DOI] [PubMed] [Google Scholar]
- Slezak BP, Diliberto JJ, Birnbaum LS. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-mediated oxidative stress in CYP1A2 knockout (CYP1A2−/−) mice. Biochem Biophys Res Commun. 1999;264:376–379. doi: 10.1006/bbrc.1999.1518. [DOI] [PubMed] [Google Scholar]
- Smith AG, Clothier B, Carthew P, Childs NL, Sinclair PR, Nebert DW, Dalton TP. Protection of the Cyp1a2(−/−) null mouse against uroporphyria and hepatic injury following exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Appl Pharmacol. 2001;173:89–98. doi: 10.1006/taap.2001.9167. [DOI] [PubMed] [Google Scholar]
- Stedman C, Liddle C, Coulter S, Sonoda J, Alvarez JG, Evans RM, Downes M. Benefit of farnesoid X receptor inhibition in obstructive cholestasis. Proc Natl Acad Sci U S A. 2006;103:11323–11328. doi: 10.1073/pnas.0604772103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stedman C, Robertson G, Coulter S, Liddle C. Feed-forward regulation of bile acid detoxification by CYP3A4: studies in humanized transgenic mice. J Biol Chem. 2004;279:11336–11343. doi: 10.1074/jbc.M310258200. [DOI] [PubMed] [Google Scholar]
- Takeda K, Kojima Y, Ikejima K, Harada K, Yamashina S, Okumura K, Aoyama T, Frese S, Ikeda H, Haynes NM, Cretney E, Yagita H, Sueyoshi N, Sato N, Nakanuma Y, Smyth MJ, Okumura K. Death receptor 5 mediated-apoptosis contributes to cholestatic liver disease. Proc Natl Acad Sci U S A. 2008;105:10895–10900. doi: 10.1073/pnas.0802702105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno S, Dalton TP, Derkenne S, Curran CP, Miller ML, Shertzer HG, Nebert DW. Oral exposure to benzo[a]pyrene in the mouse: detoxication by inducible cytochrome P450 is more important than metabolic activation. Mol Pharmacol. 2004a;65:1225–1237. doi: 10.1124/mol.65.5.1225. [DOI] [PubMed] [Google Scholar]
- Uno S, Dalton TP, Sinclair PR, Gorman N, Wang B, Smith AG, Miller ML, Shertzer HG, Nebert DW. Cyp1a1(−/−) male mice: protection against high-dose TCDD-induced lethality and wasting syndrome, and resistance to intrahepatocyte lipid accumulation and uroporphyria. Toxicol Appl Pharmacol. 2004b;196:410–421. doi: 10.1016/j.taap.2004.01.014. [DOI] [PubMed] [Google Scholar]
- Uno S, Dragin N, Miller ML, Dalton TP, Gonzalez FJ, Nebert DW. Basal and inducible CYP1 mRNA quantitation and protein localization throughout the mouse gastrointestinal tract. Free Radic Biol Med. 2008;44:570–583. doi: 10.1016/j.freeradbiomed.2007.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno S, Endo K, Ishida Y, Tateno C, Makishima M, Yoshizato K, Nebert DW. CYP1A1 and CYP1A2 expression: Comparing ‘humanized’ mouse lines and wild-type mice; comparing human and mouse hepatoma-derived cell lines. Toxicol Appl Pharmacol. 2009;237:119–126. doi: 10.1016/j.taap.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uno S, Makishima M. Benzo[a]pyrene toxicity and inflammatory disease. Curr Rheumatol Rev. 2009;5:266–271. [Google Scholar]
- Wagner M, Fickert P, Zollner G, Fuchsbichler A, Silbert D, Tsybrovskyy O, Zatloukal K, Guo GL, Schuetz JD, Gonzalez FJ, Marschall HU, Denk H, Trauner M. Role of farnesoid X receptor in determining hepatic ABC transporter expression and liver injury in bile duct-ligated mice. Gastroenterology. 2003;125:825–838. doi: 10.1016/s0016-5085(03)01068-0. [DOI] [PubMed] [Google Scholar]
- Wagner M, Halilbasic E, Marschall HU, Zollner G, Fickert P, Langner C, Zatloukal K, Denk H, Trauner M. CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology. 2005;42:420–430. doi: 10.1002/hep.20784. [DOI] [PubMed] [Google Scholar]
- Wincent E, Amini N, Luecke S, Glatt H, Bergman J, Crescenzi C, Rannug A, Rannug U. The suggested physiologic aryl hydrocarbon receptor activator and cytochrome P4501 substrate 6-formylindolo[3,2-b]carbazole is present in humans. J Biol Chem. 2009;284:2690–2696. doi: 10.1074/jbc.M808321200. [DOI] [PubMed] [Google Scholar]
- Xie W, Evans RM. Orphan nuclear receptors: the exotics of xenobiotics. J Biol Chem. 2001;276:37739–37742. doi: 10.1074/jbc.R100033200. [DOI] [PubMed] [Google Scholar]
- Zaccaro C, Sweitzer S, Pipino S, Gorman N, Sinclair PR, Sinclair JF, Nebert DW, De Matteis F. Role of cytochrome P450 1A2 in bilirubin degradation Studies in Cyp1a2 (−/−) mutant mice. Biochem Pharmacol. 2001;61:843–849. doi: 10.1016/s0006-2952(01)00568-8. [DOI] [PubMed] [Google Scholar]
- Zollner G, Marschall HU, Wagner M, Trauner M. Role of nuclear receptors in the adaptive response to bile acids and cholestasis: pathogenetic and therapeutic considerations. Mol Pharm. 2006;3:231–251. doi: 10.1021/mp060010s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.