

Abstract
A co-processed excipient was prepared from commercially available crystalline mannitol and α-chitin using direct compression as well as spray, wet, and dry granulation. The effect of the ratio of the two components, percentage of lubricant and particle size, on the properties of the prepared co-processed excipient has been investigated. α-Chitin forms non-hygroscopic, highly compactable, disintegrable compacts when co-processed with crystalline mannitol. The compaction properties of the co-processed mannitol–chitin mixture were found to be dependent upon the quantity of mannitol added to chitin, in addition to the granulation procedure used. Optimal physicochemical properties of the excipient, from a manufacturing perspective, were obtained using a co-processed mannitol–chitin (2:8, w/w) mixture prepared by wet granulation (Cop-MC). Disintegration time, crushing strength, and friability of tablets, produced from Cop-MC using magnesium stearate as a lubricant, were found to be independent of the particle size of the prepared granules. The inherent binding and disintegration properties of the compressed Cop-MC are useful for the formulation of poorly compressible, high-strength, and low-strength active pharmaceutical ingredients. The ability to co-process α-chitin with crystalline mannitol allows chitin to be used as a valuable industrial pharmaceutical excipient.
Key words: binder, chitin, co-processing, disintegrant, mannitol
INTRODUCTION
In the pharmaceutical industry, solid dosage formulations require different types of excipients to be added to the active pharmaceutical ingredient(s) in order to facilitate manufacturing and to achieve the required physicochemical properties (e.g., flowability, compressibility, disintegration, dissolution, stability, etc.). Excipients are incorporated in formulations using different techniques such as direct compression, wet or dry granulation, as well as spray or freeze drying (1).
Single-component excipients do not always provide the requisite performance/physicochemical properties to allow certain active pharmaceutical ingredients to be formulated or manufactured adequately. As a result, drug formulation scientists have relied on excipients used in combination and often introduced commercially by excipient manufacturers. Such combinations fall into two broad categories: physical mixtures and co-processed excipients. Co-processed excipients are combinations of two or more excipients that possess performance advantages that cannot be achieved using a physical admixture of the same combination of excipients (2). The use of different co-processed excipients has been investigated to overcome deficiencies arising from single-component excipients and existing formulations (3–5). For example, a co-processed excipient containing microcrystalline cellulose (MCC, Avicel PH 102) and mannitol was prepared by wet granulation with hydroxypropyl cellulose using high-shear mixing (3). The prepared excipient exhibited the necessary characteristics of a pharmaceutical filler compared with co-processed lactose- and calcium phosphate-containing formulations. Both lactose and calcium phosphate can give rise to physical and chemical problems (e.g., the Maillard reaction) (3). A novel co-processed excipient has been prepared by spray drying an aqueous slurry of MCC and mannitol to provide a system with reduced lubricant sensitivity, higher compactibility, and a lower tablet ejection force profile, relative to its individual components or the physical mixture. The prepared co-processed excipient exhibited reduced reactivity toward actives and is particularly useful as a binder for tablet formulations processed by direct compression (4).
In order to overcome the problem of the loss in compressibility of MCC when used in wet granulation formulations, silicon dioxide particles have been integrated with MCC particles using a spray drying process (5). The co-processed excipient shows better flowability, compressibility, and disintegration properties compared to commercially available MCC or a physical mixture of MCC and silicon dioxide, specifically in wet granulation formulations. However, using the aforementioned co-processed excipients in tablet formulations needs a disintegrant to enhance the disintegration and subsequent release of the active pharmaceutical ingredients from the tablets (5).
There is increasing interest in exploring new commercial uses for chitin, a polyglucosamine, which can exist in three polymorphic forms: α, β, and γ; the most abundant naturally occurring polymorph is α-chitin. Of the three polymorphic forms, α-chitin has the highest compressibility (6). Chitin is a natural, non-toxic, non-allergenic, antimicrobial, and biodegradable material and as such does not present any known health risks (7,8). The compactibility values of α-chitin powder are very similar to those of MCC and significantly higher when compared to dibasic calcium phosphate or pregelatinized starch. This is because chitin is less plastic and more elastic than microcrystalline cellulose, as well as being more plastic and elastic than dibasic calcium phosphate (9).
Recently, chitin has been used to prepare different co-processed excipients with a range of metal silicates and silicon dioxide (10,11). The excipients obtained offer formulations with the requisite physical properties (e.g., non-hygroscopic, highly compactable, and highly disintegrable). As a result of such modifications, tablet disintegration is characterized by superiority in water uptake and penetration, with no gelling hindrance effects (11). Furthermore, chitin can be used at higher concentrations than commercially available superdisintegrants without negatively affecting the disintegration properties (10,11). It is reported that commercially available superdisintgrants lose their function, as disintegrants, when their concentration exceeds certain limits (3–15%, w/w), thereby excluding their use as fillers or binders in solid dosage formulations (12).
Mannitol is a naturally occurring sugar alcohol found in animals and plants; it is present in small quantities in almost all vegetables. An acceptable daily intake of mannitol has not been specified by the World Health Organization since the amount consumed as a sweetening agent is not considered to represent a hazard to health. Mannitol is widely used in pharmaceutical formulations, and as such, it is primarily used as a diluent (10–90%, w/w) in tablet formulations where it is water-soluble, non-hygroscopic, and produces a semi-sweet, smooth, and cool taste (13). Because of its low hygroscopicity, mannitol is potentially an excellent excipient since it is compatible with the majority of active pharmaceutical ingredients. However, crystalline mannitol is excessively friable, leading to the formation of fine particles that are particularly detrimental to its flow properties. In addition, because of its compact crystal structure, mannitol obtained by crystallization from water exhibits poor solubility. This slow dissolution rate is a major disadvantage and thus restricts its use in pharmaceutical formulations (14). Recently, novel pharmaceutical formulations containing both α-chitin and crystalline mannitol have been developed in order to eliminate such disadvantages (15). It is expected that such formulations will result in the co-processed excipient displaying no adverse effect on human health because the proposed novel co-processed excipient is produced in the absence of any chemical reactions between the individual components (2). As a result, the combination of chitin with mannitol (a non-hygroscopic inert material) may offer a valuable and practical industrial choice as an excipient in terms of disintegration and compaction properties (15).
The majority of commercially available co-processed excipients are produced by spray drying (16), which is a costly and complicated technique. In the present work, an easily useable, dry or wet granulation procedure is expected to be advantageous for preparing a co-processed excipient from crystalline mannitol and α-chitin that can be used as a multipurpose superdisintegrant, filler, and binder in solid dosage formulations. From an industrial perspective, the production of co-processed excipients using classical techniques results in a less expensive method of manufacturing compared to spray drying. Furthermore, the machines used in such technique are practically available in almost every industrial facility. Consequently, the use of such technique would not imply any further cost to the manufacturer. The studies reported herein aim to test the foregoing assumption and to characterize and optimize the composition and preparation procedure for mannitol/α-chitin mixtures. The overall properties, functionality, and applications of the co-processed mixture are also investigated.
EXPERIMENTAL
Materials
Commercial chitin, average molecular mass 1,000 kDa and degree of acetylation about 0.96, was obtained from Zhejiang Jiande Biochemical (China). Crystalline grade d-mannitol (Pearlitol), with a mean particle size of 160 μm, was obtained from Roquette (France). Purified water of British Pharmacopeia grade was obtained from the Jordanian Pharmaceutical Manufacturing Co. (Jordan). Co-spray-dried microcrystalline cellulose with d-mannitol (Avicel® HFE 102) was obtained from FMC BioPolymer (Germany), magnesium stearate from Mallinckrodt (USA), and Opadry OY-1350 film coat from Colorcon (UK). All the active ingredients used were of pharmaceutical grade. These included: amlodipine besylate (Matrix Lab., India), methyldopa (Xinshiji Pharma, China), and rosuvastatin calcium (Bicon, India). Aldomet® 250 tablets (Merck & CO., Inc.), Norvasc® 10 tablets (Pfizer Inc.), and Crestor® 20 tablets (AstraZeneca UK Limited) were obtained from a market in Jordan. All other reagents used were of analytical grade.
Preparation of Co-processed Mannitol–Chitin
Three co-processed mixtures (1 kg each) of mannitol and chitin of different ratios (1:9, 2:8, and 3:7, w/w) were prepared using different processing techniques, i.e., direct mixing as well as spray, wet and dry granulation.
Direct Mixing
The components of each mixture were individually passed through a 710-μm mesh sieve (Fritsch, Germany) and then mixed together for 5 min at 10 rpm using a 7.5-L cubic blender equipped with a motor drive machine (Erweka, Germany).
Dry Granulation
The three mixtures prepared by direct mixing were compacted using a roll compactor equipped with DPS type rolls (TFC-labo, Vector Corporation, USA), set at about 5 MPa roll pressure, four rounds/minutes roll speed, and 20 rounds/minutes screw control speed. The compacted powder was collected and sieved through a 710-μm sieve using a milling machine equipped with a motor drive machine. Finally, the granules were mixed for 5 min at 10 rpm using a 7.5-L cubic blender equipped with a motor drive machine.
Spray Granulation
A 20% (w/v) aqueous mannitol solution was prepared. Chitin was sieved through a 710-μm sieve and fluidized in a fluid bed granulator vessel (Strea 1, Niro Aeromatic, Germany) using an air pressure and drying temperature of 1.5 bar and 60°C, respectively. The appropriate volumes of mannitol solution (0.5, 1, and 1.5 L for 1:9, 2:8, and 3:7 (w/w) mannitol–chitin mixtures, respectively) were sprayed into the fluidized chitin at a spray rate of 5 mL/min. The granules were then sieved and mixed using the same parameters as for the dry granulation procedure.
Wet Granulation
Different mannitol solutions (6%, 12%, and 16%, w/v) were prepared by dissolving the required quantity of mannitol to be used to prepare the mannitol–chitin mixtures (1:9, 2:8, and 3:7 (w/w) ratio, respectively) in sufficient amount of water to achieve a suitable granulation end point. The sieved chitin was placed in granulation pan (Erweka) and granulated with the mannitol solution using a mixing speed of 200 rpm. The wet mass was passed through a 9.5-mm sieve. Granule drying was performed at 60°C using a drying oven (UT6200, Heraeous, Germany). The granules were sieved and mixed using the same procedures as used for the dry granulation procedure.
Characterization of Cop-MC
Fourier Transform Infrared Spectroscopy
Fourier transform infrared spectroscopy (FT-IR) measurements were undertaken using an FT-IR instrument (Paragon 1000, Perkin Elmer, UK) using thin pellets containing 1 mg of each sample dispersed in 100 mg of KBr. The spectra were recorded at room temperature as an average of 30 scans in the 400- to 4,000-cm−1 range with a spectral resolution of 1 cm−1. In order to minimize the effects of traces of CO2 and water vapor from the atmosphere of the sample compartment, the spectrometer was purged with N2.
X-Ray Powder Diffractometry
The X-ray powder diffractometry (XRPD) profiles were measured using an X-ray diffractometer (PW1729, Philips, Holland). The radiation was generated using a 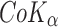 source and filtered through Ni filters; a wavelength of 1.79025 Å at 40 mA and 35 kV was used. The instrument was operated over the 2θ range of 5–60°. The range and the chart speed were set at 2 × 103 cycles/second and 10 mm/2θ, respectively.
source and filtered through Ni filters; a wavelength of 1.79025 Å at 40 mA and 35 kV was used. The instrument was operated over the 2θ range of 5–60°. The range and the chart speed were set at 2 × 103 cycles/second and 10 mm/2θ, respectively.
Scanning Electron Microscopy
Sample morphology was determined using a scanning electron microscope (Quanta 200 3D, FEI, Eindhoven/Netherlands) operated at an accelerating voltage of 1,200 V. The sample (0.5 mg) was mounted onto a 5 × 5-mm silicon wafer affixed via graphite tape to an aluminum stub. The powder was then sputter-coated for 105 s at a beam current of 20 mA/dm3 with a 100 Å layer of gold/palladium alloy.
Testing of the Cop-MC-Containing Tablets
The compressed tablets containing co-processed mannitol–chitin mixture (Cop-MC) were tested for crushing strength (6D, Schelenuiger tester, Germany), disintegration (2T31, Erweka tester), and friability (Erweka tester) following the general tests in the British Pharmacopeia (17).
Physical and Chemical Properties of Cop-MC
Bulk Density and Particle Size Distribution
The bulk density and particle size distribution were measured using a powder volumeter (SVM, tapped volumeter, Erweka) and a powder particle size analyzer (Vibratory sieve, shaker analysette 3PRO and 1,000- to 63-μm sieves, Fritsch), respectively.
pH and Water Content Measurements
The pH of a 5% (w/v) aqueous dispersion and powder water content were measured using a pH meter (Seven multi-pH meter, Mettler, Switzerland) and titrator (DL38, Karl Fischer Titrator, Mettler), respectively.
Hygroscopicity
Samples (2.5 g each) were stored in desiccators containing water-saturated salt solutions at room temperature (20°C) for 10 and 14 days. The media compositions were set according to the Handbook of Chemistry and Physics (18) to obtain relative humidities (RHs) of 52%, 62%, 75%, 84%, and 95% using Ca(NO3)⋅4H2O, NH4NO3, NaCl, KCl, and Na2HPO4⋅12H2O, respectively. The samples were withdrawn after a fixed time period and stored at 20°C for 1 and 24 h before weighing and calculating the percentage gain in weight from the original weight under the different RH conditions.
Influence of Particle Size on Compression Characteristics
Samples of Cop-MC were sieved using 710- and 853-μm sieves and individually dry mixed with 0.5% (w/w) magnesium stearate for 3 min and then compressed using a single-punch tabletting machine (SFS, Chadmach Machinery, India) at different crushing strength values of 30, 50, 70, 90, 110, 130, and 150 N using a 9-mm circular punch to produce tablets of 180-mg weight. The disintegration time and friability were measured at each tablet crushing strength point. Avicel® HFE 102 NF was treated in the same way and used as a reference.
Effect of Lubricant Quantity on Tablets’ Physical Properties
Samples of Cop-MC were physically mixed for 3 min with different percentages of magnesium stearate (0.25–5%, w/w) in a 7.5-L cubic blender equipped with a motor drive machine. The powder was compressed at an adjusted upper punch scale of 25 kN, in which a 8-mm circular punch was fitted and tablet weight was fixed at 180 mg. The disintegration time and friability were measured.
Functionality
To study the functionality of the Cop-MC, tablets of rosuvastatin calcium (RSC) were prepared by different procedures including direct mixing as well as dry and wet granulation. The tablets contained RSC (7.6%, w/w), Cop-MC (91.7%, w/w), and magnesium stearate (0.7%, w/w). For the direct mixing procedure (formula 1), RSC and Cop-MC were first mixed for 2 min and then magnesium stearate was added and further mixed for another 3 min. For the dry granulation procedure (formula 2), RSC (7.6%, w/w), Cop-MC (25% (w/w), intra-granular), and magnesium stearate (0.2%, w/w) were compacted with a DP roll type using 10-MPa roll pressure, three rounds/minutes roll speed, and 43 rounds/minutes screw control speed and then graded using a 710-μm sieve. The remaining amount of Cop-MC (66.7%, w/w) was added and mixed for 2 min; magnesium stearate (0.5%, w/w) was added. The preparation was further mixed for 2 min. For the wet granulation procedure (formula 3), RSC (7.6%, w/w) and Cop-MC (35% (w/w), intra-granular) were granulated with 50% (w/v) ethanol solution, dried at 60°C, and then sieved at 710 μm. The remaining amount of Cop-MC (56.7%) was added and mixed for 2 min; magnesium stearate (0.7%) was then added and the preparation mixed for a further 2 min. The three formulations were compressed at 290-mg tablet weight, for which 10-mm shallow concave punches and dies were used. Dissolution testing (19) (DT80, Erweka tester) was performed according to the US Food and Drug Administration published dissolution method (20) for rosuvastatin calcium tablets. The percentage release of rosuvastatin calcium was determined spectrophotometrically (Du-650i UV/visible spectrophotometer, Beckman, USA) by measuring the absorbance at a λmax of 240 nm.
Compressibility of the Cop-MC (Kawakita Equation)
Cop-MC was compressed using a universal testing machine (RKM 50, PR-F system, ABS Instruments, Germany) equipped with 12-mm round, flat face upper and lower punches as well as dies. The punch speed was fixed at 10 mm/min. Different compression forces from 80 to 390 MPa were applied. Three tablets were prepared to ensure reproducibility. Compression was carried out at 400-mg tablet weight. The compression behavior of the samples was evaluated using Kawakita analysis. The Kawakita equation (Eq. 1) is used to study powder compression using the degree of volume reduction, C. The basis for the Kawakita equation for powder compression is that particles subjected to a compressive load in a confined space are viewed as a system in equilibrium at all stages of compression so that the product of the pressure term and the volume term is a constant (21):
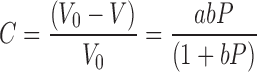 |
1 |
where V0 is the initial volume and V is the volume of powder column under an applied pressure, P. The constants a and b represent the minimum porosity before compression and plasticity of the material, respectively. The reciprocal of b defines the pressure required to reduce the powder bed by 50% (22,23). Equation 1 can be rearranged in linear form as:
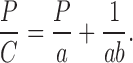 |
2 |
The expression for particle rearrangement can be affected simultaneously by the two Kawakita parameters a and b. The combination of these into a single value, i.e., the product of the Kawakita parameters a and b, may hence be used as an indicator of particle rearrangement during compression (24).
Compatibility Study of Cop-MC with Methyldopa
Differential scanning calorimetry (DSC 25, Mettler Instruments) can be used for a wide range of pharmaceutical applications ranging from the characterization of materials to the evaluation of drug excipient interactions via the appearance, shift, or disappearance of endothermic or exothermic peaks (25–27). DSC was used to study the compatibility of the methyldopa active pharmaceutical ingredient in a tabletted dosage form with Cop-MC and magnesium stearate (formula 4). The tablets were packed in either open or closed glass containers and stored at 40°C/75% RH for a 6-month period. The reference formula was prepared without methyldopa according to formula 4 using the same composition ratio of excipients. All the samples were tested before and after storage under defined conditions. DSC samples were hermetically sealed in aluminum pans and scanned over a range temperature of 0–300°C at a rate of 5°C/min. The instrument was calibrated using indium and the calorimetric data were analyzed using STAR software (version 9).
Applications
Cop-MC was used to formulate methyldopa and amlodipine besylate active ingredients into a tabletted dosage form; tablets were prepared using direct mixing. The methyldopa formulation (formula 4) comprised the active ingredient (63.3% in its hydrated form), Cop-MC (32.8%), and magnesium stearate (0.6%). Tablets were film-coated using Opadry OY-1350 (Colorcon, UK) coating material (3.3%) and sufficient water as solvent. The amlodipine besylate formulation (formula 5) comprised the active ingredient (7.0% as besylate), Cop-MC (92.5%), and magnesium stearate (0.5%). A 10-mm shallow concave and 8-mm flat face beveled edge punches were used for tabletting the methyldopa- and amlodipine besylate-containing formulations, respectively. Tablets of formulas 4 and 5 were tested for crushing strength, disintegration, and dissolution. Methyldopa tablets (250 mg) were further tested for their stability after being stored at 40°C/75% RH for 6 months using HPLC instrument equipped with a P1000 pump and a UV1000 detector (TSP/USA). A stability-indicating and validated HPLC method was used to determine methyldopa, O-3-methylmethyldopa (synthetic impurity), and other related compounds (28). A mixture of 50% (v/v) aqueous methanol and 1% (v/v) perchloric acid (350:650, v/v) was used as the mobile phase and an octyl silane column as the stationary phase (Lichrosphere 100 RP-8, 250 × 4 mm, 10 μm). UV detection at 230 nm, a flow rate of 1 mL/min, and a 20-μL injection loop were used. The HPLC method was initially tested for system suitability (i.e., peak symmetry, repeatability, and resolution) and for validation parameters (i.e., specificity, recovery, stability in solution, linearity, and limit of quantitation (LOQ)) according to USP guidelines (29). The stored tablet samples in addition to methyldopa active ingredient (as a reference) were packed in either open or close amber glass bottles. Aldomet® 250 and Norvasc® 10 tablets were used as reference materials for comparison purposes. The US FDA published dissolution method (20) for amlodipine besylate tablets was adopted for the dissolution analysis. The dissolution method states the use of USP apparatus II (Paddle), 75-rpm speed, 500 mL of 0.01 N hydrochloric acid as the dissolution medium, and 10-, 20-, 30-, 45-, and 60-min sampling times. The dissolution testing of methyldopa tablets was performed according to the United States Pharmacopeia (USP 32), following the methyldopa tablet official monograph (30). The dissolution system states the use of USP apparatus II (Paddle), 50-rpm speed, and 900 mL of 0.1 N hydrochloric acid as the dissolution medium. The percentage release of amlodipine besylate and methyldopa was determined spectrophotometrically by measuring the absorbances at a λmax of 250 (first derivative) and 280 nm, respectively.
RESULTS AND DISCUSSION
Co-processed Mannitol–Chitin with Low Lubricant Sensitivity
Lubricant sensitivity is the ratio of the un-lubricated to the lubricated compactibility of the tablet formulation. Addition of lubricant reduces the heat produced and tablet ejection force during compression, which is related to the reduction in inter-particulate interaction during compaction; as a result, powder compactibility is decreased. Lubricant sensitivity also refers to the reduction in interaction between the plastically deforming particles in the powder due to the addition of lubricant, which leads to a reduction in tablet crushing strength (31).
Chitin powder exhibits poor flow and produces fragile tablets upon compression, while pure crystalline mannitol displays undesirable compaction properties and results in tablet capping. To obtain the optimal ratio of co-processed mannitol–chitin mixture, three different ratios of mannitol/chitin were used: 10:90, 20:80, and 30:70 (w/w). These mixtures were prepared by different processing techniques, i.e., direct mixing, dry, spray, and wet granulation. The prepared granules were lubricated using different weight fractions of magnesium stearate ranging from 0.25% to 5.0% (w/w). The mixtures were compressed at a 25-kN scale of the upper punch and the tablets obtained were tested for crushing strength, friability, and disintegration time. The results indicate that improvements in the physical properties of mannitol and chitin mixtures follow the order: wet and spray granulation > dry granulation > direct mixing. The mixtures prepared by direct mixing have unacceptable physical properties (e.g., poor powder flow, powder non-uniformity (segregation), friability, and low crushing strength tablets). Additionally, the results indicate that the properties of mannitol/chitin mixtures are improved by dry granulation, but they were still not optimal. Moreover, it was noted that friability was sensitive to the fraction of magnesium stearate added. As a result, excipients produced by direct mixing and dry granulation methods were no longer used since the addition of active ingredients having critical properties (e.g., poor flow, incompressible, fragile, etc.) could have a negative impact on compressed tablets.
The main objective thus became to find the most appropriate mixture that could be used to overcome the poor flow and weak compressibility of its components. The method of integrating two components is an important factor in obtaining a suitable mixture that is needed to act as a diluent for the active ingredient.
From the preliminary results, physical mixing and direct compaction of the two components proved unsatisfactory; consequently, spray and wet granulation were used to produce the new excipient whereby the properties of the two components are able to overcome poor flow and compression properties. In addition, the compatibility of the mixture toward lubrication sensitivity was tested.
In the case of spray and wet granulations, the physical properties were improved with respect to the mannitol/chitin ratio used in the following order: 2:8 > 3:7 > 1:9 (w/w mannitol/chitin). The mixtures prepared using ratios of 1:9 and 3:7 (w/w) showed relatively high friability and sensitivity to the weight fraction of magnesium stearate added as a lubricant. The optimal ratio with respect to physical properties improvement is the 2:8 (w/w) ratio (mannitol/chitin). All the physical properties at this ratio prepared using dry granulation as well as spray and wet granulation in comparison with co-processed microcrystalline cellulose (Avicel HFE 102) as a reference are shown in Fig. 1. It is clear from the data presented in Fig. 1 that the physical properties of mixtures prepared by spray and wet granulation are not very sensitive to the amount of magnesium stearate added due to the presence of folding (e.g. high surface area) (Fig. 2), while those prepared by dry granulation and Avicel HFE 102 are relatively sensitive to the lubricant content. For further investigation, the mixture prepared by wet granulation was used and the other preparations were excluded because from a manufacturing process perspective, the former is conventional and more practical compared with using spray granulation and/or compaction.
Fig. 1.

Plots of the physical properties (crushing strength, disintegration time, and friability) of the co-processed mannitol–chitin mixture prepared by different granulation techniques versus the amount of magnesium stearate added
Fig. 2.

SEM images of Cop-MC lubricated with a 0.5% and b 3.0% (w/w) magnesium stearate; Avicel HFE 102 powder lubricated with c 0.5% and d 3.0% (w/w) magnesium stearate
Characterization of Cop-MC
Fourier Transform Infrared Spectroscopy
The FT-IR spectra of mannitol, chitin, the corresponding 2:8 physical mixture, and Cop-MC are shown in Fig. 3a–d, respectively. It is clear that the FT-IR spectra of the physical mixture of mannitol and chitin (Fig. 3c) are almost a superposition of the FT-IR profile contributed by mannitol and chitin. The dominance of the principal bands of chitin in the physical mixture is a result of its high percentage in the mixture (80%, w/w). The FT-IR spectra of Cop-MC (Fig. 3d) showed almost the same bands as the physical mixture, which suggests the absence of any detectable chemical interaction and the formation of a new solid phase, as shown by scanning electron microscopy.
Fig. 3.

FT-IR spectra of a mannitol, b chitin, c physical mixture of mannitol–chitin (2:8, w/w), and d Cop-MC
X-Ray Powder Diffractometry
Figure 4a–d shows the XRPD profiles of mannitol, chitin, the corresponding 1:1 physical mixture, and Cop-MC, respectively. It is clear that the XRPD pattern of the physical mixture of mannitol and chitin is almost a superposition of the patterns contributed by mannitol and chitin. In contrast, the diffraction pattern of Cop-MC showed a reduction in the principal diffraction peaks apparent in the XRPD patterns of the physical mixture, which suggests the formation of a new solid phase.
Fig. 4.

XRPD profiles of a mannitol, b chitin, c physical mixture of mannitol-chitin (2:8, w/w), and d Cop-MC
Differential Scanning Calorimetry
Figure 5 shows the thermal behavior of all samples investigated by DSC. Mannitol exhibits an endothermic peak at about 167°C (Fig. 5b), which is retained with high intensity in the physical mixture of mannitol and chitin (Fig. 5e), but a significant reduction in the intensity of the mannitol peak in the thermogram of the Cop-MC occurs (Fig. 5f), indicating that some of the mannitol is sequestered in the chitin pores.
Fig. 5.

DSC thermograms of a chitin, b mannitol, c treated mannitol, d non-treated physical mixture of mannitol–chitin (2:8, w/w), e treated physical mixture of mannitol–chitin (2:8, w/w) and f Cop-MC
Scanning electron microscopy
Scanning electron microscopy (SEM) was used to investigate particle surface morphology (Fig. 6). When comparing the surface of chitin (Fig. 6b) with the surface of the Cop-MC prepared using wet granulation (Fig. 6d), it is apparent that its native structure has changed from a smooth, flat surface structure, with folded edges, to three-dimensional compacts for the Cop-MC. This folding in the surface of the co-processed chitin creates a bigger surface area which can accommodate a larger quantity of lubricant. Figure 6e shows the surface of chitin–mannitol, indicating the presence of inter-particulate voids and channels. Figure 6c shows the surface of the AVICEL HFE 102, which exhibits a smaller surface area due to the solid surface and the absence of folded edges. This is the major reason underlying the clear increase in the disintegration time and reduction in the crushing strength of the tablets containing AVICEL HFE 102 as a result of increasing the quantity of magnesium stearate present. It is evident that the Cop-MC excipient is not chemically altered; consequently, it was used in further work.
Fig. 6.

SEM images of a mannitol, b chitin, c physical mixture of mannitol-chitin (2:8, w/w), d Cop-MC, and e Avicel HFE 102
Physical Properties of Cop-MC
Some of the physical properties of the mixture prepared by wet granulation and sized using two different mesh sizes, 710 and 853 μm (corresponding to mesh sizes of 22 and 18, respectively), were measured. These two sieves were selected in order to obtain Cop-MC with particle size distributions capable of improving solid formulation characteristics. Generally, in direct compression formulation, excipient flowability and compaction performance are critical; therefore, excipients used for such applications should exhibit narrow particle size distributions with moderate-to-coarse particle size (e.g., 100–200 mm) (32). The granules obtained have a bulk density of 0.2–0.3 g/cm3 with particle size distributions of 20%, 45%, and 30% in the range of <180, 180–355, >355 μm, respectively. The pH of a 5% (w/v) aqueous dispersion is 7–8 and a water content of about 6% (w/w). Cop-MC was found to be slightly hygroscopic with a high moisture uptake capacity, but only under extremely high humidity conditions (95% RH). This is expected and explains the fast disintegration of the tablets in aqueous media. The data from the water gain versus relative humidity plots (Fig. 7) clearly indicate that Cop-MC absorbs a moisture content that is up to 37% of its initial weight at 95% RH, but it loses most of this moisture after equilibration at ambient conditions of 20°C and 45% RH for a day.
Fig. 7.

Water gained by Cop-MC and Avicel HFE 102 kept in an open container at different relative humidities and 20°C
Effect of Particle Size on Tablet Disintegration Time, Crushing Strength, and Friability
The disintegration time and friability versus the crushing strength for the tablets prepared using powders with particle sizes of 710 and 853 μm are shown in Fig. 8. Generally, a reduction in particle size is associated with an increase in tablet mechanical strength. The increase in the mechanical strength of the tablets is attributed to an increase in the surface area available for inter-particulate attraction as the particles become smaller (33). However, when Cop-MC was used, it was found that varying the particle size had a minimal, or no, effect in increasing the mechanical strength of the tablets produced. Generally, for materials with a tendency to fragment, the mechanical strength of a tablet appears to be independent of particle size (34). This is likely to be the case for the Cop-MC particles which undergo fragmentation at an early stage of compression; this normally results in a limited effect on tablet crushing strength when varying the particle size.
Fig. 8.

Effect of Cop-MC particle size on tablet crushing strength, disintegration time, and friability. The tablets were 9 mm in diameter and 180 mg in weight. All samples were lubricated with 0.5% (w/w) magnesium stearate
With respect to tablet disintegration, the Cop-MC, as evidenced by the data in Fig. 8, showed a unique and distinctive characteristic whereby disintegration was independent of particle size and tablet crushing strength. Tablets produced from initial powder particle sizes of 710 and 853 μm at an upper bunch compression scale of 37–42 kN for each particle size showed a superior disintegration time, ranging from 5 to 145 s. This was achieved for tablet crushing strength values ranging from 30 to 150 N, which suggests that capillary action is the dominant mechanism for disintegration irrespective of the tablet crushing strength. Therefore, the inter-particulate voids within the chitin particle structure in the Cop-MC, as previously ascertained by scanning electron microscopy, remain intact and unchanged by varying the powder particle size and tablet crushing strength.
Processing chitin with mannitol was found to cause alterations in chitin powder compressibility and compactibility. Regarding powder compactibility, mannitol within the Cop-MC mixture provides an effective means of obtaining hard tablets with low friability while maintaining its super disintegration power, as deduced from the data in Fig. 8. While with the mannitolized microcrystalline cellulose (Avicel HFE 102), it can be clearly observed that increasing the tablet crushing strength results in a significant increase in disintegration time.
Kawakita Analysis (Compressibility)
Initial investigations of mannitol, chitin, and Cop-MC using Heckel analysis indicated non-log-linearity of compression data. Therefore, Heckel analysis was not adopted in this work as it may result in the misinterpretation of the estimated intercept and slope parameters of the Heckel plots (10). Therefore, in the present work, Kawakita analysis was adopted to linearize the compression data. Figure 9 displays representative Kawakita plots for mannitol, chitin, and Cop-MC. The Kawakita constants a, b, ab, and 1/b were calculated from the intercept and slope of the plot (Table I). The constant (a), which represents the compressibility, is nearly the same for chitin and the Cop-MC (a = 0.818 and 0.849, respectively) and is relatively low for mannitol (a = 0.576). Co-processing of chitin with mannitol has no significant increase in the compressibility of chitin. This is because mannitol (20% of the Cop-MC content) occupies only a limited area of the large surface pores of chitin. These pores are responsible for the decrease in the volume of chitin powder upon compression (35). The value of the constant (1/b) for Cop-MC (7.795), which is an inverse measure of the amount of plastic deformation occurring during the compression process (36), indicates that the presence of mannitol increases the plastic deformation of chitin. The higher degree of plastic deformation of Cop-MC during compression is most probably due to (a) the formation of intermolecular hydrogen bonding between the plastically deformed adjacent particles within the chitin particles and (b) the presence of moisture within the porous structure of chitin, which enforces the formation of hydrogen bonds and gives Cop-MC high crushing strength values upon compaction and also acts as an internal lubricant (37).
Fig. 9.

Kawakita plot for mannitol/chitin (20:80) Prepared by a wet granulation procedure. Tablets were 12 mm in diameter and 400 mg in weight
Table I.
Kawakita Parameters
| Material | Kawakita parameters | |||||
|---|---|---|---|---|---|---|
| Slope | Intercept | a | ab | b | 1/b | |
| Mannitol | 1.736 | 17.932 | 0.576 | 0.0558 | 0.0968 | 10.330 |
| Chitin | 1.223 | 12.927 | 0.818 | 0.0774 | 0.0946 | 10.570 |
| Cop-MC | 1.178 | 9.182 | 0.849 | 0.1089 | 0.1283 | 07.795 |
Cop-MC represents the co-processed mannitol–chitin (2:8, w/w) mixture
The relatively high value of ab for Cop-MC (0.1089), which is a measure of the extent of particle rearrangement, indicates that in the presence of mannitol, chitin exhibits a high degree of packing during tablet compression. This implies that co-processing reduces the surface micro-irregularities of chitin and facilitates particle rearrangement during the densification process of compaction. As a result, co-processing of chitin with mannitol has positively improved the compression characteristics of chitin as shown by Kawakita analysis.
Application
Functionality
The functionality of Cop-MC was investigated by employing different processing techniques (dry granulation, wet granulation, and direct compression) for the preparation of tablet dosage forms. RSC, a drug indicated for primary hypercholesterolemia, was selected as a model for this study. RSC is a hydrophobic drug reported to be soluble and stable in the presence of the pH modifier tribasic calcium phosphate as one of the excipients used in the tablet matrix (38). Crestor® 20 tablet commercial drug product was used as a reference. These tablets are composed of lactose and microcrystalline cellulose as fillers and tribasic calcium phosphate as alkalinizer and stabilizer in addition to crospovidone as the disintegrant (38,39). Therefore, the use of Cop-MC under neutral/alkaline conditions (pH of 7.0–8.0) is a suitable choice for RSC tablet formulation (40,41). The direct compression procedure for RSC with Cop-MC at a dilution capacity of 7.6% (w/w) resulted in hard tablets with more than 90% drug release within a 10-min time interval. The disintegration times for the RSC tablets prepared by the three different procedures and Crestor® 20 tablets were similar and lie in the 120- to 160-s range. The uncoated tablets of RSC, obtained using the three different methods of preparation, have crushing strength values in the range of 90–120 N and friability values of 0.1–0.3%. The foregoing results indicate that Cop-MC has an improved compressibility whether utilized in direct compression, dry granulation, or wet granulation formulations, thus implying that Cop-MC can be used as a single excipient acting as filler, binder, and disintegrant in tablet formulations.
Some applications using Cop-MC as the only filler, binder, and superdisintegrant were also investigated. Two antihypertensive agents, amlodipine besylate (which is a 1,4-dihydropyridine calcium channel blocker) and methyldopa (L-3-(3,4-dihydroxyphenyl)-2-methylalanine, an aromatic amino acid decarboxylase inhibitor), were selected as model drugs for this study as they have special requirements in tablet formulation design. Due to the high strength and weak compactibility of methyldopa powder, difficulties in the formulation of this active ingredient can occur, e.g., capping (i.e., “layering”) of the tablets during ejection in tabletting processing. In addition, the particle size and shape of the active ingredient have a major impact on the tablet compaction and binding properties during compression. Usually, these unfavorable characteristics mean that a granulation procedure has to be used for the preparation of the tablets (42). This is because direct compression procedures cannot overcome the aforementioned characteristics by using sufficient quantities of suitable fillers, binder, and disintegrant while maintaining a low tablet weight. The 355-mg film coated Aldomet 250 tablets contain calcium disodium edetate, cellulose powder, anhydrous citric acid, colloidal silicon dioxide, ethyl cellulose, guar gum, and magnesium stearate in the tablet core and D&C Yellow 10, hydroxypropyl methylcellulose, anhydrous citric acid, iron oxide, propylene glycol, talc, and titanium dioxide for tablet coating (43). On the other hand, lactose is incompatible with amlodipine besylate (Maillard reaction) due to the presence of a primary amine group in its chemical structure (44). Therefore, to achieve the required binding while maintaining other tablet physical properties, a multi-excipient system is employed in the originator product “Norvasc® 10 tablets” using a high dilution factor for the amlodipine besylate. The 400-mg tablets of Norvasc® 10 contain microcrystalline cellulose, dibasic calcium phosphate anhydrous, sodium starch glycolate, and magnesium stearate (38). The performance of Cop-MC in a direct compression formulation containing a poorly compressible high-strength drug, such as methyldopa, and a low-strength drug, such as amlodipine besylate, was investigated. The binding and superdisintegration properties, in addition to the drug dissolution rate of the tablets prepared, were examined and the results are summarized in Table II. Direct compression techniques were used to prepare the tablets in which Cop-MC was used as the only binder, filler, and disintegrant. Crushing strength values of the tablets obtained from both formulations showed high internal binding and fast disintegration time. Full drug release was obtained at 10-min dissolution time for both drugs. This indicates that Cop-MC functions as a binding, disintegration, and dissolution promoter. However, it is worth mentioning that although methyldopa tablets showed superdisintegrating characteristics, this did not have an influence on their coating using an aqueous coating system.
Table II.
Physical Properties of Rosuvastatin 20 mg, Methyldopa 250 mg, and Amlodipine 10-mg Tablets
| Tested parameter | Rosuvastatin 20 tablets | Methydopa 250 tablets | Amlodipine 10 tablets | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Formula 1 | Formula 2 | Formula 3 | Crestor® 20 | Formula 4 | Aldomet® 250 | Formula 5 | Norvasc® 10 | ||
| Weight (mg) | 290 | 315 | 450 | 355 | 200 | 400 | |||
| Shape (mm) | Round shallow biconvex of 9-mm diameter | Pink round of 9-mm diameter | Round shallow biconvex of 9-mm diameter | Round shallow biconvex of 9-mm diameter | Round flat faced beveled edged of 8-mm diameter | Elongated octagon flat faced beveled edged | |||
| Crushing strength (N) | 119 | 120 | 120 | –a | 140 | –a | 110 | 112 | |
| Friability (%) | 0.35 | 0.11 | 0.30 | –a | 0.25 | –a | 0.15 | 0.24 | |
| Disintegration time (s) | 120 | 160 | 120 | 180 | 35 | 120 | 30 | 10 | |
| Dissolution (%) | 10 min | 91 | 96 | 96 | 82 | 99 | 101 | 97 | 95 |
| 20 min | 91 | 95 | 97 | 88 | 101 | 102 | 98 | 98 | |
| 30 min | 92 | 96 | 96 | 92 | 101 | 101 | 97 | 99 | |
aNot performed as the tablets are film-coated
Stability of Active Ingredients in the Presence of Cop-MC
The DSC thermogram obtained for the initial analysis of pure methyldopa (Fig. 10h) shows an endothermic peak at about 124.5°C which corresponds to the loss of water of crystallization (sesquihydrate). The exothermic decomposition peak observed at about 300°C is assigned to the melting of methyldopa. The DSC thermograms (Fig. 10f, g) of a pure methyldopa sample stored for 6 months at 40°C/75% RH, in open and closed containers, showed an absence of secondary peaks corresponding to impurities. The splitting obtained in the endothermic peak of the loss of the hydrate in the 120–130°C temperature range is most likely to be due to the different melting patterns of the sesquihydrate in the methyldopa structure.
Fig. 10.

DSC scans of methyldopa 250-mg tablets (formula 4) a after 6 months at 40/75% RH—close amber glass bottles, b after 6 months at 40/75% RH—open amber glass bottles, c initial analysis, d reference formula (without active ingredient) at 40/75% RH—close amber glass bottles, e reference formula (without active ingredient) at 40/75% RH—open glass amber bottles, f methyldopa at 40/75% RH—close amber glass bottles, g methyldopa at 40/75% RH—open amber glass bottles and methyldopa initial analysis
The DSC thermograms of formula 4 (without active ingredient) samples (Fig. 10d, e) stored for 6 months at 40°C/75% RH in open and closed containers showed only one endothermic peak obtained at about 167°C, which corresponds to mannitol. The DSC scan of the samples of methyldopa 250-mg tablets (formula 4) analyzed initially (Fig. 10c) and the samples stored at 40°C/75% RH in closed (Fig. 10a) and open amber glass bottles (Fig. 10b) for 6 months showed superimposition whereby no secondary peaks were obtained upon storage under the accelerated conditions of temperature and humidity (40°C/75% RH).
Mixing methyldopa with Cop-MC lowers the decomposition peak temperature of methyldopa from 300°C to 270°C; this can be attributed to the interaction of two different crystalline structures. Such an interaction does not affect the stability of methyldopa as indicated by the absence of new/additional DSC peaks (Fig. 10a–c) (45). The absence of new/additional peaks is an indication of the thermal stability and compatibility of methyldopa in the tablet matrix containing Cop-MC.
The DSC study performed on methyldopa 250-mg tablets (formula 4) was further supported by a HPLC study. HPLC was used to investigate the chemical stability and compatibility of the tablets containing methyldopa and Cop-MC according to formula 4. The HPLC method was initially validated before use and the results showed a good separation between methyldopa and O-3-methylmethyldopa-related compound (the resolution R1 is more than 3; the relative standard deviation of replicate injections is <2%). In addition, analyses of samples obtained from a stress testing study (0.1 N NaOH, 0.1 N HCl, and 0.3% H2O2 at 25°C for 10 days) in solution indicated that the HPLC method is stability-indicating as shown by a significant decrease in percentage assay of methyldopa in the basic media and in the presence of hydrogen peroxide as oxidizing agent (35% and 89% (w/w), respectively). The accuracy of the method was investigated by testing six different sample preparations containing methyldopa at two different concentrations (5 and 25 mg/100 mL) for measuring the percentage impurities and percentage assay, respectively. The percentage recovery and RSD for six replicates are within the range of 98.1–102.9% and 0.3–1.4%, respectively. The method is linear over a concentration range of 1.3–50.0 mg/100 mL with an r2 value of >0.999. The LOQ value is 0.7 mg/100 mL. Table III shows the analytical data obtained for methyldopa 250-mg tablet (formula 4) samples stored in open and close amber glass bottles at 40°C/75% RH for 6 months. Pure methyldopa and Aldomet® 250 tablets were treated in the same manner and analyzed as a reference for this study. No significant decrease in the assay of methyldopa 250-mg tablets according to formula 4 was observed upon storage under the accelerated condition of temperature and humidity (40°C/75% RH). A small increase in the total impurities for samples of formula 4, stored in open amber glass bottles at 40°C/75% RH for 6 months, was observed when compared to the samples stored in closed amber glass bottles and Aldomet® 250 tablets (Table III, Fig. 11).
Table III.
Stability Data for Alpha Methyldopa 250-mg Film-Coated Tablets at 40°C/75% Relative Humidity
| Product | Period (months) | Assay (%) <5.0%a from initial | Impurities profile (%) | % Drug release | ||||
|---|---|---|---|---|---|---|---|---|
| O-3 MDb ≤1.0% | Total (1.0%) | 5 min | 10 min | 15 min | 20 min | |||
| Methyldopa | Initial | 99.6 | 0.01 | 0.19 | – | – | – | – |
| 6/close | 102.2 | 0.01 | 0.22 | – | – | – | – | |
| 6/open | 99.6 | 0.01 | 0.21 | – | – | – | – | |
| Formula 4 | Initial | 99.4 | 0.01 | 0.18 | 95.6 | 99.5 | 100.5 | 100.8 |
| 1/close | 97.7 | 0.01 | 0.20 | 92.2 | 98.5 | 101.5 | 102.1 | |
| 1/open | 98.1 | 0.02 | 0.16 | 75.4 | 92.6 | 96.1 | 98.0 | |
| 2/close | 97.4 | 0.01 | 0.17 | 92.9 | 97.6 | 100.0 | 100.2 | |
| 2/open | 99.9 | 0.02 | 0.16 | 75.8 | 97.8 | 100.5 | 101.1 | |
| 3/close | 98.4 | 0.01 | 0.22 | 92.0 | 98.9 | 99.8 | 102.4 | |
| 3/open | 98.6 | 0.01 | 0.17 | 79.2 | 96.8 | 99.5 | 100.0 | |
| 6/close | 99.2 | 0.01 | 0.16 | 89.3 | 96.4 | 98.4 | 99.8 | |
| 6/open | 98.2 | 0.01 | 0.31 | 88.2 | 95.5 | 98.3 | 99.4 | |
| Aldomet® 250 | Initial | 100.9 | 0.01 | 0.14 | 91.6 | 101.2 | 102.1 | 101.9 |
| 6/close | 97.8 | 0.01 | 0.29 | 83.4 | 94.7 | 99.1 | 100 | |
| 6/open | 98.0 | 0.01 | 0.27 | 57.1 | 77.5 | 90.0 | 95.3 | |
aAccording to ICH guideline for stability testing of new drug substances and products (46)
b O-3 MD represents O-3-methylmethyldopa impurity
Fig. 11.

HPLC chromatograms of a methyldopa, b methyldopa 250-mg tablets (formula 4), and c Aldomet 250-mg tablets. Where a is the initial chromatogram (no incubation), and b/c after 3 and 6 months of incubation at 40°C/75% RH, respectively, in closed containers
Conclusions
Co-processing of α-chitin with crystalline mannitol significantly improves the performance and functionality of both components. This offers a potential use of this co-processed additive as a single tablet excipient displaying superdisintegrating properties. The mechanical strength of the tablets and lubricant sensitivity were found to be dependent upon the mannitol content and the processing technique used in the preparation of the co-processed excipient. The functionality of Cop-MC is not affected by the tablet preparation procedure whether it is direct mixing or dry/wet granulation. Utilization of Cop-MC, as an excipient, in tablet formulations containing active pharmaceutical ingredients offers excellent chemical stability, binding, and disintegration properties.
References
- 1.Peck GE, Baley GJ, McCurdy VE, Banker GS. Tablet formulation and design. In: Lieberman HA, Lachman L, Schwartz JB, editors. Pharmaceutical dosage forms: tablets, vol. 1. 2. New York: Marcel Dekker; 1989. pp. 88–89. [Google Scholar]
- 2.Block LH, Moreton RC, Apte SP, Wendt RH, Munson EJ, Creekmore JR, et al. Co-processed excipients. In: Pharmacopeial forum, vol. 35 (4). Maryland, USA: United States Pharmacopeia Convention Inc;2009. p. 1026–8.
- 3.Westerhuis JA, de Haan P, Zwinkels J, Jansen WT, Coenegracht PJM, Lerk CF. Optimisation of the composition and production of mannitol/microcrystalline cellulose tablets. Int J Pharm. 1996;143:151–62. doi: 10.1016/S0378-5173(96)04699-6. [DOI] [Google Scholar]
- 4.Jian-Xin LI, Brian C, Thomas R. Co-processed microcrystalline cellulose and sugar alcohol as an excipient for tablet formulations. Applicant: FMC Corp. (US), European Patent Office, Patent No. US20080131505.
- 5.Sherwood BE, Hunter EA, Staniforth JH. Pharmaceutical excipient having improved compressibility. Applicant: Edward H Mendell Co. Inc. (US), United States Patent Office, Patent No. 5,585,115.
- 6.Muzzarelli RAA. Chitin. Oxford: Pergamon; 1977. [Google Scholar]
- 7.Safety data for chitin. http://msds.chem.ox.ac.uk/CH/chitin.html. Access date: 1/7/2010.
- 8.Uragami T, Tokura S. Material science of chitin and chitosan. Berlin: Springer; 2006. [Google Scholar]
- 9.Mir VG, Heinämäki J, Antikainen O, Revoredo OB, Colarte AI, Nieto OM, Yliruusi J. Direct compression properties of chitin and chitosan. Eur J Pharm Biopharm. 2008;69:964–8. doi: 10.1016/j.ejpb.2008.01.029. [DOI] [PubMed] [Google Scholar]
- 10.Rashid I, Daraghmeh N, Al-Remawai M, Leharne SA, Chowdhry BZ, Badwan A. Characterization of chitin–metal silicates as binding superdisintegrants. J Pharm Sci. 2009;98:4887–901. doi: 10.1002/jps.21781. [DOI] [PubMed] [Google Scholar]
- 11.Rashid I, Al-Remawi M, Eftaiha A, Badwan A. Chitin–silicon dioxide coprecipitate as a novel superdisintegrant. J Pharm Sci. 2008;97:4955–69. doi: 10.1002/jps.21354. [DOI] [PubMed] [Google Scholar]
- 12.El-Barghouthi M, Rashid I, Eftaiha A, Al-Remawi M, Badwan A. A novel superdisintegrating agent made from physically modified chitosan with silicon dioxide. Drug Dev Ind Pharm. 2008;34:373–83. doi: 10.1080/03639040701657792. [DOI] [PubMed] [Google Scholar]
- 13.Armstrong NA. Mannitol. In: Rowe RC, Sheskey PJ, Owen SC, editors. Pharmaceutical excipients. USA: Pharmaceutical Press and American Pharmacists Association; 2006. pp. 439–453. [Google Scholar]
- 14.Erik L, Philippe L, Jose L. Pulverulent mannitol and process for preparing it. Applicant: Roquette Freres, Erik L, Philippe L, Jose L, United States Patent Office, Patent No. 6,743,447.
- 15.Daraghmeh NH, Al Omari MM, Badwan AA. Pharmaceutical excipient, method for its preparation and use thereof. European Patent Office (EPO). Patent Filed.
- 16.Gupta P, Nachaegari SK, Bansal AK. Improved excipient functionality by coprocessing. In: Katdsre A, Chaubal MV, editors. Excipient development for pharmaceutical, biotechnology, and drug delivery systems. New York: Informa Healthcare; 2006. p. 123. [Google Scholar]
- 17.Disintegration, friability of uncoated tablets, resistance to crushing of tablets. In: British pharmacopeia. London: The Stationary Office. Volume IV, Appendixes XIIA, XVII G and H; 2008. p. A283, A423, A424.
- 18.Weast RC. Handbook of chemistry and physics. 55. Boca Raton: CRC; 1974–1975. [Google Scholar]
- 19.Dissolution <711>. United States Pharmacopeia and National Formulary (USP32-NF27). Rockville, MD: US Pharmacopoeia Convention. Volume 1;2009. p. 263–71.
- 20.Dissolution method. U.S. FDA, Rockville. 2010. http://www.fda.gov/scripts/cder/dissolution/index.cfm. Access date 4 Feb 2010.
- 21.Kawakita K, Ludde KH. Some considerations on powder compression equations. Powder Technol. 1971;4:61–8. doi: 10.1016/0032-5910(71)80001-3. [DOI] [Google Scholar]
- 22.Shivanand P, Sprockel OL. Compaction behaviour of cellulose polymers. Powder Technol. 1992;69:177–84. doi: 10.1016/0032-5910(92)85072-4. [DOI] [Google Scholar]
- 23.Lin C, Cham T. Compression behaviour and tensile strength of heat-treated polyethylene glycols. Int J Pharm. 1995;118:169–79. doi: 10.1016/0378-5173(94)00343-4. [DOI] [Google Scholar]
- 24.Nordström J, Klevan I, Alderborn G. A particle rearrangement index based on the Kawakita powder compression equation. J Pharm Sci. 2008;98:1053–63. doi: 10.1002/jps.21488. [DOI] [PubMed] [Google Scholar]
- 25.Giron D. Applications of thermal analysis in the pharmaceutical industry. J Pharm Biomed Anal. 1986;4:755–70. doi: 10.1016/0731-7085(86)80086-3. [DOI] [PubMed] [Google Scholar]
- 26.Botha SA, Lotter AP. Compatibility study between naproxen and tablet excipients using differential scanning calorimetry. Drug Dev Ind Pharm. 1990;16:673–83. doi: 10.3109/03639049009104410. [DOI] [Google Scholar]
- 27.Lin SY, Han RY. Differential scanning calorimetry as a screening technique to determine the compatibility of salbutamol sulfate with excipients. Pharmazie. 1992;47:266–8. [Google Scholar]
- 28.Badwan AA. The Jordanian Pharmaceutical Manufacturing Co., Unpublished data.
- 29.Validation of compendia procedures <1225>. In: United States Pharmacopeia and National Formulary (USP32-NF27). Rockville, MD: US Pharmacopoeia Convention. Volume I;2009. p. 733–6.
- 30.Methyldopa tablets. In: United States Pharmacopeia and National Formulary (USP32-NF27). 2009. Rockville, MD: US Pharmacopoeia Convention. Volume 3;2009. p. 2942.
- 31.Qiang D, Gunn J, Zong Z, Buckner I. Evaluating the effect of lubrication on powder compaction with the compression calorimeter. AAPS Journal. 2009;11(S2). http://www.aapsj.org/abstracts/AM_2009/AAPS2009-001716.PDF. Accessed 2 March 2010.
- 32.Carlson GT, Hancock BC. A comparison of physical and chemical properties of common tableting diluents. In: Katdsre A, Chaubal MV, editors. Excipient development for pharmaceutical, biotechnology, and drug delivery systems. New York: Informa Healthcare; 2006. p. 129. [Google Scholar]
- 33.Nyström C, Alderborn G, Duberg M, Karehill P-G. Bonding surface area and bonding mechanism—two important factors for the understanding of powder compactibility. Drug Dev Ind Pharm. 1993;19:2143–96. doi: 10.3109/03639049309047189. [DOI] [Google Scholar]
- 34.Alderborn G, Börjesson E, Glazer M, Nyström C. Studies on direct compression of tablets. XIX. The effect of particle size and shape on the mechanical strength of sodium bicarbonate tablets. Acta Pharm Suec. 1988;25:31–40. [PubMed] [Google Scholar]
- 35.Paronen P, Iilla J. Porosity–pressure functions. In: Alderborn G, Nyström C, editors. Pharmaceutical powder compaction technology. New York: Marcel Dekker; 1996. pp. 55–75. [Google Scholar]
- 36.Adetunji OA, Odeniyi MA, Itiola OA. Compression, mechanical and release properties of chloroquine phosphate tablets containing corn and trifoliate yam starches as binders. Trop J Pharm Res. 2006;5:589–96. [Google Scholar]
- 37.Zhang Y, Law Y, Chakrabarti S. Physical properties and compact analysis of commonly used direct compression binders. AAPSPharmSciTech 2005;4(4), Article 62. [DOI] [PMC free article] [PubMed]
- 38.Summary of product characteristics (SPC) for Crestor® 20 and Istin® 10 tablets. In: Electronic Medicines Compendium. 2009 edition, Datapharm Communications Ltd, Leatherhead, United Kingdom. http://emc.medicines.org.uk/default.aspx. Accessed 2 March 2010.
- 39.Richard CJ, Alfred WN. Pharmaceutical composition comprising A HMG COA reductase inhibitor. Applicant: Astrazeneca AB (SE), European Patent Office. Patent No. EP1223918.
- 40.Fritz B, Adriaan DSP, Martin S. Crystalline forms of rosuvastatin calcium salt. Applicants: Ciba SC Holding AG, Fritz B, Adriaan DSP, Martin S, European Patent Office, Patent No. WO2006079611.
- 41.Shlomit W, Valerie N-H, Shalom S. Crystalline rosuvastatin calcium. Applicant: Teva Pharma, Shlomit W, Valerie N-H, Shalom S, European Patent Office, Patent No. WO2008036286.
- 42.Koichi W, Hikaru F. Tablet formulation. Applicant: NOVO NORDISK AS. (DK), European Patent Office, Patent No. US2009252790.
- 43.The Internet Drug Index (RxList). 2010. http://www.rxlist.com/aldomet-drug.htm. Accessed 4 Feb 2010.
- 44.Abdoh A, Al-Omari MM, Badwan AA, Jaber AMY. Amlodipine besylate–excipients interaction in solid dosage form. Pharmaceut Dev Tech. 2004;9:15–24. doi: 10.1081/PDT-120027414. [DOI] [PubMed] [Google Scholar]
- 45.Agatonovic-Kustrin S, Markovic N, Ginic-Markovic M, Mangan M, Glass BD. Compatibility studies between mannitol and omeprazole sodium isomers. J Pharm Biomed Anal. 2008;48:356–60. doi: 10.1016/j.jpba.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 46.ICH Topic Q1 A (R2) Stability testing of new drug substances and products. European Medicine Agency (EMEA). 2003. http://www.ema.europa.eu/pdfs/human/ich/273699en.pdf. Accessed 4 Feb 2010.