

Abstract
Etodolac (ET) is a nonsteroidal anti-inflammatory drug with proved potential antitumor and uric acid lowering effects. It shows dissolution rate-dependent bioavailability. This work was carried out to improve the dissolution rate of etodolac using three carriers of known potential to improve solubility and hence dissolution rate of poorly soluble drugs through coevaporation technique. The polymeric surfactant inutec, 2-hydroxypropyl-β-cyclodextrin, and tromethamine were used at three different drug/carrier ratios. The dissolution rate of ET at pH 1.2 and 6.8 is improved in all of the solid dispersion systems compared to that of the pure drug and physical mixtures. DSC of coevaporates at 1:5 drug/carrier ratio providing the fastest dissolution rate suggested loss of ET crystallinity which was further confirmed by X-ray diffraction. Inutec-based coevaporate was chosen for the formulation of ET chewable tablets. Chewable tablets (F3) that met the USP monograph specifications for ET tablets, with 86% dissolved amount within 15 min, was chosen for in vivo absorption study in comparison with pure ET-filled hard gelatin capsules. The results showed significantly higher mean Cmax and shorter mean Tmax (about 2 h earlier) and about 1.32-fold higher mean AUC0–24 values for the F3 chewable tablets compared to ET-filled capsules.
Key words: chewable tablets, etodolac, hydroxypropyl-β-cyclodextrin, inutec, in vivo absorption study, solid dispersion
INTRODUCTION
Oral route is perhaps the most widely used route for drug delivery systems. It is well established that the active ingredient in a solid dosage form must undergo successive rate processes, before it is available for absorption from the gastrointestinal tract. These processes are disintegration, release of the drug, and dissolution of the drug in an aqueous environment. The rate at which drug reaches the circulatory system is determined by the slowest step in the sequence of rate processes (1,2).
Depending on the biopharmaceutical classification system (3), dissolution rate is the rate-controlling step in the absorption process for drugs possessing high membrane permeability but low aqueous solubility (class II drugs).
Today, 35–40% of all new chemical entities suffer from poor aqueous solubility. The poor dissolution characteristics of water-insoluble drugs, which lead to a poor and/or varying bioavailability after oral administration, are a major challenge for pharmaceutical scientists (4,5).
Several approaches have been developed over years to enhance the drug dissolution rate and hence bioavailability. These approaches include producing solid dispersion of the drug with additives (6), making liquisolid formulations (7), increase surface area of drug particles via micronization and nanosizing (8–10), complex formation with cyclodextrin (11), water-soluble prodrug formation (12), and water-soluble salt formation (13).
Etodolac (ET; (1,8-diethyl-1,3,4,9-tetrahydropyrano[3,4-6]indol-1-yl) acetic acid) is a nonsteroidal anti-inflammatory agent prescribed for the treatment of acute pain, osteoarthritis, and rheumatoid arthritis at an oral dose of 200 mg twice daily; up to 1,200 mg daily may be given if necessary (14,15). It has also been reported to be effective in the treatment of gout by lowering uric acid blood levels in humans (16,17). The drug shows high therapeutic index between gastric irritation and anti-inflammatory effects. Recent studies have proved that etodolac has antitumor effect on different human cancer cells (18,19). ET is one of the selective COX-2 inhibitors; it possesses 10-fold COX-2 selectivity over COX-1 (20) responsible for the production of prostaglandins involved in cytoprotection of gastric mucosa and regulation of the renal blood flow. ET thus safely treats inflammatory disorders without causing gastric irritation, ulceration, or bleeding.
ET is practically water insoluble; its oral bioavailability is expected to be limited by its dissolution rate, which might be increased using solid dispersion technology.
The main objectives of this work are to investigate the possibility of improving dissolution rate of etodolac via simple solid dispersion method (coevaporation) using three carriers, namely the polymeric surfactant inutec ®SP 1 (inutec is a graft copolymer based on a naturally occurring polysaccharide, namely inulin (polyfructose) that has been hydrophobically modified by introducing several alkyl groups on the linear polyfructose chain (6)), 2-hydroxypropyl-β-cyclodextrin (HPβCD), and tromethamine, at three drug-to-carrier ratios, formulation of etodolac chewable tablets to get a benefit from the expected increase in etodolac solubility at pH of the buccal cavity (21); in addition to that, chewable tablets provide a mean for administration of large tablets to children and adults who found difficulty in swallowing solid dosage forms (22), and finally to assess the in vivo absorption of selected formula in comparison to pure ET-filled hard gelatin capsules. The market lacks etodolac dosage form which is suitable for geriatrics and pediatrics administration; therefore, formulation of etodolac as chewable tablets could be beneficial in improving compliance and convenience of these patients.
MATERIALS AND METHODS
Materials
Etodolac was kindly supplied by Pharco Pharmaceuticals, Alexandria, Egypt; HPβCD by Alfa Aesar, Germany; Inutec® SPI by Beneo Biobased Chemicals, Belgium; tromethamine by MP Biomedical, LLC, Germany; sodium lauryl sulfate, mannitol, ethanol, and methanol by El-Nasr Company for Pharmaceuticals, Cairo, Egypt; emedex and sodium stearyl fumarate; PRUV® by JRS Pharma GmbH, Rosenberg, Germany; Galen IQTM 720 by Beneo-Palatinit, GmbH, Germany; and high-performance liquid chromatography (HPLC) grade Acetonitrile by Merk Co, Hohenbrunn, Germany. All other reagents and chemicals were of analytical grade.
Preparation of Physical Mixtures
The physical mixtures (PM) of etodolac and different carriers were prepared by mixing both components in a mortar for 5 min followed by sieving (<500 μm). The following ratios of etodolac/different carriers (w/w) were prepared: 1:1, 1:2, and 1:5.
Preparation of Solid Dispersions (Coevaporates)
Etodolac and the carrier at the same ratios used for preparation of physical mixtures were dissolved in the least amount of ethanol (inutec was dissolved in methanol); the solvent was evaporated under reduced pressure at 40°C. The dried mass was pulverized and sieved (<500 μm), and the collected mass was used for further investigation.
Physicochemical Characterization
Differential Scanning Calorimetry
The differential scanning calorimetry (DSC) measurements were performed using a Shimadzu DSC-60 (Kyoto, Japan). Samples (5 mg) were sealed in aluminum pans and analyzed in an atmosphere of nitrogen at a flow rate of 30 ml/min. A temperature range of 0°C to 200°C was used and the heating rate was 10°C/min.
X-ray Diffraction
The X-ray diffraction (XRD) was obtained using Scintag XGEN-4000 X-ray diffractometer (Advanced Diffraction system, Scintag Inc., USA). The samples were exposed to Cu-Ka radiation (40 kV × 30 mA) at a scan rate of 2°/min over the 2θ range of 4–50°; the output is given as intensity versus 2θ.
Determination of Etodolac Content
An amount of etodolac physical mixtures or coevaporates, equivalent to 50 mg of etodolac, was dissolved in 100-ml simulated saliva fluid (pH 6.8), the solution was filtered, and the absorbance of the filtrate was measured spectrophotometrically at 279 nm.
Stability Testing
The selected ET/inutec (1:5) coevaporate was stored in an aluminum package under ambient conditions of temperature and humidity to evaluate its physical stability (drug recrystallization); the samples were analyzed after 6 and 12 months of storage using DSC and XRD.
Preparation of ET Chewable Tablets
The ET/inutec (1:5) coevaporate, 10% NaHCO3 (microenvironment alkaline pH modifier), 15% microcrystalline cellulose, 2% aspartame (sweetening agent), 2% strawberry flavor, 0.5% Pruv® (lubricant), and 22.5% diluent of either mannitol (F1); galen IQTM 720 (F2); or emedex (F3) were blended thoroughly using a pestle and mortar. Tablets were compressed using a single punch tablet press (17 mm punches and die set, model TDP, Shanghai Tianhe, Pharmaceutical Machinery Factory, China); the hardness of the tablets was kept constant at approximately 4 kg (HDT-300, Logan Instruments Corp., Somerset, NJ, USA). The content of etodolac was maintained at 200 mg/tablet.
Evaluation of the Prepared Tablets
Drug Content Uniformity
Ten tablets were weighed individually, crushed, and dissolved in 100-ml simulated saliva fluid (pH 6.8). The solution was filtered and the absorbance of the filtrate was measured spectrophotometrically at 279 nm.
Tablet Friability
A sample of whole tablets corresponding to 6 g was placed in the drum of a tablet friability test apparatus (FAB-2, Logan Instruments Corp., Somerset, NJ, USA). The drum was adjusted to rotate at 25 rpm for a period of 4 min; then, the tablets were removed from the drum, dedusted, and accurately weighed. The percent weight loss was calculated.
Dissolution Testing
Dissolution profiles of pure etodolac, physical mixtures, and coevaporates (equivalent to 50 mg of etodolac) were determined at 37 ± 0.5°C at a stirring rate of 100 rpm using the USP 30 dissolution apparatus II in 900 ml of simulated gastric fluid (0.1 N HCl, pH 1.2) or simulated saliva fluid (Sorensen’s phosphate buffer, pH 6.8). Three milliliters aliquot samples were withdrawn with replacement at 5, 10, 15, 30, 45, and 60 min. Samples were filtered using 0.45 μm Millipore filters and properly diluted prior to measuring their absorbance at either λmax 274 nm (pH 1.2) or λmax 279 nm (pH 6.8).
Dissolution efficiency at 30 min (DE30) was calculated from the area under the dissolution curve at time t (measured using trapezoidal rule) and expressed as a percentage of the area of the rectangle described by 100% dissolution at the same time (23). Amount of drug (in micrograms) dissolved per minute that presented by each tablet formulation during 30 min (dissolution rate, DR30) was calculated as follows (24):
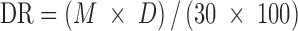 |
1 |
where M is the total amount of ET in each tablets and D denotes percentage of drug dissolved in 30 min.
Dissolution studies of chewable tablets (crushed tablets to simulate chewing) were carried out in 900 ml of simulated gastric fluid containing 0.5% (w/v) sodium lauryl sulfate or 900 ml of simulated saliva fluid. Samples were collected at 5, 10, 15, and 30 min; the procedures were continued as mentioned above.
In Vivo Absorption Study
Study Design
The study was approved by the Cairo University Protection of Human Subjects Committee and the protocol complies with the declarations of Helsinki and Tokyo for humans. The study was carried out to compare the pharmacokinetics of etodolac from chewable tablets (F3), treatment A, to pure etodolac powder filled in hard gelatin capsules, treatment B, following administration of single doses of 200 mg each using open-label, two-treatment, two-period, randomized crossover design with a washout period of 14 days. Three healthy volunteers in ages from 25 to 32 years participated in the study. All subjects were prohibited from taking medicines and smoking for 1 week before the beginning of the studies to the end of the test. All subjects fasted for at least 10 h before the study day (25). Subjects taking etodolac chewable tablets (F3) were asked to chew the tablets for 10 min and swallow them without water. Pure etodolac powder-filled capsules were taken with 180 ml of water.
Blood samples for pharmacokinetic analysis were obtained immediately before drug intake and at 0.17, 0.33, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, and 24 h after dosing. Blood samples were collected in heparinized tubes and were centrifuged for 10 min at 3,000 rpm at room temperature within 1 h of collection. Separated plasma was aspirated and transferred into plastic tubes and was stored at −20°C until assayed.
HPLC Analysis
Plasma concentrations of etodolac were determined using an HPLC procedure described by Nahla S. Barakat with slight modification (26). The column was ODS3, 5 μm, 4 × 150 mm (Shimadzu, Japan). The mobile phase consisted of acetonitrile/phosphate buffer (pH 7.4; 40:60, v/v), and the flow rate was 0.8 ml/min. The effluent was monitored at 278 nm using a SPD-10AVP, UV visible detector.
Primary standard solution (100 μg/ml) of etodolac was prepared in methanol. Blank plasma samples were spiked with etodolac stock solution to contain 0.5–20 μg/ml. Spiked plasma sample were mixed with methanol/acetonitrile (1:1) mixture, and the tubes were vortexed for 30 s and centrifuged for 10 min at 3,000 rpm; 50 μl of the supernatants was injected onto HPLC column. Pioglitazone was used as internal standard (5 μg/ml).The retention time was 4.8 and 8 min for etodolac and pioglitazone, respectively. A standard curve was constructed by plotting the peak area ratio of etodolac to pioglitazone against etodolac concentrations in plasma. All assays were performed in triplicate. The linearity, precision, accuracy, and specificity of the method were demonstrated.
Pharmacokinetic and Statistical Calculations
Peak concentration (Cmax) and time to peak concentration (Tmax) were derived directly from the individual plasma concentration–time curves. The other pharmacokinetic parameters were computed by noncompartmental analysis using WinNonlin® software (version 1.5, scientific consulting, Inc, Cary, NC, USA).
The pharmacokinetic parameters of the two tested formulae were compared by two-way analysis of variance using the software SPSS (SPSS Inc., Chicago, IL, USA). The significance of the difference was determined at α = 0.05.
RESULTS AND DISCUSSION
Physical Characterization and Evaluation of ET Processed Systems
The results of dissolution studies are presented in Fig. 1a, b and summarized in Table I, expressed in terms of dissolution efficiency (DE30), dissolution percentage (DP30), and dissolution rate (DR30) at 30 min. Since the 1:5 mixing ratio showed the most significant effect on dissolution rate, only the profiles of the 1:5 ratio were depicted.
Fig. 1.

Dissolution profiles of pure etodolac and etodolac/carrier (1:5 ratio) systems at pH 1.2 (a) and pH 6.8 (b); ET etodolac, PM physical mixture, CE coevaporate
Table I.
Dissolution Efficiency, Dissolution Percentage, and Dissolution Rate Values at 30 Min for Etodolac PM and Etodolac CE Systems at pH 1.2
| Parameters | Inutec | HPβCD | Tromethamine | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1:1 | 1:2 | 1:5 | 1:1 | 1:2 | 1:5 | 1:1 | 1:2 | 1:5 | ||||||||||
| PM | CE | PM | CE | PM | CE | PM | CE | PM | CE | PM | CE | PM | CE | PM | CE | PM | CE | |
| DP30 | 25.8 | 47 | 26.1 | 42 | 38 | 81 | 8 | 35 | 9 | 44 | 15 | 71 | 27 | 35 | 25 | 40 | 34 | 59 |
| DE30 | 19.10 | 32.75 | 16.92 | 30.16 | 24.2 | 66.75 | 6.83 | 26.75 | 7.08 | 34.11 | 12.91 | 63.25 | 22.72 | 28.08 | 20.83 | 35.83 | 30.33 | 50.41 |
| DR30 (μg/min) | 430 | 783.33 | 435 | 700 | 633.33 | 1,350 | 133.3 | 583.33 | 150 | 733.33 | 250 | 1,183.3 | 450 | 583.33 | 416.67 | 666.67 | 566.66 | 983.33 |
Etodolac DP30 = 3 μg/min, DE30 = 2.41 μg/min, and DR30 = 50 μg/min
DP 30 dissolution percentage at 30 min, DE 30 dissolution efficiency at 30 min, DR dissolution rate at 30 min, PM physical mixtures, CE coevaporated, HPβCD 2-hydroxypropyl-β-cyclodextrin
As shown in Fig. 1, pure ET and all preparations showed markedly higher amount dissolved at pH of buccal cavity (6.8) compared with that at pH of gastric fluid (1.2); ET is a weakly acidic drug having a pKa value of 4.65; its solubility dramatically increases at pH values above its pKa (27,28). Pure ET percent dissolved at pH 6.8 (52%) in 30 min is 17.33 times higher compared to that at pH 1.2 (3%). However, the amount dissolved is less than that specified by USP 30 monograph for ET tablets which states that not less 80% of ET should be dissolved in 30 min, which confirms the need for enhancing of ET dissolution rate particularly at pH 1.2.
As shown in Fig. 1 and Table I, all used carriers were effective in enhancing ET DR and their performance is dependent on both their content in the mixture and the system preparation method. The obtained dissolution rates could be ranked as follows: coevaporated systems > physical mixtures > pure drug.
The improvement in the DR caused by PM could be attributed to reduction of the interfacial tension between the hydrophobic drug particles and the dissolution medium owing to the presence of the hydrophilic polymers, in addition to local solubilizing effect acting during the early stages of the dissolution process in the microenvironment surrounding the particles (29). Interestingly, it was found that DR of ET from PM with tromethamine (alkalinizing agent) was higher than that of PM with other two carriers which is thought to be due to wetting of particles and instantaneous increase in pH in the diffusion layer of the dissolution medium (30).
The evident improvement obtained with coevaporated systems was due to the closer contact between the components and the better dispersion of the drug into the hydrophilic carrier obtained through the coevaporation technique (31); in addition to reduction in the drug particle size, increased wettability of the drug particles (32) and in situ increase in pH in the diffusion layer of the dissolution medium when tromethamine is in intimate contact with ET particles (30).
Generally, all drug dissolution parameters progressively improved with increasing carrier amount in the mixture and reached the highest values at 1:5 drug/carrier ratio except with inutec where dissolution improvement produced by 1:1 ratio was comparable to that obtained by 1:2 ratio.
The physical structure of drug coevaporates showing best performance (with inutec and 2-hydroxypropyl-β-cyclodextrin at 1:5 ratio) was investigated by DSC and X-ray powder diffraction.
Figure 2a, b shows DSC curves of ET/inutec and ET/2-hydroxypropyl-β-cyclodextrin coevaporates, respectively, in addition to pure ET and pure excipients. The DSC curve of ET is a typical of crystalline anhydrous substance, showing a sharp endothermic peak at 149.75°C corresponding to melting point of the drug. Complete disappearance of the drug thermal profile in the coevaporated systems indicates homogenous dispersion of the drug into carriers and complete drug amorphization.
Fig. 2.

DSC curves of etodolac/inutec (1:5) coevaporate (a) and etodolac/2- hydroxypropyl-β-cyclodextrin (1:5) coevaporate (b). ET etodolac, CE coevaporate
X-ray powder analysis was performed to confirm the results of DSC study (Fig. 3), and the coevaporated systems diffractograph showed the disappearance of ET characteristic diffraction peaks pointing to loss of drug crystallinity which is in agreement with DSC results.
Fig. 3.

X-ray diffraction patterns of etodolac/inutec and etodolac/2-hydroxypropyl-β-cyclodextrin coevaporated systems at 1:5 ratio
After 12 months of storage, DSC analysis (Fig. 4a) of stored ET/inutec (1:5) coevaporated system demonstrated the absence of characteristic endothermic peak of ET. However, a trace of one ET characteristic peak was detectable in XRD analysis (Fig. 4b). No endotherm corresponding to melting of pure crystalline ET was observed because thermal energy in the DSC caused traces of ET crystals to dissolve completely in inutec below the melting temperature of the crystalline ET (30). It can be concluded that the stability of ET in the selected coevaporated system is satisfactory with no appreciable change in the amorphous state of ET in the coevaporate, which could be attributed to crystal growth inhibiting properties of inutec (33). Also, ET/inutec (1:5) coevaporated system (81% DP30 at pH 1.2 and 99% DP30 at pH 6.8) satisfied the compendial requirement (80% ET in 30 min); therefore, it was chosen for the preparation of ET chewable tablets.
Fig. 4.

Solid-state characterization of etodolac/inutec (1:5) coevaporate after 12 months of storage under ambient conditions, DSC (a) and XRD (b) analysis
Evaluation of ET Chewable Tablets
The effect of NaHCO3, MgO, and tromethamine as microenvironmental pH modifiers on percent ET dissolved was investigated; insignificant difference was found between different alkalinizers (81%, 80.8%, and 81.8% after 15 min for NaHCO3, MgO, and tromethamine, respectively). Therefore, NaHCO3 was chosen because it is widely used in edible products. Dissolution profiles of ET from different chewable tablets are presented in Fig. 5. The prepared tablets showed 88%, 91%, and 94.6% percent dissolved at 5 min for F1, F2, and F3, respectively, at pH 6.8 and 81.7%, 76%, and 86.7% percent dissolved at 15 min for F1, F2, and F3, respectively, at pH 1.2. However, the obtained dissolution profiles of all prepared tablets were insignificantly different either at pH 1.2 or at pH 6.8. Drug content uniformity results were found to conform to pharmacopoeial limits; 85–115% of the label claim. The mean percentage of drug content was 101.56 ± 0.89, 98.21 ± 0.22, and 96.11 ± 0.25 for F1, F2, and F3, respectively. The percentage weight loss for all the formulations was below 1%, indicating that the friability is within the compendial limits. F3 chewable tablet was chosen for subsequent in vivo study in comparison to pure ET-filled hard gelatin capsules.
Fig. 5.

Dissolution profiles of etodolac from different chewable tablets at pH 1.2 and pH 6.8
In Vivo Absorption Study
The calibration curve of etodolac was linear over the range from 0.5 to 20 μg/ml. The calibration curve had the regression equation of y = 0.4745x + 0.1164 with the determination coefficient (R2) of 0.9941. The assay method showed acceptable precision with CV% <6% (5.22% (low concentration; 2 μg/ml), 2.71% (medium concentration; 10 μg/ml), and 3.1% (high concentration; 20 μg/ml)) and CV% <8.5% (8.15% (2 μg/ml), 1.67% (10 μg/ml), and 0.89% (20 μg/ml)) for the inter-day assay and the intra-day assay, respectively. Also, the assay method showed acceptable accuracy with percentage relative error not exceeding 7.1% (0.29 (2 μg/ml), 2.29 (10 μg/ml), and −3.64 (20 μg/ml)) and percentage recovery range within 94.65–107.09% of the actual values. No endogenous plasma components were observed at the retention times corresponding to both the drug and internal standard which proves the specificity of the method.
Chewable tablets showed significantly higher plasma levels starting from the first sampling time and up to 2 h (p < 0.001–0.006) expressed in 2.5 times higher AUC0–2 compared to that of pure drug-filled capsules. Chewable tablets showed nearly six times higher plasma level (3.03 μg/ml) compared to filled capsules (0.54 μg/ml) during first 10 min (T0.17) following drug administration which could be attributed to pregastric absorption (34,35) of etodolac fraction that may dissolve at pH of the buccal cavity (6.8) during chewing process; etodolac aqueous solubility gradually increases with increasing pH up to 5 followed by 30-fold increase in solubility as the pH increases from 5 to 7 (21).
The mean plasma concentration–time profiles for the etodolac chewable tablets and pure etodolac-filled capsules are shown in Fig. 6. Remarkable differences in the shape of the plasma concentration–time courses between the two treatments were found, expressed by significantly higher mean Cmax and shorter mean Tmax (about 2 h earlier) values for the chewable tablets. The mean AUC0–24 estimate for treatment A which measures the total amount of drug absorbed over 24 h time period was significantly higher (about 1.32-fold higher) compared to treatment B; this reflects higher amount of drug absorption over 24 h period. It can be concluded that etodolac chewable tablets improved both rate and extent of drug absorption. The bioavailability parameters for both formulae in addition to the statistical analysis comparing the two formulae are summarized in Table II
Fig. 6.

Plasma concentration–time profiles of etodolac after oral administration of etodolac chewable tablets (F3) and pure etodolac-filled capsules to three healthy human volunteers (mean ± SD)
Table II.
Pharmacokinetic Parameters of Etodolac After Oral Administration of Chewable Tablets (F3) and Pure Etodolac-Filled Capsules to Three Healthy Human Volunteers Under Fasted Condition (Mean ± SD)
| Parameters | Chewable tablets | Pure ET | Statistical test (p) |
|---|---|---|---|
| C max (μg/ml) | 21.10 ± 2.8 | 15.80 ± 1.5 | 0.029 |
| AUC0–24 (μg h/ml) | 152.05 ± 11.42 | 115.13 ± 3.16 | 0.014 |
| AUC0–∞ (μg h/ml) | 176.23 ± 7.92 | 137.21 ± 8.70 | 0.006 |
| T max (h) | 1.66 ± 0.28 | 3.66 ± 0.57 | 0.022 |
| t 1/2 (h) | 8.25 ± 1.91 | 9.49 ± 2.63 | 0.659 |
ET etodolac, AUC area under the plasma concentration–time curve, C max peak concentration, T max time to peak concentration
CONCLUSION
Formulation of etodolac chewable tablets not only improved its dissolution rate-dependent bioavailability but also provides a useful tool to improve patient convenience and compliance. Chewable tablets are suitable for administration of large tablets to geriatrics and pediatrics that have difficulty in swallowing solid dosage forms.
Acknowledgments
Authors are grateful to Beneo Biobased Chemicals, Belgium and JRS CO., Germany for supplying us with the necessary chemicals to participate in the field of research.
References
- 1.Martin A, Swarbrick J, Cammarta A. Physical pharmacy, physical chemical principles in the pharmaceutical sciences. 4. Philadelphia: Lea and Febiger; 1993. pp. 324–512. [Google Scholar]
- 2.Shargel L, Yu ABC. Applied biopharmaceutics and pharmacokinetics. 3. New York: Prentice-Hall International; 1993. pp. 136–7. [Google Scholar]
- 3.Yu LX, Amidon GL, Polli JE, Zhao H, Mehta MU, Conner DP, et al. Biopharmaceutics classification system: the scientific basis for biowaiver extensions. Pharm Res. 2002;19(7):921–5. doi: 10.1023/A:1016473601633. [DOI] [PubMed] [Google Scholar]
- 4.Friedrich H, Fussnegger B, Kolter K, Bodmeier R. Dissolution rate improvement of poorly water-soluble drugs obtained by adsorbing solutions of drugs in hydrophilic solvents onto high surface area carriers. Eur J Pharm Biopharm. 2006;62:171–7. doi: 10.1016/j.ejpb.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 5.Biradar SV, Patil AR, Sudarsan GV, Pokharkar VB. A comparative study of approaches used to improve solubility of roxithromycin. Powder Technol. 2006;169:22–32. doi: 10.1016/j.powtec.2006.07.016. [DOI] [Google Scholar]
- 6.Van den Mooter G, Weuts I, De Ridder T, Blaton N. Evaluation of Inutec SP1 as a new carrier in the formulation of solid dispersions for poorly soluble drugs. Int J Pharm. 2006;316:1–6. doi: 10.1016/j.ijpharm.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 7.Javadzadeh Y, Jafari-Navimipour B, Nokhodchi A. Liquisolid technique for dissolution rate enhancement of a high dose water-insoluble drug (carbamazepine) Int J Pharm. 2007;341:26–34. doi: 10.1016/j.ijpharm.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 8.Rasenack N, Müller BW. Dissolution rate enhancement by in situ micronization of poorly water-soluble drugs. Pharm Res. 2002;19(12):1894–900. doi: 10.1023/A:1021410028371. [DOI] [PubMed] [Google Scholar]
- 9.Zhong J, Shen Z, Yang Y, Chen J. Preparation and characterization of uniform nanosized cephradine by combination of reactive precipitation and liquid anti-solvent precipitation under high gravity environment. Int J Pharm. 2005;30:286–93. doi: 10.1016/j.ijpharm.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 10.Merisko-Liversidge E, Liversidge GG, Cooper ER. Nanosizing: a formulation approach for poorly-water-soluble compounds. Eur J Pharm Sci. 2003;18:113–20. doi: 10.1016/S0928-0987(02)00251-8. [DOI] [PubMed] [Google Scholar]
- 11.Brewster ME, Vandecruys R, Peeters J, Neeskens P, Verreck G, Loftsson T. Comparative interaction of 2-hydroxypropyl cyclodextrin and sulfobutylether-cyclodextrin with itraconazole: phase-solubility behavior and stabilization of supersaturated drug solutions. Eur J Pharm Sci. 2008;34(2–3):94–103. doi: 10.1016/j.ejps.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 12.Mäntylä A, Rautio J, Nevalainen T, Keski-Rahkonen P, Vepsälainen J, Järvinen T. Design, synthesis and in vitro evaluation of novel water-soluble prodrugs of buparvaquone. Eur J Pharm Sci. 2004;23:151–8. doi: 10.1016/j.ejps.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 13.Zu YG, Li QY, Fu YJ, Wang W. Synthesis and cytotoxicity of water soluble quaternary salt derivatives of camptothecin. Bioorg Med Chem Lett. 2004;14:4023–6. doi: 10.1016/j.bmcl.2004.05.032. [DOI] [PubMed] [Google Scholar]
- 14.Balfour JA, Buckley MM. Etodolac a reappraisal of its pharmacology and therapeutic use in rheumatic diseases and pain states. Drugs. 1991;42:274–99. doi: 10.2165/00003495-199142020-00008. [DOI] [PubMed] [Google Scholar]
- 15.Reynolds JEF. Martindale, the extra pharmacopoeia. 31. London: Royal Pharmaceutical Society; 1996. [Google Scholar]
- 16.Maccagno A, Di Giorgio E, Romanowicz A. Effectiveness of etodolac (Lodine) compared with naproxen in patients with acute gout. Curr Med Res Opin. 1991;12(7):423–9. doi: 10.1185/03007999109111513. [DOI] [PubMed] [Google Scholar]
- 17.Mullane JF. Etodolac for treatment of gout, United States Patent 4663345, 1987.
- 18.Okamotoa A, Shirakawaab T, Bitoc T, Shigemurab K, Hamadad K, Gotohe A, et al. Etodolac, a selective cyclooxygenase-2 inhibitor, induces upregulation of e-cadherin and has antitumor effect on human bladder cancer cells in vitro and in vivo. Urology. 2008;71(1):156–60. doi: 10.1016/j.urology.2007.09.061. [DOI] [PubMed] [Google Scholar]
- 19.Tsuneoka N, Tajima Y, Kitazato A, Fukuda K, Kitajima T, Kuroki T, et al. Chemopreventative effect of a cyclooxygenase-2-specific inhibitor (etodolac) on chemically induced biliary carcinogenesis in hamsters. Carcinogenesis. 2005;26(2):465–9. doi: 10.1093/carcin/bgh311. [DOI] [PubMed] [Google Scholar]
- 20.Glaser KA. Cyclooxygenase selectivity and NSAIDs: cyclooxygenase-2 selectivity of etodolac (Lodine) Inflammopharmacol. 1995;3:335–45. doi: 10.1007/BF02668029. [DOI] [Google Scholar]
- 21.Raghuvanshi RS, Rampal A, Sen H. Extended release formulation of etodolac, United States Patent 6586005, 2003.
- 22.Allen LV, Popovich NG, Ansel HC. Ansel’s pharmaceutical dosage forms and drug delivery systems, chapter 8. 8. Baltimore: Lippincott Williams & Wilkins; 2005. p. 230. [Google Scholar]
- 23.Khan KA. The concept of dissolution efficiency. J Pharm Pharmacol. 1975;27:48–9. doi: 10.1111/j.2042-7158.1975.tb09378.x. [DOI] [PubMed] [Google Scholar]
- 24.Javadzadeh Y, Siahi-Shadbad MR, Barzegar-Jalali M, Nokhodchi A. Enhancement of dissolution rate of piroxicam using liquisolid compacts. Farmaco. 2005;60:361–5. doi: 10.1016/j.farmac.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 25.FDA . Guidance of industry, food-effect bioavailability and bioequivalence studies. Rockville: FDA; 2002. [Google Scholar]
- 26.Barakat NS. Enhanced oral bioavailability of etodolac by self-emulsifying systems: in-vitro and in-vivo evaluation. J Pharm Pharmacol. 2010;62(2):173–80. doi: 10.1211/jpp.62.02.0004. [DOI] [PubMed] [Google Scholar]
- 27.Brittain HG. Analytical profiles of drug substances and excipients, vol 29. San Diego: Academic; 2002. p. 111. [Google Scholar]
- 28.Herzfeldi C, Kummel R. Dissociation constants, solubilities and dissolution rates of some selected nonsteroidal anti inflammatories. Drug Dev Ind Pharm. 1983;9:767–93. doi: 10.3109/03639048309039887. [DOI] [Google Scholar]
- 29.Najib NM, Suleiman MS. Characterization of a diflunisal polyethylene glycol solid dispersion system. Int J Pharm. 1989;51:225–32. doi: 10.1016/0378-5173(89)90195-6. [DOI] [Google Scholar]
- 30.Abdelkader H, Abdallah OY, Salem HS. Comparison of the effect of tromethamine and polyvinylpyrrolidone on dissolution properties and analgesic effect of nimesulide. AAPS PharmSciTech. 2007;8(3):E1–8. doi: 10.1208/pt0803065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mura P, Zerrouk N, Mennini N, Maestrelli F, Chemtob C. Development and characterization of naproxen–chitosan solid systems with improved drug dissolution properties. Eur J Pharm Sci. 2003;19(1):67–75. doi: 10.1016/S0928-0987(03)00068-X. [DOI] [PubMed] [Google Scholar]
- 32.Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50(1):47–60. doi: 10.1016/S0939-6411(00)00076-X. [DOI] [PubMed] [Google Scholar]
- 33.Mishra PR, Al Shaal L, Müller RH, Keck CM. Production and characterization of Hesperetin nanosuspensions for dermal delivery. Int J Pharm. 2009;371(1-2):182–9. doi: 10.1016/j.ijpharm.2008.12.030. [DOI] [PubMed] [Google Scholar]
- 34.Habib W, Khankari R, Hontz J. Fast-dissolving drug delivery systems, critical review in therapeutics. Drug Carrier Systems. 2000;17(1):61–72. [PubMed] [Google Scholar]
- 35.Clarke A, Brewer F, Johnson ES, Kelly EA. Proceeding of the 122nd annual meeting of the American Neurological Association, 1997. M69.