

Summary
Kv7.x (KCNQ) voltage-gated potassium channels form the cardiac and auditory IKs current and the neuronal M-current. The five Kv7 subtypes have distinct assembly preferences encoded by a C-terminal cytoplasmic assembly domain, the A-domain Tail. Here, we present the high-resolution structure of the Kv7.4 A-domain Tail together with biochemical experiments that show that the domain is a self-assembling, parallel, four-stranded coiled coil. Structural analysis and biochemical studies indicate conservation of the coiled coil in all Kv7 subtypes and that a limited set of interactions encode assembly specificity determinants. Kv7 mutations have prominent roles in arrhythmias, deafness, and epilepsy. The structure together with biochemical data indicate that A-domain Tail arrhythmia mutations cluster on the solvent-accessible surface of the subunit interface at a likely site of action for modulatory proteins. Together, the data provide a framework for understanding Kv7 assembly specificity and the molecular basis of a distinct set of Kv7 channelopathies.
Introduction
Members of the Kv7 (KCNQ) potassium channel family play important roles in the function of the heart, brain, auditory and vestibular organs, and epithelia (Jentsch, 2000; Jespersen et al., 2005). These proteins belong to the voltage-gated ion channel superfamily (Hille, 2001) and constitute the pore-forming subunits of the IKs current that is present in cardiac myocytes, vestibular dark cells, and marginal cells of the stria vascularis (Barhanin et al., 1996; Jentsch, 2000; Sanguinetti et al., 1996; Wang et al., 1996), and the classically studied neuronal M-current (Delmas and Brown, 2005; Wang et al., 1998). Kv7 channels have a prominent role in human disease and harbor mutations that are linked to cardiac arrhythmias, deafness, and epilepsy (Jentsch, 2000). While there has been great effort in the functional characterization of Kv7 channels, little is known about their underlying structure. This is a particularly important problem because many disease mutations cause amino acid substitutions in the cytoplasmic regions of the protein where their direct functional consequences are not immediately obvious.
There are five mammalian Kv7 subtypes (Kv7.1–Kv7.5) (Gutman et al., 2003). Functional studies indicate that each has distinct assembly preferences (Friedrich et al., 2002; Kubisch et al., 1999; Lerche et al., 2000; Schroeder et al., 2000). The subunit responsible for the IKs current, Kv7.1 (KCNQ1), does not coassemble with other Kv7 subunits. In contrast, Kv7.3 (KCNQ3) can form heterotetramers with all subunits except for Kv7.1 and is poorly expressed at the plasma membrane as a homotetramer (Schwake et al., 2000). The other family members, Kv7.2 (KCNQ2), Kv7.4 (KCNQ4), and Kv7.5 (KCNQ5), can form functional homotetramers and functional heterotetramers with Kv7.3. Different combinations of Kv7 subunits display different biophysical properties that produce functional diversity (Hadley et al., 2000; Kubisch et al., 1999; Schwake et al., 2000; Selyanko et al., 2000; Wang et al., 1998).
The molecular mechanisms that direct Kv7 subunit assembly selectivity are incompletely understood. Kv7 subunits contain a C-terminal region of ∼100 amino acids, called the A-domain, that appears to function as an assembly domain (Figure 1A) (Maljevic et al., 2003; Schmitt et al., 2000; Schwake et al., 2003, 2000) and that carries all information for directing subtype-specific assembly properties (Friedrich et al., 2002). Sequence alignment of the five Kv7 subtype A-domains indicates that the A-domain has three subdomains (Figure 1B). Two of these, the Head (Jenke et al., 2003) and the Tail (Jenke et al., 2003; Schwake et al., 2006), have a high probability for forming coiled coils. The Head domain on its own appears incapable of supporting multimerization (Schmitt et al., 2000). Surface expression seems to be closely tied to the integrity of the A-domain Tail (Kanki et al., 2004). The A-domain Tail shows significant sequence variation among the subtypes. There is a clear role for this subdomain as the primary determinant of assembly specificity (Maljevic et al., 2003; Schwake et al., 2006, 2003), but how exactly this domain encodes specificity determinants remains unknown.
Figure 1. Structure of the Kv7 Coiled-Coil Assembly Domain.

(A) Topology cartoon for one Kv7 subunit. The S1-S6 transmembrane segments and pore-forming “P region” are labeled. The voltage-sensor helix, S4, is indicated by the plus symbols. The A-domain is shown as an oval.
(B) Sequence alignment of A-domains from all five Kv7 family members, indicating the positions of the Head, Linker, and Tail regions. Yellow highlighting and symbols below alignments indicate sequence conservation (asterisk, identical residues; colon, conservative substitution; period, weakly conservative substitution). In the Tail region, the positions of the coiled-coil heptad repeat (abcdefg) are indicated below the alignment. Coiled-coil residues occupying hydrophobic “a” and “d” positions are denoted by blue and pink, respectively.
(C) (Left) Kv7.4 A-domain Tail coiled-coil model used for molecular replacement and calculated 2Fo-Fc map after initial rigid body refinement. F639, for which there is clear electron density, was absent from the model. (Right) Final refined structure and composite-omit 2Fo-Fc map built by random omission of 5% of model. Both maps are contoured at 1.2 σ. The positions of F639 and Y640 from one subunit are indicated.
(D) Ribbon diagram of the Kv7.4 A-domain Tail coiled coil with side chains shown in ball-and-stick representation. Chains from individual subunits are colored green, orange, purple, and gray. The N- and C-terminal ends of the green and orange subunits are indicated. The positions of F639 and Y640 of the orange subunit are also indicated.
(E) Ribbon diagram looking down the helical axis from the N terminus. Subunits are colored as in (D).
Understanding the nature of Kv7 A-domains has important consequences for elucidating the relationship between Kv7 channels and disease, as one class of disease mutations falls within the A-domain (Cooper and Jan, 2003; Jentsch, 2000). Some of these mutations prevent channel assembly and impair channel function by a simple truncation of the A-domain (Schmitt et al., 2000; Schwake et al., 2000). However, a number of A-domain Kv7.1 Long-QT (LQT) mutations in Romano Ward syndrome (RWS) and Jervell and Lange-Nielsen syndrome (JNLS) are missense mutations that reside in the A-domain Tail. One of the LQT mutants, G589D, has been shown to disrupt β-adrenergic receptor modulation of Kv7.1 by interfering with the interaction of the channel with the scaffold protein yotiao. This interaction is necessary for anchoring protein kinase A and protein phosphatase 1 to the channel complex (Marx et al., 2002). The exact mechanisms by which the other mutations, T587M (Itoh et al., 1998; Neyroud et al., 1999), A590T (Novotny et al., 2006; Tester et al., 2005), R591H (Neyroud et al., 1999), and R594Q (Splawski et al., 2000), act remain unclear. Elucidation of Kv7 A-domain architecture should provide an important framework for understanding how such disease mutations act.
Results
Structure of the Kv7 Assembly Specificity Domain
The A-domain Tail carries the major determinants of channel assembly specificity (Maljevic et al., 2003; Schwake et al., 2006, 2003) and exhibits periodic heptad repeats, denoted (a-b-c-d-e-f-g)n, (Figure 1B) in which the “a” and “d” positions are hydrophobic residues. This sequence motif is characteristic of coiled coils (Jenke et al., 2003; Schwake et al., 2006), common protein-protein interaction motifs (Lupas and Gruber, 2005; Woolfson, 2005). While it is relatively straightforward to discern likely coiled coils based on the presence of multiple heptad repeats, it is difficult to determine from the sequence alone which oligomeric state might be encoded, whether the sequence makes homomers or heteromers, what the heteromeric partners might be, and where the specificity determinants lie (Lupas and Gruber, 2005). Thus, we were interested in defining the structure of the Kv7 A-domain Tail to begin to understand the structural basis for Kv7 assembly specificity.
Sequence analysis suggests that the Kv7 Tail contains as many as four complete heptad repeats (Figure 1B). We expressed, purified, and crystallized a Kv7.4 A-domain Tail construct, residues 610–645, that was slightly longer than the identified heptads and that formed tetramers in solution (see below). The crystals grew in space group P4212 and diffracted synchrotron X-rays to 2.10Å. Despite extensive effort, we were unable to determine phases by selenomethionine anomalous experiments, heavy metal derivatives, or by molecular replacement using parallel left-handed four-stranded structures from SNARE complexes (Antonin et al., 2002; Sutton et al., 1998), Sendai virus (Tarbouriech et al., 2000), and designed coiled coils (Harbury et al., 1993). Therefore, we decided to exploit the fact that coiled-coil backbone structure can be described by a simple parameterization (Crick, 1953; Harbury et al., 1998, 1995) to build a library of de novo search models that could be used for molecular replacement.
We generated a library of 300 polyalanine search models in which the superhelix radius of the coiled-coil bundle, R0, the supercoil pitch (residues/superhelical turn), and the total number of residues were varied near their expected values for parallel left-handed four-stranded coiled coils (see Experimental Procedures). Each model was used as a molecular replacement search model in EPMR (Kissinger et al., 2001). The best solution had two 28 residue peptides in the asymmetric unit, R0 = 5.0 Å, and a pitch = 100 residues/superhelical turn. We were able to use this solution to build residues 612–642; however, we could not refine the structure to acceptable R/Rfree values (Table 1). Because the electron density clearly showed the appropriate side chains within the coiled-coil core but had poor density for the C-terminal part of the bundle, we suspected that some degree of crystallographic disorder might be the source of the refinement difficulties. Therefore, we tested a series of truncation constructs to try to find one that would crystallize and yield data that were more amenable to structure solution and refinement.
Table 1. Data Collection and Refinement Statistics.
| Data Collection | Kv7.4 (610–645) | Kv7.4 (610–640) |
|---|---|---|
| Space group | P 4212 | I4 |
| Cell dimensions | ||
| a, b, c (Å) | 30.53, 30.53, 139.94 | 33.18, 33.18, 55.28 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 50–2.10 (2.18–2.10) | 50–2.07 (2.14–2.07) |
| Rsym | 11.3 (52.7) | 5.3 (9.6) |
| I/σI | 18.7 (2.5) | 28.4 (12.6) |
| Completeness (%) | 96.7 (89.5) | 99.6 (100.0) |
| Redundancy | 12.5 (7.2) | 7.3 (6.1) |
| Refinement | ||
| Resolution (Å) | 2.10 | 2.07 |
| No. reflections | 53,620 | 13,350 |
| Rwork/Rfree | 32.1 / 36.1 | 19.6 / 22.4 |
| Total protein atoms | 249 | |
| Water molecules | 18 | |
| Average B factors: protein (Å2) | 21.1 | |
| Average B factors: water (Å2) | 36.5 | |
| RMSD in bond lengths (Å) | 0.014 | |
| RMSD in bond angles (°) | 1.59 | |
Values in parentheses are for the highest-resolution shell.
Rfree = Rwork calculated using 5% of the reflection data chosen randomly and omitted from the start of refinement.
Crystals of a C-terminal truncation, Kv7.4 residues 610–640, grew in space group I4 and diffracted X-rays to 2.07 Å. We were able to solve the structure by molecular replacement using the program CNS (Brunger et al., 1998) and the model from the P4212 crystals truncated to residue 638. Clear electron density was visible in the initial maps for portions of the coil that were absent from the search model (Figure 1C). We were able to build all the atoms in the sequence except those of the three N-terminal residues and refine the structure to an acceptable level (R/Rfree = 19.6/22.4%) (Table 1).
The overall structure of the A-domain Tail assembly domain is that of a tightly twisted left-handed four-stranded coiled coil that is ∼24 Å wide and 40 Å long (Figures 1D and 1E). The last three C-terminal residues (Gly 638, Phe 639, and Tyr 640) splay outward to form a broad base (31 Å from Cα to Cα of opposite subunits) and participate in crystal contacts with neighboring molecules in the crystal lattice.
The Kv7.4 coiled-coil heptad repeat “a” and “d” positions interact by classical “knobs into holes” packing (Crick, 1953; Lupas and Gruber, 2005) to form alternating layers of the hydrophobic core of the coiled coil (Figures 2A and 2B). Two-, three-, and four-stranded coiled coils each have different characteristic “knobs into holes” packing geometries (Harbury et al., 1993). In four-stranded coiled coils, the Cα-Cβ vector of each “a” position knob makes a perpendicular angle with the Cα-Cα vector of the “hole” formed by the “g” and “a” residues of the helix to the left (looking down the superhelical axis from the N terminus), whereas the Cα-Cβ vector of each “d” position knob runs parallel to the Cα-Cα vector of its “hole” formed by the “d” and “e” position residues of the helix to the right. The Kv7.4 A-domain Tail core packing uniformly corresponds to the canonical four-stranded coiled-coil geometry, having perpendicular “a” layers and parallel “d” layers (Figure 2B). Comparison of the superhelical parameters of the Kv7.4 A-domain Tail with previously determined four-stranded coiled-coil structures shows that the particular combination of supercoil radius, pitch, and radius of curvature found in the A-domain Tail is unique and highlights the structural diversity that can be obtained from such a seemingly simple protein fold (Table 2).
Figure 2. Hydrophobic and Electrostatic Contacts in the Kv7.4 Coiled-Coil Domain.

(A) Hydrophobic layers of the coiled-coil core. Van der Waals spheres depicting the side chains of the “a” (blue) and “d” (pink) layers on a ribbon backbone (gray) are shown. The N- and C-terminal ends of the coiled coil are indicated.
(B) Geometry of individual coiled-coil “a” and “d” layers. Top pictograms represent “a” (right) and “d” layers (left). Arrows show the direction from the N to C terminus, open circles represent the Cα atoms, and black circles the Cβ atoms. Ball-and-stick representations show each layer of the core. Van der Waals spheres indicate core residues, colored as in (A).
(C) Intra- and intermolecular electrostatic interactions. Ribbon diagram of tetramer with helices colored as in Figure 1D shows network 1 and network 2 interactions between the side chains (shown as sticks) of the green and orange subunits. Salt bridges (black lines) and hydrogen bonds (dotted lines) are indicated. Side chain labels are color coded to indicate the subunit of origin.
Table 2. Coiled-Coil Parametersa.
| Superhelical parameters | Kv7.4 (613–637) | GCN4-pLI (1–31)b | SeVP (67–111)c | nSNARE (32–83)d | eSNARE (12–63)e | Phe-14M (13–54)f |
|---|---|---|---|---|---|---|
| Supercoil radius (Å) | 7.30 ± 0.15 | 7.54 ± 0.08 | 7.77 ± 0.51 | 6.99 ± 0.24 | 7.09 ± 0.25 | 8.24 ± 0.22 |
| Residues per superhelix turn | 105 ± 8 | 133 ± 8 | 183 ± 54 | 107 ± 13 | 120 ± 14 | 123 ± 13 |
| Supercoil pitch (Å) | 152 ± 12 | 197 ± 13 | 273 ± 81 | 152 ± 19 | 175 ± 22 | 178 ± 21 |
| Radius of curvature (Å) | 87.4 ± 11.9 | 139 ± 12 | 267 ± 155 | 91 ± 20 | 118 ± 27 | 106 ± 21 |
| Helix-crossing angle (°) | 33.8 ± 2.1 | 27.1 ± 1.5 | 21.6 ± 4.9 | 32.6 ± 2.8 | 28.8 ± 2.8 | 32.8 ± 3.1 |
| α-Helical parameters | ||||||
| Residues per turn | 3.61 ± 0.03 | 3.59 ± 0.02 | 3.61 ± 0.07 | 3.62 ± 0.03 | 3.61 ± 0.03 | 3.61 ± 0.03 |
| Rise per residue (Å) | 1.52 ± 0.02 | 1.52 ± 0.02 | 1.52 ± 0.05 | 1.48 ± 0.02 | 1.50 ± 0.02 | 1.50 ± 0.01 |
| α-Helix radius (Å) | 2.28 ± 0.04 | 2.24 ± 0.03 | 2.27 ± 0.05 | 2.25 ± 0.03 | 2.26 ± 0.03 | 2.29 ± 0.03 |
| Helix-crossing angle (°) | 23.7 ± 1.5 | 19.0 ± 1.1 | 15.2 ± 3.4 | 22.9 ± 1.9 | 20.3 ± 2.0 | 23.0 ± 2.1 |
| Interhelix distance (Å) | 10.3 ± 0.2 | 10.7 ± 0.1 | 11.0 ± 0.7 | 9.9 ± 0.3 | 10.0 ± 0.4 | 11.7 ± 0.3 |
Helical and superhelical parameters obtained by fitting the Cα backbone to the supercoil parameterization suggested by Crick (Crick, 1953).
Designed parallel four-stranded coiled coil based on the GCN4-p1 dimeric leucine zipper peptide (Harbury et al., 1993).
Sendai virus phosphoprotein oligomerization domain (Tarbouriech et al., 2000).
Neuronal SNARE core complex; numbering corresponds to synaptobrevin 2 (Sutton et al., 1998).
Endosomal SNARE core complex; numbering corresponds to endobrevin (Antonin et al., 2002).
Designed parallel four-stranded “Phe-zipper” coiled-coil based on the Major Outer Membrane Lipoprotein (Liu et al., 2006).
The surface of the Kv7.4 A-domain Tail complex is predominantly polar and has two distinct networks of side chain salt bridges and hydrogen bonds, which we term “network 1” and “network 2,” that make interhelical contacts across helix interfaces (Figure 2C). In network 1, Glu623, an “e” position in the heptad repeat, makes salt bridges to Arg618, a “g” position from the neighboring helix, and Lys624 from the “c” position of its own chain. Network 2 involves five side chains. The two central players are residues from neighboring helices: Glu630, an “e” position, and Lys632, a “g” position. Glu630 makes hydrogen bonds to Gln625 and Ser628 and a salt bridge to Lys632, three side chains from the adjacent helix. Lys632 further participates in cross-subunit interactions by making a salt bridge to Asp634 from the neighboring chain. The Gln627 side chain is positioned between network 1 and network 2 and could form a bridge between the networks by making hydrogen bonds to Glu623 and Gln625. However, our structural data suggest that neither the geometry nor distances are optimal for this interaction, despite the proximity of Gln627 to Glu623 and Gln625. Notably, this potential link between the networks is absent from Kv7.1 and Kv7.2 (Figure 1B). It is striking that the amino acids that form interactions in both network 1 and network 2 of the Kv7.4 A-domain Tail are changed in the Kv7 subtypes with specific assembly preferences, Kv7.1 and Kv7.3 (see Discussion).
Kv7.4 Is a Stable Tetramer in Solution
We used a number of biochemical and biophysical measures to probe the structure and assembly properties of the Kv7.4 A-domain Tail in aqueous solution. The circular dichroism (CD) spectrum of Kv7.4 residues 610–645 shows minima at 208 and 222 nm that are characteristic of a protein with high helix content (Figure 3A). Estimation of helical content fraction (Chen et al., 1974) indicates that the peptide is 66.0% helical. This value is in good agreement with the structure where each helix contains 26 residues (Glu612–Leu637) that comprise 68.4% of the 38 residue peptide. The CD spectra of the complex at pH 3.0, 7.5, and 9.0 were superimposable and indicate that the Kv7.4 A-domain Tail retains its secondary structure over a wide range of pH conditions.
Figure 3. Solution Properties of the Kv7.4 A-Domain Tail.

(A) Circular dichroism (CD) spectrum of the Kv7.4 A-domain Tail is insensitive to pH over a wide range. Overlaid CD spectra of 25 μM Kv7.4 A-domain Tail residues 610–645 at pH 3.0 (purple diamonds), 7.5 (gray triangles), and 9.0 (pink squares).
(B) Superdex75 (Amersham Biosciences) size exclusion chromatography of 500 μM Kv7.4 residues 610–642 monitored at 280 nm. Horizontal axis shows elution volume VE corrected for void elution volume by subtracting that of blue dextran (V0). Vertical dotted lines indicate predicted elutions of tetrameric (red) and monomeric (blue) peptides. (Inset) Standard curve used to calculate molecular weight of eluted peptides on the Superdex75 column. Molecular weights are 17.1 kD, observed; 3.98 kD, expected monomer; and 15.9 kD, expected tetramer.
(C) Sedimentation equilibrium of Kv7.4 residues 610–645. Equilibrium distribution of peptide (gray circles) measured by its absorbance at 275 nm is plotted as a function of radial distance at 33,800 rpm at 4°C. Initial protein concentration was 600 μM. Raw data shown relative to predicted curves for tetrameric (red), trimeric (orange), dimeric (green), and monomeric (blue) species. (Inset) Random distribution of residuals as a function of radial distance.
Both size exclusion chromatography and analytical ultracentrifugation corroborate the crystallographic observation that the Kv7.4 A-domain Tail is tetrameric. Size exclusion chromatography shows that the Kv7.4 A-domain Tail migrates as a single peak with an apparent molecular mass that is consistent with a tetramer (Figure 3B). Because size exclusion chromatography relies on the hydrodynamic radius, comparison of an elongated protein such as a coiled coil with globular protein standards could be misleading. Thus, we also used sedimentation equilibrium, a method that unlike gel filtration provides shape-independent mass information (Laue, 1995), to obtain a precise measurement of the oligomeric state. The equilibrium sedimentation data were well fit by a single-species model that corresponded to the molecular mass of a tetramer and had random residuals (Figure 3C). Together, these data establish unambiguously that the Kv7.4 A-domain Tail is a helical tetramer in solution that mirrors the crystal structure and the expected stoichiometry of Kv7 channels.
Biochemical and Structural Comparisons of Kv7 A-Domain Tails
Sequence comparisons of Kv7 A-domain Tails show that the coiled-coil motif is conserved in all Kv7s (Figure 1B). To investigate self-assembly properties of the other Kv7 members, we expressed and purified fusion proteins of A-domain Tails from each subtype. Each fusion protein contained from N to C terminus: a hexahistidine tag, maltose binding protein, and a specific protease site (termed “HMT,” see Experimental Procedures) followed by the Kv7 coiled coil. The first two elements serve as orthogonal affinity purification tags. Gel filtration chromatography experiments show that purified Kv7.1, Kv7.2, Kv7.4, and Kv7.5 fusion proteins elute with apparent molecular weights corresponding to tetramers, and all behave similarly (Figure 4A). In contrast, the Kv7.3 A-domain Tail fusion protein elutes as a broad peak that suggests a mixture of monomers and dimers (Figure 4A). Thus, the A-domain Tails from the four subtypes (Kv7.1, Kv7.2, Kv7.4, and Kv7.5) in which the channels are known to form homotetramers retain the ability to oligomerize as independent domains while the A-domain Tail from the channel isoform that does not form robust homotetramers, Kv7.3 (Wang et al., 1998), is impaired in its self-association ability.
Figure 4. Comparing Interactions in Alternate Kv7 Subtypes.
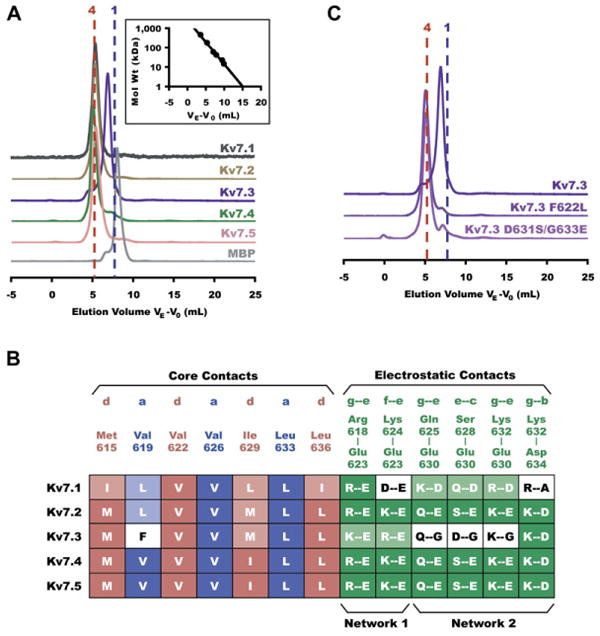
(A) Stoichiometry of coiled-coil assembly domains in all five Kv7 subtypes shown by Superdex200 (Amersham Biosciences) size exclusion chromatography. Normalized absorbance is plotted against elution volume VE corrected for void elution volume V0 as in Figure 3B. All samples were loaded at a concentration of 50 μM. Vertically displaced chromatograms show traces for, from top to bottom, Kv7.1 (black), Kv7.2 (orange), Kv7.3 (purple), Kv7.4 (green), Kv7.5 (pink), and MBP (gray). Vertical dotted lines indicate the predicted elution volumes of tetrameric (red) and monomeric (blue) fusion proteins. (Inset) Standard curve used to calculate molecular weight of eluted proteins on the Superdex200 column. Molecular weights for each are as follows (observed ± SD, expected monomer, expected tetramer); Kv7.1 (180 ± 2 kD, 49.4 kD, 198 kD); Kv7.2 (203 ± 6 kD, 49.3 kD, 197 kD); Kv7.3 (90.3 ± 2 kD, 49.9 kD, 200 kD); Kv7.4 (207 ± 6 kD, 48.8 kD, 195 kD); Kv7.5 (191 ± 6 kD, 48.9 kD, 196 kD).
(B) Comparative interaction mapping in all subtypes. Column labels identify residue types involved in hydrophobic “a” (blue) and “d” (pink) layer contacts and electrostatic interactions (green) observed in the Kv7.4 coiled-coil structure. Filled boxes in table indicate entirely conserved interactions; shaded boxes indicate nonconserved residues that are still capable of interacting as predicted; white boxes indicate unfavorable contacts. Electrostatic interactions involved in networks 1 and 2 are indicated below the alignment.
(C) Stoichiometry of mutant coiled-coil assembly domains as determined by size exclusion. Kv7.3 A-domain Tail mutants F622L and D631S/G633E restore tetramerization. Molecular weights for each are as follows (observed, expected monomer, expected tetramer); Kv7.3 (90.3 kD, 49.9 kD, 200 kD); Kv7.3 F622L (212 kD, 49.9 kD, 200 kD); Kv7.3 D631S/G633E (208 kD, 49.9 kD, 200 kD). All samples were loaded onto the column at a concentration of 50 μM.
Structure-based sequence comparisons indicate two sets of Kv7.3 amino acids that could impair assembly: there is a large aromatic residue, Phe622, substituted at an “a” position and there are two amino acid substitutions, D631 and G633, that disrupt the interhelical salt bridge and hydrogen bond interactions of network 2 (Figure 4B). The recent structure determination of a tetrameric coiled coil bearing phenylalanine at most “a” and “d” positions (Liu et al., 2006) indicates that accommodation of phenylalanine side chains in a coiled-coil hydrophobic core requires a superhelix radius that is significantly larger than that of the Kv7.4 A-domain Tail (Table 2). Thus, incorporation of four phenyalanines into the core of a homomeric Kv7.3 tetrameric coiled coil should incur a substantial steric and energetic penalty in the context of the coiled-coil core formed by the smaller alkyl side chains at the other “a” and “d” positions. To test whether the presence of Phe622 was a major factor preventing tetramer formation by the Kv7.3 A-domain Tail, we mutated this position to a residue that was smaller, commonly found at four-stranded coiled-coil “a” positions, and also found at the equivalent position of Kv7.2 (F622L). Gel filtration experiments demonstrate that this single mutation was sufficient to convert the Kv7.3 A-domain Tail into a form that was predominantly tetrameric (Figure 4C). This result highlights the critical importance of the hydrophobic core in controlling Kv7 A-domain Tail assembly.
We also investigated the effects of restoring the network 2 interactions into the Kv.7.3 coiled coil. Similar to the F622L mutation, the double mutant D631S/G633E was sufficient to endow the Kv7.3 A-domain Tail with robust tetramerization properties (Figure 4C). Taken together, the biochemical and mutational analysis suggest that two factors prevent Kv7.3 A-domain Tail self-assembly: the incompatibility of making a stable tetramer bearing four phenylalanines in the coiled-coil core and the lack of interstrand stabilizing interactions provided by network 2. These factors are likely to be important for determining the Kv7.3 preference for heteromer versus homomer formation. Experiments in Xenopus oocytes failed to show a functional enhancement of Kv7.3 channels bearing mutations that impart homotetramerization to the Kv7.3 Tail (data not shown). This observation is consistent with the poor expression of Kv7.3 homomeric chimeras bearing A-domains or A-domain portions from other Kv7 channels (Maljevic et al., 2003; Schwake et al., 2003) and suggests that other yet to be defined factors besides the assembly defects in the A-domain Tail play a role in impairing Kv7.3 homotetramer surface expression.
Structural Insight into Cardiac Arrhythmia Mutations
The Kv7.1 A-domain Tail bears five mutations that are associated with cardiac Long QT syndromes (LQT) known as Romano Ward syndrome (RWS) and Jervell and Lange-Nielsen syndrome (JNLS): T587M (Chen et al., 2003; Itoh et al., 1998; Neyroud et al., 1999), G589D (Piippo et al., 2001), A590T (Novotny et al., 2006; Tester et al., 2005), R591H (Neyroud et al., 1999), and R594Q (Splawski et al., 2000). The exact mechanisms by which these mutations cause arrhythmia remain unclear. Prior work has shown that G589D is able to abolish sympathetic regulation of cardiac IKs currents by disrupting the assembly of a macromolecular complex of Kv7.1 and the scaffolding protein yotiao (Marx et al., 2002). Three of the mutants, T587M (Yamashita et al., 2001), R591H (Grunnet et al., 2005), and R594Q (Huang et al., 2001), appear to be incapable of making functional homomeric channels. T587M (Yamashita et al., 2001) and R591H (Grunnet et al., 2005) cannot coassemble with wild-type Kv7.1 subunits. Four mutations, T587M (Kanki et al., 2004; Yamashita et al., 2001), G589D (Kanki et al., 2004), R591H (Grunnet et al., 2005; Kanki et al., 2004), and R594Q (Kanki et al., 2004), compromise the ability of the channel to reach the plasma membrane. R594Q appears to coassemble with wild-type and has a weak dominant-negative effect (Huang et al., 2001). No functional characterization of A590T has yet been reported.
To examine the impact of LQTS mutations on assembly of the Kv7.1 A-domain Tail, we made and tested the effects of each of them in the background of Kv7.1 HMT-fusion protein. Gel filtration experiments show that none of the LQT mutations disrupt Kv7.1 A-domain Tail assembly (Figure 5A). While it is possible that there are subtle effects on tetramerization affinity caused by the mutations, the observation that there are no substantial changes in assembly properties suggests that some other mechanism must underlie their arrhythmogenic effects.
Figure 5. Mapping of Long-QT Syndrome Mutations.
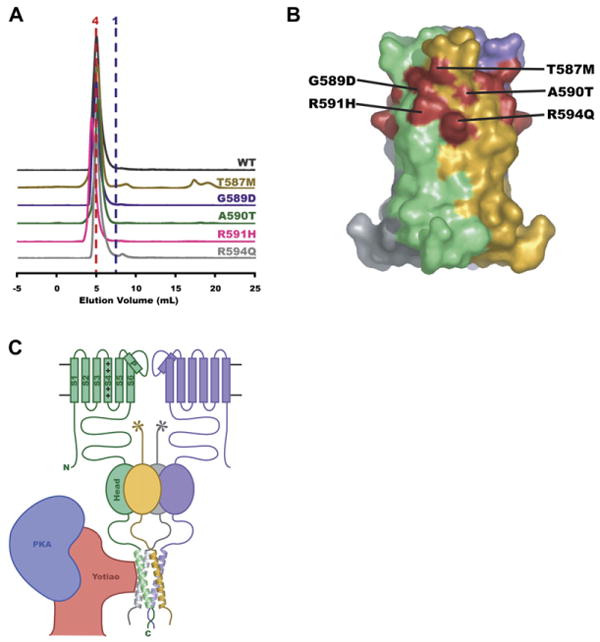
(A) Stoichiometry of Kv7.1 disease mutants. Normalized size exclusion chromatograms for Kv7.1 MBP-fusions: wild-type (black), T587M (orange), G589D (purple), A590T (green), R591H (pink), and R594Q (gray).
(B) Surface representation of Kv7.4 coiled-coil domain with helices colored separately. LQTS mutation sites have been mapped onto equivalent positions (red) in Kv7.4.
(C) Cartoon of a Kv7 channel regulatory complex. Two complete Kv7 pore-forming subunits are shown (green and purple). S1-S6 transmembrane segments and the pore helix (P) are indicated for the green subunit. “+”s indicate the S4 voltage sensor segment. All four subunits of the Kv7 A-domain are shown. The Head domain is shown as an oval. The Tail domain helices are shown. The Linker domain connects the Head and Tail. An interaction between a portion of the scaffolding protein yotiao (red) and the channel is shown bringing protein kinase A “PKA” (blue) near the channel. The stoichiometry of the yotiao/channel complex is not known.
Comparison of the positions of the Kv7.1 mutations with their equivalent positions on the Kv7.4 A-domain Tail structure reveals a striking pattern. The sites cluster around a common surface pocket at the interhelix cleft that defines a three-dimensional mutational “hotspot” (Figure 5B). Notably, we observe that this pocket is the site of protein-protein contacts within the Kv7.4 A-domain Tail crystal lattice (see Figure S1 in the Supplemental Data available online). The observation that none of the A-domain Tail disease mutants disrupt the self-assembly of this part of the channel and that a mutation in the center of the cluster, G589D, disrupts the binding of a necessary regulatory complex (Kanki et al., 2004; Marx et al., 2002) suggests that all members of this family of LQTS mutations may act by a similar mechanism.
Discussion
The minimal functional unit of any voltage-gated potassium channel is a homotetramer or heterotetramer of pore-forming subunits. Because different combinations of pore-forming subunits can have profound effects on functional properties, it is critical for all members of this channel class to encode assembly determinants and assembly specificity preferences to direct association with the appropriate partners (Deutsch, 2002, 2003; Hille, 2001; Papazian, 1999). Despite the fundamental nature of this problem, the mechanisms that drive channel assembly and assembly specificity remain imperfectly understood.
Voltage-gated ion channel family members are most similar within the membrane domain but exhibit considerable diversity within their extramembranous domains (Hille, 2001). While contacts between membrane-spanning regions may provide some stability, the more diverse cytoplasmic domains appear to play a major role in driving assembly and assembly specificity. The best understood case involves the Shaker-type KV family (Kv1-Kv4 subunits), where an independently folded, N-terminal intracellular module, called the “T1 domain,” drives tetramerization and assembly specificity (Bixby et al., 1999; Li et al., 1992; Shen and Pfaffinger, 1995). T1 domains are unique to Shaker-type Kv channels (Hille, 2001) and pose an important question: do other voltage-gated potassium channels use modular intracellular domains to drive assembly and assembly specificity?
A number of Kv7 A-domain functional properties mirror those of T1 and suggest that the A-domain is another example of an independent channel intracellular assembly module: the isolated A-domain forms stable multimeric complexes (Schmitt et al., 2000); Kv7 channels bearing A-domain deletions, including those that are a consequence of certain disease mutations, fail to assemble into functional channels (Schmitt et al., 2000; Schwake et al., 2000); and coexpression of Kv7.1 A-domain sequences act as dominant-negatives on Kv7.1 channel assembly (Schmitt et al., 2000). The A-domain and T1 domain amino acid sequences are unrelated and have different locations relative to the pore-forming transmembrane domain (C-terminal versus N-terminal, respectively). These differences suggest that voltage-gated channel superfamily members have exploited diverse types of protein-protein interaction domains to direct channel assembly.
Kv7 A-Domain Tails and Assembly Specificity
Our structural data demonstrate that the A-domain Tail is a self-assembling, four-stranded, parallel coiled coil (Figures 1D and 1E) that is conserved among Kv7 subtypes (Figures 1B and 4) and establish a framework for understanding Kv7 assembly. Sequence variations among the Kv7 subtypes at both the hydrophobic core of the coil and the polar network at the subunit interfaces appear as candidates for sites of specificity. Kv7.2, Kv7.4, and Kv7.5 can form functional channels as either homomers or as heteromers with Kv7.3 (Schwake et al., 2003). Comparison of the core and electrostatic contacts that make protein-protein contacts shows that Kv7.4 and Kv7.5 are identical (Figure 4B). Kv7.4 and Kv7.2 differ by only two conservative variations, V619L and I629M, that are at “a” and “d” positions, respectively (for simplicity, all comparisons use Kv7.4 amino acid numbers). In contrast, Kv7.1, the channel that does not coassemble with any of the other subtypes (Schwake et al., 2003), has changes at both the core positions (M615I, V619L, I629L, and L636I) and in network 1 (K624D) and network 2 (E360D and K632A) relative to Kv7.4. The K624D Kv7.1 change in network 1 alters the network surrounding Glu623. The Kv7.1 network 2 would be completely rearranged as one of the central players in the ionic and hydrogen bond network is shortened by a methylene group, E630D, and the other is eliminated, K632A. It is striking that the remaining positions of the Kv7.1 network 2 are changed in a way that could still form a hydrogen bond and ionic network of side chain interactions around E630D. These observations suggest the hypothesis that the differences in network 2 play a role in preventing Kv7.1 A-domain Tails from associating with other Kv7 subtypes.
Kv7.3 does not express robustly as a homomeric channel (Schwake et al., 2000) but does show robust channel activity when coassembled with Kv7.2 (Schroeder et al., 1998; Wang et al., 1998), Kv.7.4 (Kubisch et al., 1999), and Kv7.5 (Schroeder et al., 2000). Our work indicates that the Kv7.3 Tails are divergent at a number of critical positions (Figure 4B). There are changes at two coiled-coil core positions: V619F, an “a” position, and I629M, a “d” position. The changes in the interhelical interface networks include a reversal in the relative positions of the basic residues that surround Glu623 in network 1, R618K and K624R, and a relocation of the carboxylate of one of the central side chains in network 2, E630G, to a position at a similar altitude on the adjacent subunit by the S628D change (Figure 2C). These alterations in the core and the interface networks appear to be critical for the unique assembly properties of the Kv7.3 Tail.
Our biochemical experiments suggest a rationale for the poor self-assembly and promiscuous heteromer formation properties of Kv7.3. The Kv7.3 A-domain Tail is prevented from making stable tetramers by both the incompatability of placing four phenylalanines within the core of the tetramer at the “a” position of the coiled coil and by the disruption of the interhelical hydrogen bond and salt bridge interactions made by network 2. Although the combination of both factors is important for impairing homotetramerization, neither factor alone is incompatible with homotetramer formation (i.e., Kv7.3 Tails bearing the core phenylalanine can make tetramers provided network 2 is intact, and tetramers lacking network 2 interactions can form provided the steric problems in the core caused by the “a” position phenylalanine are relieved). Thus, it appears that the Kv7.3 A-domain Tails are poised to form tetramers provided that the destabilizing factors can be overcome.
This property of being poised for assembly is particularly interesting when considered in the context of the promiscuous assembly behavior of Kv7.3 with Kv7.2, Kv7.4, and Kv7.5. Each non-Kv7.3 subunit would bring smaller hydrophobic residues at the Kv7.3 F622 “a” level of the coiled-coil core (Figure 4B) that would relieve some of the steric penalty for including the F622 from each Kv7.3 subunit and promote heteromer formation. Network 1 and network 2 residues are identical in Kv7.2, Kv7.4, and Kv7.5 (Figure 4B). Consideration of the amino acid combinations that would be present in both networks in the context of a heterotetramers suggests that these interfaces should remain compatible across the Kv7.2/Kv7.3, Kv7.4/Kv7.3, and Kv7.5/Kv7.3 A-domain Tail interface and could contribute to the stability of a heteromeric complex. The precise arrangements of interactions at such interfaces are not evident from the current structure and will require further study.
Structural Consequences for Kv7.1 Arrhythmia Mutations and Implications for Coiled Coils as Sites of Ion Channel Regulatory Complex Assembly
In addition to providing insight into the likely sources of assembly specificity, the Kv7.4 coiled-coil structure suggests a new hypothesis as to how certain Kv7.1 LQTS mutants act. One of the LQTS mutations, G589D, is known to interfere with the recruitment of a macromolecular complex that responds to β-adrenergic receptor stimulation and includes the scaffolding protein yotiao, protein kinase A, and protein phosphatase 1 (Marx et al., 2002). Both G589D and simultaneous mutation of two “d” positions to alanine (L602A/I609A) in the A-domain Tail prevent yotiao binding. Based on these mutations, Marx et al. suggested that that the A-domain heptad repeats interact directly with similar repeats in yotiao through a leucine-zipper-like mechanism (Marx et al., 2002). Our structural and biochemical data show that the position of G589D is on the coiled-coil surface and that the G598D mutation does not affect tetramerization. Together, these data suggest a different scenario for how G598D and other LQTS mutations within the A-domain Tail act (Figure 5C). We propose that yotiao interacts with the exterior surface of the helical bundle and that disease mutations such as G589D interfere with yotiao binding to the intact A-domain Tail coiled-coil. In support of this idea, it is striking that all of the known A-domain Tail disease mutations map to a single hotspot on the coiled-coil surface. This clustering of mutations to a single area on the surface of the assembled domain suggests that this site may be the binding site for yotiao and that all four Long-QT syndrome mutations cause disease by a similar mechanism to G589D. It is notable that this hotspot is composed of residues from adjacent subunits and is only present when the domain is intact and assembled as a coiled coil. Because the integrity of the A-domain Tail appears to be critical for proper channel trafficking (Kanki et al., 2004) and T587M (Kanki et al., 2004; Yamashita et al., 2001), G589D (Kanki et al., 2004), R591H (Grunnet et al., 2005; Kanki et al., 2004), and R594Q (Kanki et al., 2004) compromise the ability of the channel to reach the plasma membrane, binding of accessory proteins in this region may be a mechanism of quality control for correctly assembled subunits.
Recent work indicates that a variety of voltage-gated ion channel superfamily members employ C-terminal cytoplasmic coiled-coil domains as a modular means for directing subunit assembly (Jenke et al., 2003). Coiled coils are one of the most widespread and versatile protein-protein interaction domains and are found in diverse types of proteins (Lupas and Gruber, 2005; Woolfson, 2005). The characteristic coiled-coil heptad repeat (a-b-c-d-e-f-g)n in which “a” and “d” are usually hydrophobic side chains makes coiled-coil identification from protein sequences straightforward; however, the oligomeric state, homomeric versus heteromeric preferences, and parallel or antiparallel orientation specified by a particular sequence remain difficult to discern from sequence information alone (Lupas and Gruber, 2005). To date, there is experimental evidence for coiled-coil assembly domains in varied types of voltage-gated ion channel superfamily members, including Kv7 channels (Schwake et al., 2006) (and this work), eag channels (Jenke et al., 2003), cyclic nucleotide gated channels (Zhong et al., 2003, 2002), intermediate conductance channels (Syme et al., 2003), and TRPM channels (Tsuruda et al., 2006). Thus, the coiled-coil motif appears to represent a general strategy for directing channel assembly in this diverse superfamily of channels.
Interactions between coiled-coil sequences and regulatory proteins are an emerging theme in channel regulation and disease (Kass et al., 2003; Marx et al., 2002). The presence of coiled-coil assembly domains in a wide variety of voltage-gated ion channels and the insights provided from the structural analysis presented here suggest that beyond channel assembly these domains may be important sites of nucleation for protein complexes that regulate channel activity (Figure 5C). Furthermore, disruption of protein-protein interaction regulatory networks that are assembled on such intracellular channel domains is likely to be an important mechanism of disregulation and channelopathies such as Long QT syndrome.
Experimental Procedures
Protein Cloning, Expression, and Purification
DNA fragments of Kv7.1 (residues 583–623), Kv7.2 (residues 614–654), Kv7.3 (residues 613–653), Kv7.4 (residues 610–645), Kv7.4 (residues 610–640), and Kv7.5 (residues 597–633) were amplified by PCR and ligated into the NarI/HindIII or NarI/XhoI sites of a pET27 (Novagen) derived vector (pSV272) denoted “HMT” (Van Petegem et al., 2004) that contains, in sequence, a hexahistidine tag, maltose binding protein, and a cleavage site for the Tobacco Etch Mosaic Virus (TEV) protease. Point mutations were made using mutated oligonucleotide extension (Pfu Turbo Polymerase, Strategene) from plasmid templates harboring the HMT fusion of interest, digested with DpnI (New England Biolabs), and transformed into DH5α cells. All constructs were verfied by complete DNA sequencing of the HMT fusion.
HMT fusion proteins were expressed in Escherichia coli [BL21(DE3)-pLysS] grown in 2YT media at 37°C and induced at OD600nm = 0.4–0.8 with 0.4 mM IPTG for 4 hr. Cells were harvested by centrifugation at 5000 × g for 20 min at 4°C, and cell pellets were frozen at −20°C. Thawed cell pellets were lysed by sonication in lysis buffer (100 mM Tris pH 7.6, 200 mM KCl, 10% sucrose, 25 mM β-octylglucoside, 20 μg mr−1 lysozyme, 25 μg ml−1 DNaseI, 5 mM MgCl2, 1 mM PMSF). Insoluble material was precipitated by centrifugation for 20 min at 12,000 × g at 4°C. The resulting soluble fraction, which contained the HMT fusion protein, was applied to a 45 ml Poros20MC (Perseptive Biosystems) nickel-charged column, washed in wash buffer (10 mM PO42−, pH 7.3, 250 mM KCl, 1 mM PMSF), and eluted on a linear gradient to 300 mM imidazole in the same buffer on an ÄKTA-FPLC system (Pharmacia). Imidazole was removed using a Centriprep YM-10 concentrator (Millipore). Fusion proteins were then applied to a 60 ml Amylose (New England Biolabs) column, washed in wash buffer, and eluted in maltose buffer (10 mM PO42−, pH 7.3, 250 mM KCl, 10 mM maltose). To prepare purified Kv7.4 A-domain Tails, the HMT-fusion protein was cleaved with TEV protease (Kapust et al., 2001). Coiled-coil peptides were collected in the flow-through from another Poros20MC nickel column and concentrated in a Centriprep YM-4. Protein concentration was determined by absorbance (Edelhoch, 1967).
Crystallization and Data Collection
Crystals of two Kv7.4 coiled-coil constructs, one comprising residues 610–645 and the other 610–640, were obtained at 16°C by sitting-drop vapor-diffusion. For the longer construct, 1 μl peptide (4 mg ml−1 in 10 mM PO42−, pH 7.3, 250 mM KCl) was mixed with 1 μl of a reservoir solution containing 0.1 M citric acid pH 3.0, 23% (w/v) PEG 1000 and 10% (v/v) n-propanol. For the shorter construct, 1 μl peptide (0.1 mg ml−1 in 10 mM PO42−, pH 7.3, 250 mM KCl) was mixed with 1 μl 0.1 M Bis-Tris, pH 5.5, 0.2 M NH4 acetate and 45% (v/v) MPD. For data collection, crystals of the longer construct were transferred to a solution containing 0.1 M citric acid, pH 3.0, 23% PEG 1000 and 15% n-propanol and flash-cooled in liquid nitrogen. Crystals of the shorter construct were frozen directly out of the drop in liquid nitrogen. Data were collected at ≤ 100 K at Advanced Lightsource Beamline 8.3.1 (Lawrence Berkeley National Laboratory, Berkeley, CA) equipped with a Quantum 210 CCD detector (Area Detector Systems). All data were processed using HKL2000 (HKL Research) (Otwinowski and Minor, 1997).
Structure Determination
A library of 300 polyalanine parallel four-stranded left-handed coiled-coil models was generated based on the coiled-coil geometric parameterization (Crick, 1953) and the helix generator code (Harbury et al., 1998) (helix generator code is available at http://bl831.als.lbl.gov/∼jamesh/scripts/supertwist.awk). Models varied by number of residues per monomer (28–40), superhelical radius R0 (4.0–8.0 Å), and residues per superhelical turn (90–190). Each model in the library was used for molecular replacement with EPMR (Kissinger et al., 2001) in a dataset collected from crystals of Kv7.4 (residues 610–645). The de novo model giving the best correlation coefficient (0.641; second best = 0.635) and R factor (77.6%; second best = 76.2%) was used for a second round of molecular replacement with PHASER (Storoni et al., 2004). This initial refinement led to a model with a superhelical radius of ∼7 Å for each molecule in the asymmetric unit. Refinement and iterative model building were performed using ARP/wARP (Perrakis et al., 1999) and CNS (Brunger et al., 1998). The structure could not be refined fully. The best model was used as a molecular replacement model in data collected from crystals of Kv7.4 (residues 610–640) using CNS (Brunger et al., 1998). The resulting solution was built manually and refined using CNS (Brunger et al., 1998) and the CCP4 program suite (Collaborative Computational Project, Number 4, 1994). Structure quality was monitored with PROCHECK. Structure and cavity volumes were calculated in SwissPDB (Schwede et al., 2003). Coiled-coil parameters were calculated using the FITCC script (M. Sales and T. Alber, personal communication: http://ucxray.berkeley.edu/∼mark/fitcc.html). Table 1 summarizes the data collection, refinement, and model quality. Figures were prepared with PyMOL (DeLano, 2002).
CD Spectroscopy
Twenty-five μM purified Kv7.4 coiled-coil peptide in a buffer of 250 mM NaCl and 2 mM each Na acetate, borate, citrate, and phosphate, pH as indicated in Figure 3A was analyzed with an Aviv Model 215 spectropolarimeter (Aviv Biomedical) equipped with a peltier device. Wavelength scans from 315 nm to 190 nm were taken at 2°C in a cuvette of 1 mm path length. Molar ellipticity per residue of the buffer-subtracted CD spectrum was calculated as a function of concentration, path length, and number of residues per monomer:
where [θ]MRD is the molar ellipticity per residue in deg●cm2 (dmol●res)−1, θ is the experimental ellipticity in millidegrees, M is the molecular mass of the peptide, c is the protein concentration in μM, l is the cuvette path length in cm, and NR is the number of residues in the peptide. The percent helicity was estimated by:
where [θ]222 is the experimental molar ellipticity per residue at 222 nm, is the molar ellipticity for a helix of infinite length at 222 nm (i.e., −39,500 deg●cm2 dmol−1), i is the number of helices, κ is a wavelength-specific constant with a value of 2.57 at 222 nm, and N is the number of residues in the peptide (Chen et al., 1974).
Size Exclusion Chromatography
For the isolated Kv7.4 coiled coil, 100 μl peptide (500 μM peptide in 250 mM KCl, 10 mM PO42−, pH 7.3) was passed through a Superdex75 HR 10/30 column (Amersham Biosciences) in HEPES buffer (250 mM KCl, 1 mM EDTA, 10 mM maltose, 20 mM HEPES, pH 7.3) on an ÄKTA-FPLC system (Pharmacia) at 4°C. For HMT fusion proteins, 100 μl at 2 μg ml−1 was loaded on a Superdex200 column (Amersham Biosciences) equilibrated with high-salt buffer (400 mM KCl, 1 mM EDTA, 10 mM PO42, pH 7.3). On both columns, eluates were monitored at 280 nm over a flow rate of 0.3 ml min−1. Each column was calibrated using at least four standard protein molecular mass markers. Elution volumes from at least three runs were averaged.
Equilibrium Sedimentation
Sedimentation equilibrium experiments were performed at 4°C in a Beckman Optima XL-A analytical ultracentrifuge (Beckman Coulter). Kv7.4 coiled-coil peptide (700 μM in 10 mM PO42−, pH 7.3, 250 mM KCl) was loaded in a six-chamber analytical ultracentrifuge cuvette, using buffer in the adjacent chamber as a blank. The molecular mass was calculated from a single-species exponential fit (Excel) to the distribution of concentration over the radius of the chamber:
where M is the molecular weight in g/mol, R is the gas constant [8.314 J (mol●K)−1], T is the temperature in K, ν is the partial specific volume of the protein in ml g−1, ρ is the density of the solvent in g/mL, ω is the angular velocity of centrifugation in rad s−1, and r is the distance in cm from the center of the rotor to a given position in the cell (Laue, 1995). Partial specific volume was calculated from the sum of the volumes of individual residues in the protein. Solvent density was calculated from the components of the buffer. Residuals were calculated as the difference between the measured absorbance value and the predicted value extrapolated from the calculated molecular mass.
Supplementary Material
Crystal lattice contacts between Kv7.4 tetramers. One complete Kv7.4 Tail is shown in color with subunits from neighboring molecules shown in grey. Positions of the corresponding Kv7.1 LQT mutations are shown.
Acknowledgments
The authors would like to thank T. Jentsch for the Kv7 clones, F. Van Petegem for help with data collection and processing, J.M. Berger and A. Moroni for comments on the manuscript, and members of the Minor laboratory for support at all stages of this work. This work was supported by awards to D.L.M. from the Alfred P Sloan Foundation, March of Dimes Basil O'Connor Scholar Program, Sandler Family Supporting Foundation, McKnight Foundation for Neuroscience, and the NIH. R.J.H. is supported by predoctoral fellowships from the NSF and AHA Western States Affiliate. D.L.M. is a Alfred P. Sloan Research Fellow, Basil O'Connor Scholar, and a McKnight Foundation Scholar. The coordinates and structure factors have been deposited with PDB structure code 2OVC.
Footnotes
Supplemental Data: The Supplemental Data for this article can be found online at http://www.neuron.org/cgi/content/full/53/5/663/DC1/.
References
- Antonin W, Fasshauer D, Becker S, Jahn R, Schneider TR. Crystal structure of the endosomal SNARE complex reveals common structural principles of all SNAREs. Nat Struct Biol. 2002;9:107–111. doi: 10.1038/nsb746. [DOI] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. KVLQT1 and IsK (minK) proteins associate to form the IKs cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Bixby KA, Nanao MH, Shen NV, Kreusch A, Bellamy H, Pfaffinger PJ, Choe S. Zn2+-binding and molecular determinants of tetramerization in voltage-gated K+ channels. Nat Struct Biol. 1999;6:38–43. doi: 10.1038/4911. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Chen YH, Yang JT, Chau KH. Determination of the helix and beta form of proteins in aqueous solution by circular dichroism. Biochemistry. 1974;13:3350–3359. doi: 10.1021/bi00713a027. [DOI] [PubMed] [Google Scholar]
- Chen S, Zhang L, Bryant RM, Vincent GM, Flippin M, Lee JC, Brown E, Zimmerman F, Rozich R, Szafranski P, et al. KCNQ1 mutations in patients with a family history of lethal cardiac arrhythmias and sudden death. Clin Genet. 2003;63:273–282. doi: 10.1034/j.1399-0004.2003.00048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Cooper EC, Jan LY. M-channels: neurological diseases, neuromodulation, and drug development. Arch Neurol. 2003;60:496–500. doi: 10.1001/archneur.60.4.496. [DOI] [PubMed] [Google Scholar]
- Crick FHC. The packing of α-helices: Simple coiled-coils. Acta Crystallogr. 1953;6:689–697. [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Deutsch C. Potassium channel ontogeny. Annu Rev Physiol. 2002;64:19–46. doi: 10.1146/annurev.physiol.64.081501.155934. [DOI] [PubMed] [Google Scholar]
- Deutsch C. The birth of a channel. Neuron. 2003;40:265–276. doi: 10.1016/s0896-6273(03)00506-3. [DOI] [PubMed] [Google Scholar]
- Edelhoch H. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1967;6:1948–1954. doi: 10.1021/bi00859a010. [DOI] [PubMed] [Google Scholar]
- Friedrich T, Schwake M, Bamberg E, Jentsch TJ. A KCNQ1 construct contaning a C-terminal heteromerization domain from KCNQ3 functionally interacts with KCNQ2 and KCNQ3. Biophys J. 2002;82:577A. [Google Scholar]
- Grunnet M, Behr ER, Calloe K, Hofman-Bang J, Till J, Christiansen M, McKenna WJ, Olesen SP, Schmitt N. Functional assessment of compound mutations in the KCNQ1 and KCNH2 genes associated with long QT syndrome. Heart Rhythm. 2005;2:1238–1249. doi: 10.1016/j.hrthm.2005.07.025. [DOI] [PubMed] [Google Scholar]
- Gutman GA, Chandy KG, Adelman JP, Aiyar J, Bayliss DA, Clapham DE, Covarriubias M, Desir GV, Furuichi K, Ganetzky B, et al. International Union of Pharmacology. XLI. Compendium of voltage-gated ion channels: potassium channels. Pharmacol Rev. 2003;55:583–586. doi: 10.1124/pr.55.4.9. [DOI] [PubMed] [Google Scholar]
- Hadley JK, Noda M, Selyanko AA, Wood IC, Abogadie FC, Brown DA. Differential tetraethylammonium sensitivity of KCNQ1-4 potassium channels. Br J Pharmacol. 2000;129:413–415. doi: 10.1038/sj.bjp.0703086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbury PB, Zhang T, Kim PS, Alber T. A switch between two-, three-, and four-stranded coiled coils in GCN4 leucine zipper mutants. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
- Harbury PB, Tidor B, Kim PS. Repacking protein cores with backbone freedom: structure prediction for coiled coils. Proc Natl Acad Sci USA. 1995;92:8408–8412. doi: 10.1073/pnas.92.18.8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbury PB, Plecs JJ, Tidor B, Alber T, Kim PS. High-resolution protein design with backbone freedom. Science. 1998;282:1462–1467. doi: 10.1126/science.282.5393.1462. [DOI] [PubMed] [Google Scholar]
- Hille B. Ion Channels of Excitable Membranes. Third. Sunderland, MA: Sinauer Associates, Inc.; 2001. [Google Scholar]
- Huang L, Bitner-Glindzicz M, Tranebjaerg L, Tinker A. A spectrum of functional effects for disease causing mutations in the Jervell and Lange-Nielsen syndrome. Cardiovasc Res. 2001;51:670–680. doi: 10.1016/s0008-6363(01)00350-9. [DOI] [PubMed] [Google Scholar]
- Itoh T, Tanaka T, Nagai R, Kikuchi K, Ogawa S, Okada S, Yamagata S, Yano K, Yazaki Y, Nakamura Y. Genomic organization and mutational analysis of KVLQT1, a gene responsible for familial long QT syndrome. Hum Genet. 1998;103:290–294. doi: 10.1007/s004390050819. [DOI] [PubMed] [Google Scholar]
- Jenke M, Sanchez A, Monje F, Stuhmer W, Weseloh RM, Pardo LA. C-terminal domains implicated in the functional surface expression of potassium channels. EMBO J. 2003;22:395–403. doi: 10.1093/emboj/cdg035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ. Neuronal KCNQ potassium channels: Physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- Jespersen T, Grunnet M, Olesen SP. The KCNQ1 potassium channel: from gene to physiological function. Physiology (Bethesda) 2005;20:408–416. doi: 10.1152/physiol.00031.2005. [DOI] [PubMed] [Google Scholar]
- Kanki H, Kupershmidt S, Yang T, Wells S, Roden DM. A structural requirement for processing the cardiac K+ channel KCNQ1. J Biol Chem. 2004;279:33976–33983. doi: 10.1074/jbc.M404539200. [DOI] [PubMed] [Google Scholar]
- Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 2001;14:993–1000. doi: 10.1093/protein/14.12.993. [DOI] [PubMed] [Google Scholar]
- Kass RS, Kurokawa J, Marx SO, Marks AR. Leucine/isoleucine zipper coordination of ion channel macromolecular signaling complexes in the heart. Roles in inherited arrhythmias. Trends Cardiovasc Med. 2003;13:52–56. doi: 10.1016/s1050-1738(02)00211-6. [DOI] [PubMed] [Google Scholar]
- Kissinger CR, Gehlhaar DK, Smith BA, Bouzida D. Molecular replacement by evolutionary search. Acta Crystallogr D Biol Crystallogr. 2001;57:1474–1479. doi: 10.1107/s0907444901012458. [DOI] [PubMed] [Google Scholar]
- Kubisch C, Schroeder BC, Friedrich T, Lütjohan B, El-Amaraoui A, Marlin S, Petit C, Jentsch TJ. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell. 1999;96:437–446. doi: 10.1016/s0092-8674(00)80556-5. [DOI] [PubMed] [Google Scholar]
- Laue TM. Sedimentation equilibrium as a thermodynamic tool. Methods Enzymol. 1995;259:427–453. doi: 10.1016/0076-6879(95)59055-2. [DOI] [PubMed] [Google Scholar]
- Lerche C, Scherer CR, Seebohm G, Derst C, Wei AD, Busch AE, Steinmeyer K. Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J Biol Chem. 2000;29:22395–22400. doi: 10.1074/jbc.M002378200. [DOI] [PubMed] [Google Scholar]
- Li M, Jan YN, Jan LY. Specification of subunit assembly by the hydrophillic amino-terminal domain of the Shaker potassium channel. Science. 1992;257:1225–1230. doi: 10.1126/science.1519059. [DOI] [PubMed] [Google Scholar]
- Liu J, Zheng Q, Deng Y, Kallenbach NR, Lu M. Conformational transition between four and five-stranded phenylalanine zippers determined by a local packing interaction. J Mol Biol. 2006;361:168–179. doi: 10.1016/j.jmb.2006.05.063. [DOI] [PubMed] [Google Scholar]
- Lupas AN, Gruber M. The structure of alphα-helical coiled coils. Adv Protein Chem. 2005;70:37–78. doi: 10.1016/S0065-3233(05)70003-6. [DOI] [PubMed] [Google Scholar]
- Maljevic S, Lerche C, Seebohm G, Alekov AK, Busch AE, Lerche H. C-terminal interaction of KCNQ2 and KCNQ3 K+ channels. J Physiol. 2003;548:353–360. doi: 10.1113/jphysiol.2003.040980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR, Kass RS. Requirement for a macromolecular signaling complex for β adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295:496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- Neyroud N, Richard P, Vignier N, Donger C, Denjoy I, Demay L, Shkolnikova M, Pesce R, Chevalier P, Hainque B, et al. Genomic organization of the KCNQ1 K+ channel gene and identification of C-terminal mutations in the long-QT syndrome. Circ Res. 1999;84:290–297. doi: 10.1161/01.res.84.3.290. [DOI] [PubMed] [Google Scholar]
- Novotny T, Kadlecova J, Papousek I, Chroust K, Bittnerova A, Florianova A, Ceskova E, Weislamplova M, Palensky V, Sisakova M, et al. Mutational analysis of LQT genes in individuals with drug-induced QT interval prolongation. Vnitr Lek. 2006;52:116–118. [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Papazian DM. Potassium channels: some assembly required. Neuron. 1999;23:7–10. doi: 10.1016/s0896-6273(00)80746-1. [DOI] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6:458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- Piippo K, Swan H, Pasternack M, Chapman H, Paavonen K, Viitasalo M, Toivoven L, Kontula K. A founder mutation of the potassium channel KCNQ1 in Long QT syndrome. J Am Coll Cardiol. 2001;37:562–568. doi: 10.1016/s0735-1097(00)01124-4. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Meating MT. Coassembly of KVLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Schmitt N, Schwarz M, Peretz A, Abitbol A, Attali B, Pongs O. A recessive C-terminal Jervell and Lange-Nielsen mutation of the KCNQ1 channel impairs subunit assembly. EMBO J. 2000;19:332–340. doi: 10.1093/emboj/19.3.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BC, Kubisch C, Stein V, Jentsch TJ. Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature. 1998;396:687–690. doi: 10.1038/25367. [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Hechenberger M, Weinriech F, Kubisch C, Jentsch TJ. KCNQ5, novel potassium channel broadly expressed in brain, mediates M-type currents. J Biol Chem. 2000;275:24089–24095. doi: 10.1074/jbc.M003245200. [DOI] [PubMed] [Google Scholar]
- Schwake M, Pusch M, Kharkovets T, Jentsch TJ. Surface expression and single channel properties of KCNQ2/KCNQ3, M-type K+ channels invovled in epilepsy. J Biol Chem. 2000;275:13343–13348. doi: 10.1074/jbc.275.18.13343. [DOI] [PubMed] [Google Scholar]
- Schwake M, Jentsch TJ, Friedrich T. A carboxy-terminal domain determines the subunit specificity of KCNQ K+ channel assembly. EMBO Rep. 2003;4:76–81. doi: 10.1038/sj.embor.embor715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwake M, Athanasiadu D, Beimgraben C, Blanz J, Beck C, Jentsch TJ, Saftig P, Friedrich T. Structural determinants of M-type KCNQ (Kv7) K+ channel assembly. J Neurosci. 2006;26:3757–3766. doi: 10.1523/JNEUROSCI.5017-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyanko AA, Hadley JK, Wood IC, Abogadie FC, Jentsch TJ, Brown DA. Inhibition of KCNQ1-4 potassium channels expressed in mammalian cells via M1 muscarinic acetylcholine receptors. J Physiol. 2000;522:349–355. doi: 10.1111/j.1469-7793.2000.t01-2-00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen NV, Pfaffinger PJ. Molecular recognition and assembly sequences involved in the subfamily-specific assembly of voltage-gated K+ channel subunit proteins. Neuron. 1995;14:625–633. doi: 10.1016/0896-6273(95)90319-4. [DOI] [PubMed] [Google Scholar]
- Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT. Spectrum of mutations in Long-QT syndrome genes KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr D Biol Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- Syme CA, Hamilton KL, Jones HM, Gerlach AC, Giltinan L, Papworth GD, Watkins SC, Bradbury NA, Devor DC. Trafficking of the Ca2+-activated K+ channel, hIK1, is dependent upon a C-terminal leucine zipper. J Biol Chem. 2003;278:8476–8486. doi: 10.1074/jbc.M210072200. [DOI] [PubMed] [Google Scholar]
- Tarbouriech N, Curran J, Ruigrok RW, Burmeister WP. Tetrameric coiled coil domain of Sendai virus phosphoprotein. Nat Struct Biol. 2000;7:777–781. doi: 10.1038/79013. [DOI] [PubMed] [Google Scholar]
- Tester DJ, Will ML, Haglund CM, Ackerman MJ. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm. 2005;2:507–517. doi: 10.1016/j.hrthm.2005.01.020. [DOI] [PubMed] [Google Scholar]
- Tsuruda PR, Julius D, Minor DL., Jr Coiled coils direct assembly of a cold-activated TRP channel. Neuron. 2006;51:201–212. doi: 10.1016/j.neuron.2006.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem F, Clark KA, Chatelain FC, Minor DL., Jr Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature. 2004;429:671–675. doi: 10.1038/nature02588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet. 1996;12:17–23. doi: 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-Channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Woolfson DN. The design of coiled-coil structures and assemblies. Adv Protein Chem. 2005;70:79–112. doi: 10.1016/S0065-3233(05)70004-8. [DOI] [PubMed] [Google Scholar]
- Yamashita F, Horie M, Kubota T, Yoshida H, Yumoto Y, Kobori A, Ninomiya T, Kono Y, Haruna T, Tsuji K, et al. Characterization and subcellular localization of KCNQ1 with a heterozygous mutation in the C terminus. J Mol Cell Cardiol. 2001;33:197–207. doi: 10.1006/jmcc.2000.1300. [DOI] [PubMed] [Google Scholar]
- Zhong H, Molday LL, Molday RS, Yau KW. The heteromeric cyclic nucleotide-gated channel adopts a 3A:1B stoichiometry. Nature. 2002;420:193–198. doi: 10.1038/nature01201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Lai J, Yau KW. Selective heteromeric assembly of cyclic nucleotide-gated channels. Proc Natl Acad Sci USA. 2003;100:5509–5513. doi: 10.1073/pnas.0931279100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal lattice contacts between Kv7.4 tetramers. One complete Kv7.4 Tail is shown in color with subunits from neighboring molecules shown in grey. Positions of the corresponding Kv7.1 LQT mutations are shown.