

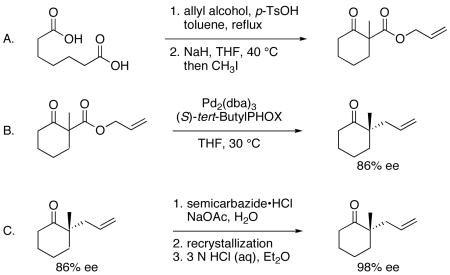
1. Procedure
Caution! This procedure should be carried out in an efficient fume hood due to the evolution of hydrogen gas during the reaction.
A. 1-Methyl-2-oxo-Cyclohexanecarboxylic acid 2-propenyl ester
A 500 mL single-neck round-bottom flask equipped with a large magnetic stir bar was charged with 50.00 g of pimelic acid (312.2 mmol, 1.0 equiv), 156 mL of toluene, and 63.7 mL of allyl alcohol (936.5 mmol, 3.0 equiv) (Note 1). The mixture was stirred vigorously to create a uniform suspension, and 296.9 mg of p-toluenesulfonic acid monohydrate (1.56 mmol, 0.005 equiv) was added. A Dean–Stark trap, a water-cooled condenser, and a nitrogen inlet were affixed to the flask, and the resulting suspension was heated to reflux (120 °C oil bath temperature). The mixture in the flask soon became homogeneous. After 12 h at reflux, approximately 11 mL of water had accrued in the Dean–Stark trap. The vessel was cooled to ambient temperature and the solution was transferred to a separatory funnel. The organic solution was washed successively with saturated aqueous sodium bicarbonate (3 × 15 mL) and brine (2 × 15 mL) and then dried over anhydrous magnesium sulfate. After filtration through cotton, the organic solution was concentrated by rotary evaporation under vacuum (1 torr) and then the last traces of solvent were removed under high vacuum (0.5 torr) overnight to yield 74.74 g of diallyl pimelate (311.0 mmol, 99.6% yield) as a slightly yellow-colored, free-flowing liquid. GC analysis indicates 98% purity (Note 2).
A flame-dried three-neck 1 L flask equipped with a glass stopper, a water-cooled reflux condenser, a rubber septum, and a magnetic stir bar was charged with 13.69 g of 60% sodium hydride (342.1 mmol, 1.1 equiv) and tetrahydrofuran (260 mL, distilled from 9-fluorenone ketyl). The flask was immersed in a water bath (22 °C) and a solution of 74.74 g of crude diallyl pimelate (311.0 mmol, 1 equiv) in 50 mL of tetrahydrofuran was added in a steady stream via syringe. Some moderate bubbling (molecular hydrogen evolution) of the reaction mixture was observed during the addition. Following the addition, the mixture was stirred at 22 °C for 7 h, and then heated to 40 °C for 4 h to complete cyclization (Note 3). Once the starting material was consumed (based on TLC, Note 3), 25.17 mL of neat iodomethane (404.3 mmol, 1.3 equiv) was added to the mixture. After an additional 8 h at 40 °C, the mixture was cooled to ambient temperature (22 °C) and water (60 mL) was added carefully via syringe over the course of 5 min. The mixture was transferred to a separatory funnel and ethyl acetate (100 mL) was added. The phases were separated and then the aqueous phase was extracted with ethyl acetate (3 × 75 mL). The combined organic extracts were washed with brine (1 × 50 mL) and then dried over anhydrous magnesium sulfate. After filtration through cotton, the organic solution was concentrated by rotary evaporation under vacuum (1 torr) to a yield a yellow liquid. The material was purified by short-path distillation to give 41.13 g (209.6 mmol, 67% yield) of a clear, colorless liquid boiling from 92–95 °C/0.3 torr. GC analysis found 92% product purity which is suitable for use in the next step (Note 4).
B. (2S)-2-Methyl-2-(2-propen-1-yl)-cyclohexanone
A flame-dried 50 mL conical flask equipped with a rubber septum was charged with a portion of allyl 1-methyl-2-oxocyclohexanecarboxylate, placed under vacuum (1 torr) for 20 min to remove any dissolved gases, and then backfilled with argon. After this process, the mass of allyl 1-methyl-2-oxocyclohexanecarboxylate in the flask was 17.47 g (89.03 mmol, 1.0 equiv). A 1 L three-neck round-bottom flask was equipped with a stir bar, two rubber septa, and a gas inlet. The apparatus was flame dried under vacuum and backfilled with dry argon (three cycles). After cooling, 435 mL of anhydrous tetrahydrofuran (dried by passage through a column of activated alumina under argon) (Note 5) was added and the flask was immersed in a 30 °C oil bath. A twelve-inch needle was inserted through one of the septa and used to bubble dry argon gas through the liquid for 30 min. The needle was removed and then 1.02 g of tris(dibenzylideneacetone) dipalladium(0) (Pd2(dba)3, 1.11 mmol, 0.0125 equiv) and 1.03 g of (S)-tert-ButylPHOX (2.67 mmol, 0.030 equiv) (Note 1) were added. The mixture immediately became opaque and took on a golden-brown color. This mixture was stirred at 30 °C for 30 min (Note 6). A cannula was attached to connect the conical flask containing allyl 1-methyl-2-oxocyclohexanecarboxylate to the three-neck flask, and the neat substrate was then transferred in a rapid dropwise fashion to the catalyst mixture by applying positive pressure to the conical flask. When the transfer was complete (about 10 min), the conical flask was rinsed successively with two 5 mL portions of anhydrous tetrahydrofuran and the washes added to the reaction mixture through the cannula. Upon addition of the substrate to the catalyst mixture, the color changed to olive green. The mixture was maintained at 30–32 °C for 26 h, when TLC indicated complete consumption of the starting material (Note 7). The olive green-colored mixture was then passed through a pad of silica gel (5 cm diameter × 5 cm height) and eluted with diethyl ether (200 mL). The bright yellow filtrate was concentrated by rotary evaporation under vacuum (150 torr) (Note 8). The liquid was then transferred to a 50 mL round-bottom flask and distilled through a short path apparatus into a receiving flask immersed in an ice water bath to provide 10.34 g (67.95 mmol, 76% yield) of (S)-2-allyl-2-methylcyclohexanone as a clear, colorless liquid boiling from 104–105 °C/26 torr that was analytically pure based on standard techniques (Note 9). Analysis of this material by chiral GC found 86% enantiomeric excess (Note 10). Additional product was obtained by subjecting the material remaining in the distillation pot to flash chromatography on silica gel (Note 11), which provided an additional 1.49 g of product (9.79 mmol, 11% yield), also of 86% ee, for a combined yield of 11.83 g (77.74 mmol, 87% yield).
C. Enrichment of (2S)-2-methyl-2-(2-propen-1-yl)-cyclohexanone via (2E)-2-[(2S)-2-methyl-2-(2-propenyl)cyclohexylidene]-hydrazinecarboxamide
A 200 mL pear-shaped flask containing 5.57 g of sodium acetate (67.95 mmol, 1.0 equiv), 8.34 g of semicarbazide hydrochloride (74.75 mmol, 1.1 equiv), 75 mL of purified water (Note 1), and a magnetic stir bar was stirred until all of the solids had dissolved. At this point, 10.34 g of neat 2-allyl-2-methylcyclohexanone (67.95 mmol, 1.0 equiv) was added via pipette. When the addition was complete, a nitrogen inlet was attached and the mixture was heated to 60 °C for 14 h (Note 12). The thick slurry was vacuum (water aspirator) filtered directly through filter paper on a porcelain Büchner funnel and rinsed with water (2 × 20 mL). The white solid was dried for 15 min on the funnel, and then transferred to a round-bottom flask and dried under vacuum (1 torr) until a constant mass of 12.15 g (58.05 mmol, 85% yield) was achieved (about 10 h). At this point, the semicarbazone was found to have 91% ee (measured by reverting to the ketone, Note 13).
A stir bar was added to the flask and the solids were suspended in 150 mL of toluene with mixing at approximately 400 rpm. The mixture was then heated to 110 °C (bath temperature) in an oil bath. After a few minutes at this temperature, the solids dissolved completely to afford a clear, colorless solution (Note 14). Heating was discontinued and the stirred mixture was allowed to cool to ambient temperature (20 °C) while still immersed in the oil bath (Note 15). After 8 h had elapsed, the cooled heterogeneous mixture was vacuum (water aspirator) filtered through filter paper on a porcelain Büchner funnel. The solids were rinsed with toluene (2 × 10 mL) and then dried on the filter for 15 min (Note 16). The solids were transferred to a 200 mL pear-shaped flask and dried under vacuum (1 torr) until a constant mass of 10.96 g (52.36 mmol, 90% recovery) was observed. This material was found to have 98% enantiomeric excess (measured by reverting to the ketone, Note 13).
A 200 mL pear-shaped flask containing a magnetic stir bar and 10.96 g of semicarbazone (52.34 mmol) and 40 mL of diethyl ether was stirred to suspend the solids. To the suspension was added 20 mL of 3 N aqueous hydrochloric acid. No appreciable heat evolution was observed. The mixture was stirred vigorously for 4.5 h at ambient temperature (20 °C), at which time all of the solids had disappeared and two clear colorless phases were observed. The biphasic mixture was transferred to a 125 mL separatory funnel and the phases were separated. The aqueous phase was extracted with diethyl ether (3 × 10 mL). The combined organic layers were then washed successively with saturated sodium bicarbonate (2 × 5 mL), water (1 × 5 mL), and brine (2 × 5 mL). The organic phase was dried over anhydrous magnesium sulfate and then filtered through cotton and concentrated by rotary evaporation under vacuum (150 torr, first at 20 °C, then at 30 °C to remove the last traces of solvent) to provide 7.89 g (51.82 mmol, 99% yield) of (S)-2-allyl-2-methylcyclohexanone of 98% ee (Note 10). GC analysis found >99% product purity (Note 17).
2. Notes
Pimelic acid (≥99%, Fluka), allyl alcohol (99+%, Aldrich), p-toluenesulfonic acid monohydrate (99%, Sigma), toluene (ACS reagent grade, Fisher), solid sodium bicarbonate (ACS reagent grade, Mallinckrodt), magnesium sulfate (anhydrous powder, 99.5%, Alfa Aesar), sodium hydride (60% dispersion in mineral oil, Acros), iodomethane (99%, Aldrich), ethyl acetate (HPLC grade, Fisher), tris(dibenzylideneacetone)dipalladium (Pd2(dba)3, Strem), sodium acetate (99%, Aldrich), semicarbazide hydrochloride (99%, Alfa Aesar), hydrochloric acid (37 wt%, EMD), and diethyl ether (ACS reagent grade, Fisher) were purchased and used as received. Tetrahydrofuran (HPLC grade, Fisher) was distilled from sodium 9-fluorenone ketyl2 or passed through an activated alumina column under argon prior to use.3 The ligand (S)-tert-ButylPHOX was prepared using our accompanying procedure in Organic Syntheses.4,5 Purified water was obtained using a Barnstead NANOpure Infinity UV/UF system.
The esterification product, diallyl pimelate, may be distilled (bp 134–135 °C/0.2 torr), but this is not necessary for this application. Distillation of a separate sample of diallyl pimelate led to significant loss of material to unidentified polymeric byproducts formed in the distillation flask, and distillation is therefore not recommended. Product purity was measured by achiral GC using an Agilent 6850 GC equipped with an Agilent DB-WAX column (30.0 m × 0.25 mm) and a flame ionization detector using a method of 100 °C isothermal for 5 min, then ramp 13 °C/min to 240 °C, then 240 °C isothermal for 5 min with a 1.0 mL/min He carrier gas flow. The retention time for the product was 14.70 min, with a predominant but unidentified impurity with a retention time of 14.30 min. The product exhibited the following characteristics: 1H NMR (300 MHz, CDCl3) δ 5.92 (ddt, J = 17.3, 10.4, 5.8 Hz, 2H), 5.31 (app. dq, J = 17.3, 1.7 Hz, 2H), 5.23 (app. dq, J = 10.4, 1.1 Hz, 2H), 4.57 (ddd, J = 5.8, 1.4, 1.4 Hz, 4H), 2.34 (t, J = 7.4 Hz, 4H), 1.66 (app. quintet, J = 7.7 Hz, 4H), 1.43–1.31 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 173.3, 132.3, 118.3, 65.1, 34.1, 28.7, 24.6; IR (Neat Film NaCl) 3086, 3020, 2943, 2867, 1736, 1649, 1457, 1421, 1379, 1272, 1174, 1086, 992, 932, 738 cm−1; HRMS (EI) m/z calc’d for C13H20O4 [M]+: 240.1362, found 240.1355.
The progression of the cyclization may be monitored by TLC analysis using 20% ethyl acetate in hexanes as eluent with p-anisaldehyde staining: Rf diallyl pimelate = 0.45 (stains dark blue), Rf cyclized intermediate = 0.35–0.50 (broad, stains deep purple, also UV active), Rf alkylation product = 0.40 (stains brown). The detection of diallyl pimelate is often obscured by the cyclized intermediate.
Using the GC method described in Note 2, allyl 1-methyl-2-oxocyclohexanecarboxylate has a retention time of 11.79 min. The distilled material contains a small amount (6% by GC) of uncyclized diallyl pimelate and 2% of an unidentified byproduct (retention time of 14.39 min) that do not significantly affect the subsequent step. GC response factors between allyl 1-methyl-2-oxocyclohexanecarboxylate and diallyl pimelate were calculated with further purified materials to confirm these ratios, however assuming a 1:1 response factor gave the same ratios. Analytically pure material may be obtained by flash chromatography on silica gel using a gradient of 1.5 → 4% diethyl ether in hexanes as eluent. The product showed the following characterization data: 1H NMR (300 MHz, CDCl3) δ 5.85 (dddd, J = 17.1, 10.2, 5.9, 5.9 Hz, 1H), 5.24 (m, 2H), 4.59 (d, J = 5.7 Hz, 2H), 2.58–2.34 (comp. m, 3H), 2.08–1.88 (m, 1H), 1.80–1.54 (comp. m, 3H), 1.52–1.37 (m, 1H), 1.27 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 207.9, 172.6, 131.4, 118.7, 65.6, 57.1, 40.5, 38.1, 27.4, 22.5, 21.1; IR (Neat Film NaCl) 3086, 2939, 2867, 1715, 1452, 1259, 1211, 1159, 1084, 976 cm−1; HRMS (EI) m/z calc’d for C15H16O3 [M]+: 196.1099, found 196.1096.
The substrate concentration (0.2 M) described herein yields product of slightly lower enantiomeric excess (about 1% lower) than our previously reported, optimized conditions (0.033 M in substrate). For smaller scale where overall quantity of solvent is less important, the lower substrate concentration is recommended.
The complexation time prior to adding substrate is important to the overall reaction. Shorter or longer complexation times led to lower product yield and incomplete substrate conversion.
Although the reaction produces an equivalent of carbon dioxide, the evolution of this byproduct is not visually apparent during the reaction. The reaction is readily evaluated by TLC analysis using 10% Et2O in pentane as eluent with p-anisaldehyde staining: Rf dibenzylideneacetone = 0.14 (stains orange, also UV active), Rf β-ketoester = 0.25 (stains brown), Rf product = 0.35 (stains yellow).
Care should be taken to ensure that the moderately volatile product is not lost during concentration of the filtrate. However, if a substantial amount of solvent remains, distillation of the product does not occur smoothly. At 150 torr, tetrahydrofuran and diethyl ether are easily removed and product is not lost.
The distilled material was analytically pure based on standard spectral techniques. The following properties were observed: 1H NMR (500 MHz, CDCl3) δ 5.69 (dddd, J = 16.6, 10.7, 7.3, 7.3 Hz, 1H), 5.05 (dddd, J = 10.7, 2.2, 1.0, 1.0 Hz, 1H), 5.04 (dddd, J = 16.6, 2.2, 1.5, 1.2 Hz, 1H), 2.39 (app. t, J = 6.8 Hz, 2H), 2.36 (dddd, J = 13.9, 7.6, 1.2, 1.0 Hz, 1H), 2.23 (dddd, J = 13.9, 7.3, 1.2, 1.0 Hz, 1H), 1.90–1.65 (comp. m, 5H), 1.62–1.55 (m, 1H), 1.07 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 215.4, 133.7, 117.9, 48.4, 41.9, 38.8, 38.5, 27.4, 22.6, 21.0; IR (Neat Film NaCl) 2934, 2865, 1707, 1451, 912 cm−1; HRMS (EI) m/z calc’d for C10H16O [M]+: 152.1201, found 152.1204; [α]D21.0 −49.64 (c 2.38, dichloromethane, 98% ee).
GC analyses were performed with an Agilent 6850 GC equipped with a Chiraldex G-TA column (30.0 m × 0.25 mm) and a flame ionization detector. The assay conditions for 2-allyl-2-methylcyclohexanone are 100 °C isothermal, 1.0 mL/min He carrier gas flow, retention times: major (S) enantiomer = 14.40 min, minor (R) enantiomer = 16.72 min. The absolute configuration was established by X-ray crystallographic analysis of a semicarbazone derivative bearing a substituent with known absolute configuration.6
Column conditions: 5 cm diameter × 10 cm height, eluting with a gradient of 2 → 5% diethyl ether in petroleum ether, collecting 60 mL fractions. See Note 6 for TLC conditions. For smaller scale preparations, it is often convenient to perform chromatography directly rather than distilling the product.
Semicarbazone formation begins before the addition of ketone is complete, however, conversion at room temperature is sluggish.
To ensure an accurate ee value, the powder was mixed thoroughly prior to measurement. The enantiomeric excess was determined by suspending a small amount of semicarbazone (approximately 5 mg) in a biphasic mixture of diethyl ether (1 mL) and 2 N aqueous hydrochloric acid (1 mL) at ambient temperature. After 2 h, all of the solids had dissolved and the organic layer was separated, dried briefly over anhydrous magnesium sulfate, filtered through cotton, and the filtrate concentrated by rotary evaporation. The residue was then dissolved in diethyl ether and analyzed by GC (see Note 10 for separation conditions). The semicarbazone appears to be a single geometric isomer exhibiting the following properties: mp 188–189 °C (toluene, 98% ee); 1H NMR (300 MHz, CDCl3) δ 7.93 (br s, 1H), 5.73 (m, 1H), 5.05 (s, 1H), 5.00 (app. d, J = 3.3 Hz, 1H), 2.40–2.11 (comp. m, 4H), 1.71–1.44 (comp. m, 6H), 1.10 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 158.1, 156.8, 134.6, 117.2, 42.9, 41.5, 38.6, 25.9, 24.5, 22.5, 21.0; IR (Neat Film NaCl) 3465, 3195, 1693, 1567, 1478 cm−1; HRMS (CI, CH4) m/z calc’d for C11H20N3O [M + H]+: 210.1606, found 210.1599; [α]D21.0 −43.73 (c 1.94, methanol, 96% ee).
At the reported concentration, the hot toluene solution is not saturated. The additional solvent helps maintain efficient stirring as the crystallization progresses and the viscosity of the mixture increases. The additional solvent does not significantly affect the efficiency of product recovery.
Stirring during the crystallization process is very important to the efficiency of the ee improvement. For example, two separate 300 mg portions of semicarbazone with 89% ee were recrystallized from hot toluene (about 3 mL) with and without stirring. Although product recovery was comparable for either procedure (81% and 80%, respectively), the unstirred crystallization provided semicarbazone of 93% ee while the stirred crystallization provided semicarbazone of 96% ee. Either procedure yields the product as very fine needles.
Concentration of the filtrate by rotary evaporation provided an additional 1.15 g (9.5% recovery) of semicarbazone. GC analysis of the corresponding ketone found 28% ee for this material (see Note 13).
Using the GC method described in Note 2, 2-allyl-2-methylcyclohexanone has a retention time of 8.62 min.
Waste Disposal Information
All hazardous materials should be handled and disposed of in accordance with “Prudent Practice in the Laboratory”; National Academy Press; Washington, D. C., 1995.
3. Discussion
The Dieckmann cyclization protocol employed here is a modification of a similar procedures developed by Tsuji and coworkers7 and Fuchs and coworkers.8 This improved procedure allows preparation of racemic allyl β-ketoester substrates in two steps with a single purification. Importantly, the single-pot cyclization/alkylation is an improvement over our previously reported method that required solvent exchange.9 The Dieckmann protocol is useful for the preparation of a number of substituted allyl β-ketoesters by varying the electrophile. Possible substituents include alkyl, benzyl, substituted benzyl, and alkenyl.9 Other more sensitive substituents may be introduced by quenching the intermediate β-ketoester enolate with aqueous acid and then alkylating the resulting β-ketoester under more mild conditions (e.g., K2CO3, acetone, 50 °C).9 In this manner, the β-ketoester enolate may undergo conjugate addition, aldol, or fluorination reactions with appropriate electrophilic components.9 The β-ketoester substrates are useful not only for enantioselective decarboxylative allylation, but also for enantioselective decarboxylative protonation reactions generating α-tertiary cycloalkanones.10 Alternative methods for synthesis of β-ketoester substrates include acylation of ketones with diallyl carbonates,9 allyl cyanoformates,9,11 allyl chloroformates,12 or allyl 1H-imidazole-1-carboxylates.13
The enantioselective decarboxylative allylation method from allyl β-ketoesters,9,14 based on non-enantioselective transformations pioneered by Tsuji and Saegusa,15 represents a substantial advance in asymmetric allylation since prior methods16 required that the putative prochiral enolate intermediate17 be stabilized by an electron-withdrawing group (e.g., esters or aryl groups) or contain only a single acidic site.18,19 To highlight the previous deficiency in the literature, 2-allyl-2-methylcyclohexanone had not been prepared in high enantiomeric excess prior to our work since few alternative synthetic methods are available.20 Related enantioselective transformations for the conversion of allyl enol carbonates and silyl enol ethers to α-quaternary cycloalkanones, also based on earlier work by Tsuji,21 have been developed by our group6 and others.22 However, β-ketoester substrates are often preferable due to the ease of synthesis and ease of substrate handling. The procedure reported herein has been optimized for large-scale preparation and features lower catalyst loading and higher substrate concentration than our previously reported work. These changes have minimal impact on the efficiency and selectivity observed in the reaction. Improvements to purification include conditions for distillation of the product and an improved protocol for conversion to the corresponding semicarbazone derivative. Conditions for recrystallization of the semicarbazone derivative are also reported, and provide access to highly enantioenriched 2-allyl-2-methylcyclohexanone.
The scope of this transformation9 and the related transformation of allyl enol carbonates and enol silanes6 has been demonstrated to include alkyl, alkenyl, aryl, ethereal, siloxy, halogen,23 ketone, ester, and nitrile substituents. Additionally, the ring may be appended, unsaturated, enlarged, or substituted with a heteroatom. The delivered allyl group may be substituted at the internal position. Cascade allylation has also been performed to generate two quaternary stereocenters. Good levels of enantioselectivity are observed throughout these variations and products may be obtained in 55–99% yield and 80–92% ee (Table 1).6,9
Table 1.
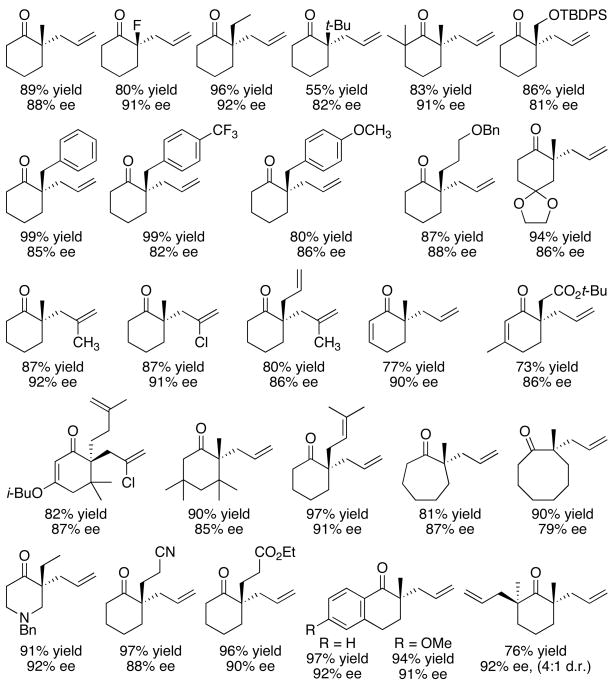
|
The non-enantioselective Tsuji allylation reaction has been used sparingly in total synthesis efforts.24 Since the development of asymmetric variants, however, enantioselective decarboxylative allylation has functioned as a key asymmetric step in the synthesis of the natural products (+)-dichroanone,25 (+)-elatol,26 (+)-laurencenone B,26 and (−)-cyanthiwigin F,27 and in an approach to the natural product zoanthenol28 (Table 2). Other useful transformations of the product (S)-2-allyl-2-methyl cyclohexanone include elaboration to various [6.5]- and [6.6]-fused bicycles and oxidation to a caprolactone derivative (Table 3a).6 Spirocyclic systems are accessible by employing Grubbs’ olefin metathesis catalysts29 with α,ω -dienef8,26 (Table 3b).
Table 2.
Synthetic targets accessed via enantioselective decarboxylative allylation.
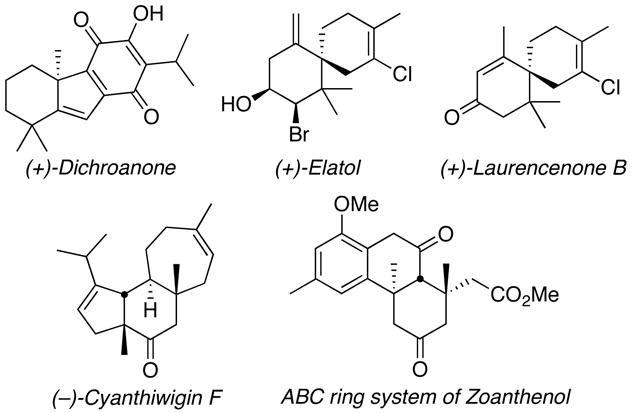
|
Table 3.
(a) Derivatives of 2-allyl-2-methylcyclohexanone.6 (b) Spirocycles accessible via ring-closing metathesis.18,26b
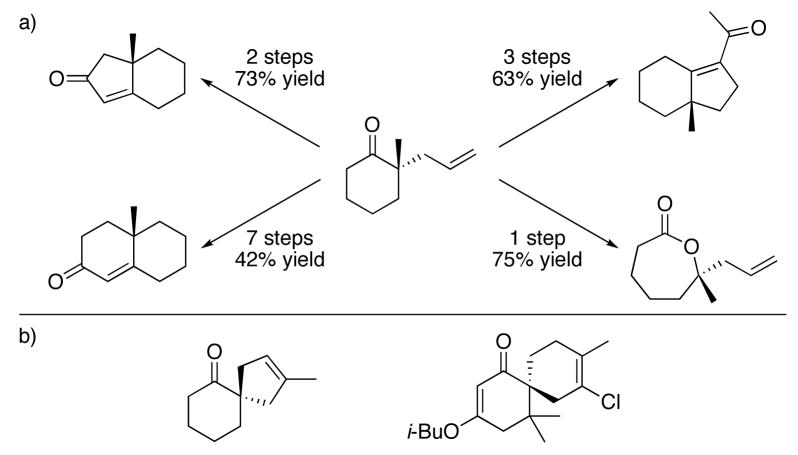
|
Appendix Chemical Abstracts Nomenclature; (Registry Number)
Pimelic acid: Heptanedioic acid; (111-16-0)
Toluene: Benzene, methyl-; (108-88-3)
Nitrogen; (7727-37-9)
Argon; (7440-37-1)
Water; (7732-18-5)
Allyl alcohol: 2-Propen-1-ol; (107-18-6)
p-Toluenesulfonic acid monohydrate: Benzenesulfonic acid, 4-methyl-, hydrate (1:1); (6192-52-5)
Sodium bicarbonate: Carbonic acid sodium salt (1:1); (144-55-8)
Magnesium sulfate: Sulfuric acid magnesium salt (1:1); (7487-88-9)
Diallyl pimelate: Pimelic acid, diallyl ester; (91906-66-0)
Sodium hydride; (7646-69-7)
Tetrahydrofuran: Furan, tetrahydro-; (109-99-9)
Iodomethane: Methane, iodo-; (74-88-4)
Ethyl acetate: Acetic acid ethyl ester; (141-78-6)
Allyl 1-methyl-2-oxocyclohexanecarboxylate: Cyclohexanecarboxylic acid, 1-methyl-2-oxo-, 2-propenyl ester; (7770-41-4)
Tris(dibenzylideneacetone) dipalladium(0): Palladium, tris[μ-[(1,2-η:4,5-η)- (1E,4E)-1,5-diphenyl-1,4-pentadien-3-one]]di-; (51364-51-3)
(S)-tert-ButylPHOX: Oxazole, 4-(1,1-dimethylethyl)-2-[2-(diphenylphosphino)phenyl]-4,5-dihydro-, (4S)-; (148461-16-9)
(S)-2-Allyl-2-methylcyclohexanone: Cyclohexanone, 2-methyl-2-(2-propen-1-yl)-, (2S)-; (812639-07-9)
Sodium acetate: Acetic acid, sodium salt (1:1); (127-09-3)
Semicarbazide hydrochloride: Hydrazinecarboxamide, hydrochloride (1:1); (563-41-7)
(S)-2-(2-Allyl-2-methylcyclohexylidene)hydrazinecarboxamide: Hydrazinecarboxamide, 2-[(2S)-2-methyl-2-(2-propenyl)cyclohexylidene]-, (2E)-; (812639-25-1)
Diethyl ether: Ethane, 1,1′-oxybis-; (60-29-7)
Hydrochloric acid; (7647-01-0)
References
- 1.Department of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California, 91125; E-mail: stoltz@caltech.edu.
- 2.Kamaura M, Inanaga J. Tetrahedron Lett. 1999;40:7347–7350. [Google Scholar]
- 3.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 4.Krout, M. R.; Mohr, J. T.; Stoltz, B. M. Submitted for publication.
- 5.Tani K, Behenna DC, McFadden RM, Stoltz BM. Org Lett. 2007;9:2529–2531. doi: 10.1021/ol070884s. [DOI] [PubMed] [Google Scholar]
- 6.Behenna DC, Stoltz BM. J Am Chem Soc. 2004;126:15044–15045. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]
- 7.Tsuji J, Nisar M, Shimizu I, Minami I. Synthesis. 1984;12:1009. [Google Scholar]
- 8.Pariza RJ, Kuo F, Fuchs PL. Synth Commun. 1983;13:243–254. [Google Scholar]
- 9.Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew Chem, Int Ed. 2005;44:6924–6927. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]
- 10.(a) Mohr JT, Nishimata T, Behenna DC, Stoltz BM. J Am Chem Soc. 2006;128:11348–11349. doi: 10.1021/ja063335a. [DOI] [PubMed] [Google Scholar]; (b) Marinescu SC, Nishimata TN, Mohr JT, Stoltz BM. Org Lett. 2008;10:1039–1042. doi: 10.1021/ol702821j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Mander LN, Sethi SP. Tetrahedron Lett. 1983;24:5425–5428. [Google Scholar]; (b) Donnelly DMX, Finet JP, Rattigan BA. J Chem Soc, Perkin Trans 1. 1993:1729–1735. [Google Scholar]
- 12.Trost BM, Bream RN, Xu J. Angew Chem, Int Ed. 2006;45:3109–3112. doi: 10.1002/anie.200504421. [DOI] [PubMed] [Google Scholar]
- 13.Trost BM, Xu J. J Org Chem. 2007;72:9372–9375. doi: 10.1021/jo7016313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakamura M, Hajra K, Endo K, Nakamura E. Angew Chem, Int Ed. 2005;44:7248–7251. doi: 10.1002/anie.200502703.(b) For a related method using a bis(phosphine) ligand, see: ref 12.
- 15.(a) Shimizu I, Yamada T, Tsuji J. Tetrahedron Lett. 1980;21:3199–3202. [Google Scholar]; (b) Tsuda T, Chujo Y, Nishi S-i, Tawara K, Saegusa T. J Am Chem Soc. 1980;102:6381–6384. [Google Scholar]
- 16.(a) Hayashi T, Kanehira K, Hagihara T, Kumada M. J Org Chem. 1988;53:113–120. [Google Scholar]; (b) Sawamura M, Nagata H, Sakamoto H, Ito Y. J Am Chem Soc. 1992;114:2586–2592. [Google Scholar]; (c) Sawamura M, Sudoh M, Ito Y. J Am Chem Soc. 1996;118:3309–3310. [Google Scholar]; (d) Trost BM, Radinov R, Grenzer EM. J Am Chem Soc. 1997;119:7879–7880. [Google Scholar]; (e) Trost BM, Ariza X. Angew Chem, Int Ed. 1997;36:2635–2637. [Google Scholar]; (f) Kuwano R, Ito Y. J Am Chem Soc. 1999;121:3236–3237. [Google Scholar]; (g) Trost BM, Schroeder GM. J Am Chem Soc. 1999;121:6759–6760. [Google Scholar]; (h) You SL, Hou XL, Dai LX, Cao BX, Sun J. Chem Commun. 2000:1933–1934. [Google Scholar]; (i) You SL, Hou XL, Dai LX, Zhu XZ. Org Lett. 2001;3:149–151. doi: 10.1021/ol0067033. [DOI] [PubMed] [Google Scholar]; (j) Trost BM, Schroeder GM, Kristensen J. Angew Chem, Int Ed. 2002;41:3492–3495. doi: 10.1002/1521-3773(20020916)41:18<3492::AID-ANIE3492>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; (k) Kuwano R, Uchida K, Ito Y. Org Lett. 2003;5:2177–2179. doi: 10.1021/ol034665s. [DOI] [PubMed] [Google Scholar]; (l) Trost BM, Schroeder GM. Chem–Eur J. 2005;11:174–184. doi: 10.1002/chem.200400666. [DOI] [PubMed] [Google Scholar]
- 17.For a computational investigation of the mechanism of the allylation reaction, see: Keith JA, Behenna DC, Mohr JT, Ma S, Marinescu SC, Oxgaard J, Stoltz BM, Goddard WA., III J Am Chem Soc. 2007;129:11876–11877. doi: 10.1021/ja070516j.
- 18.For a review of the development of these asymmetric allylation methods, see: Mohr JT, Stoltz BM. Chem–Asian J. 2007;2:1476–1491. doi: 10.1002/asia.200700183. and references therein.
- 19.For recent reviews of allylic alkylation of ketone enolates, see: Braun M, Meier T. Angew Chem, Int Ed. 2006;45:6952–6955. doi: 10.1002/anie.200602169.You SL, Dai LX. Angew Chem, Int Ed. 2006;45:5246–5248. doi: 10.1002/anie.200601889.Braun M, Meier T. Synlett. 2006:661–676.Kazmaier U. Curr Org Chem. 2003;7:317–328.
- 20.For notable alternative catalytic methods for the generation of α-quaternary cycloalkanones, see: Yamashita Y, Odashima K, Koga K. Tetrahedron Lett. 1999;40:2803–2806.Doyle AG, Jacobsen EN. J Am Chem Soc. 2005;127:62–63. doi: 10.1021/ja043601p.Doyle AG, Jacobsen EN. Angew Chem, Int Ed. 2007;46:3701–3705. doi: 10.1002/anie.200604901.
- 21.(a) Tsuji J, Minami I, Shimizu I. Chem Lett. 1983:1325–1326. [Google Scholar]; (b) Tsuji J, Minami I, Shimizu I. Tetrahedron Lett. 1983;24:1793–1796. [Google Scholar]
- 22.(a) Trost BM, Xu J. J Am Chem Soc. 2005;127:2846–2847. doi: 10.1021/ja043472c. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Xu J. J Am Chem Soc. 2005;127:17180–17181. doi: 10.1021/ja055968f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Trost BM, Xu J, Reichle M. J Am Chem Soc. 2007;129:282–283. doi: 10.1021/ja067342a. [DOI] [PMC free article] [PubMed] [Google Scholar]; Schulz SR, Blechert S. Angew Chem, Int Ed. 2007;129:3966–3970. doi: 10.1002/anie.200604553. [DOI] [PubMed] [Google Scholar]
- 23.For examples of the utility of the allylation reaction to generate tertiary fluoride stereocenters, see: (a) Ref 9. (b) Ref 14a. Burger EC, Barron BR, Tunge JA. Synlett. 2006:2824–2826.Bélanger É, Cantin K, Messe O, Tremblay M, Paquin JF. J Am Chem Soc. 2007;129:1034–1035. doi: 10.1021/ja067501q.
- 24.(a) Ohmori N. J Chem Soc, Perkin Trans 1. 2002:755–767. [Google Scholar]; (b) Nicolaou KC, Vassilikogiannakis G, Mägerlein W, Kranich R. Angew Chem, Int Ed. 2001;40:2482–2486. doi: 10.1002/1521-3773(20010702)40:13<2482::AID-ANIE2482>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]; (c) Herrinton PM, Klotz KL, Hartley WM. J Org Chem. 1993;58:678–682. [Google Scholar]; (d) Burns AC, Forsyth CJ. Org Lett. 2008;10:97–100. doi: 10.1021/ol7024058. [DOI] [PubMed] [Google Scholar]
- 25.McFadden RM, Stoltz BM. J Am Chem Soc. 2006;128:7738–7739. doi: 10.1021/ja061853f. [DOI] [PubMed] [Google Scholar]
- 26.White DE, Stewart IC, Grubbs RH, Stoltz BM. J Am Chem Soc. 2008;130:810–811. doi: 10.1021/ja710294k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Enquist JA, Jr, Stoltz BM. Nature. 2008 doi: 10.1038/nature07046. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Behenna DC, Stockdill JL, Stoltz BM. Angew Chem, Int Ed. 2007;46:4077–4080. doi: 10.1002/anie.200700430. [DOI] [PubMed] [Google Scholar]
- 29.(a) Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]; (b) Stewart IC, Ung T, Pletnev AA, Berlin JM, Grubbs RH, Schrodi Y. Org Lett. 2007;9:1589–1592. doi: 10.1021/ol0705144. [DOI] [PubMed] [Google Scholar]