

Abstract
The title Schiff base compound, C10H10Cl3NO, was prepared by the condensation of 1-(3,5-dichloro-2-hydroxyphenyl)ethanone with chloroethylamine. The imine adopts an E configuration with respect to the C=N bond. The H atom of the phenolic OH group is disordered over two positions with site occupation factors of 0.52 (7) and 0.48 (7), respectively, and the major occupancy component is involved in an intramolecular N—H⋯O hydrogen bond. The compound therefore exists in an iminium–phenolate as well as in the imino–phenol form. In the crystal, molecules are connected by C—H⋯O and C—H⋯Cl hydrogen bonds and Cl⋯Cl interactions [3.7864 (9) Å] into a three-dimensional network. In addition, intermolecular π–π stacking interactions [centroid–centroid distance = 4.4312 (9) Å] are observed.
Related literature
For a related structure, see: Wang et al. (2010 ▶). For applications of Schiff base ligands, see: Yin et al. (2004 ▶); Böhme & Günther (2007 ▶).
Experimental
Crystal data
C10H10Cl3NO
M r = 266.54
Monoclinic,
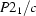
a = 14.5710 (2) Å
b = 10.2323 (2) Å
c = 7.7384 (1) Å
β = 94.376 (1)°
V = 1150.39 (3) Å3
Z = 4
Mo Kα radiation
μ = 0.77 mm−1
T = 296 K
0.38 × 0.19 × 0.07 mm
Data collection
Bruker APEXII CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2005 ▶) T min = 0.762, T max = 0.951
8253 measured reflections
2625 independent reflections
1940 reflections with I > 2σ(I)
R int = 0.023
Refinement
R[F 2 > 2σ(F 2)] = 0.033
wR(F 2) = 0.091
S = 1.02
2625 reflections
146 parameters
2 restraints
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.25 e Å−3
Δρmin = −0.21 e Å−3
Data collection: APEX2 (Bruker, 2005 ▶); cell refinement: SAINT (Bruker, 2005 ▶); data reduction: SAINT; program(s) used to solve structure: SIR97 (Altomare et al., 1999 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: SHELXTL (Sheldrick, 2008 ▶); software used to prepare material for publication: publCIF (Westrip, 2010 ▶).
Supplementary Material
Crystal structure: contains datablocks I, global. DOI: 10.1107/S160053681004448X/im2238sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S160053681004448X/im2238Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1N⋯O1 | 0.88 (2) | 1.68 (2) | 2.479 (2) | 150 (4) |
| C10—H10B⋯O1i | 0.97 | 2.48 | 3.416 (2) | 159 |
| C10—H10A⋯Cl3ii | 0.97 | 2.86 | 3.621 (2) | 136 |
Symmetry codes: (i) 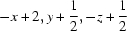 ; (ii)
; (ii) 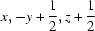 .
.
Acknowledgments
This study was supported by the Natural Science Foundation of Shandong Province (Z2008B10).
supplementary crystallographic information
Comment
Schiff-base ligands have attracted much attention over the years, e.g. as ligands in organotin(IV) compounds owing to their anti-tumour activities (Yin et al., 2004), and in silicon complexes applied to the field of photovoltaic applications, as coloring material and due to their antimicrobial activity. (Böhme & Günther, 2007). We report here the crystal structure of the title Schiff-base ligand (Fig. 1).
The molecular structure of the ligand is represented in Fig. 1. The bond lengths and angles are in eligible range. The C7—N1 and C9—N1 bond lengths of 1.290 (2), 1.460 (2) Å, respectively, conform to the value for a double and single bonds and they are comparable with the corresponding bond lengths in similar Schiff-base compounds (Wang et al., 2010). The hydrogen atom of the phenolic OH group is disordered over two positions with site occupation factors of 0.52 and 0.48, respectively. The compound therefore exists in an iminium-phenolate as well as in an imino-phenol form. The situation may be interpreted as the intramolecular protonation of the basic imine nitrogen by the acidic phenol group. In the crystal, molecules are connected by C—H···O, C—H···Cl (Table 1) and Cl···Cl interactions [Cl1···Cl2 distance = 3.7864 (9) Å for symmetry operation -x + 1, -y + 1, -z; Cl2···Cl3 distance = 3.7709 (7) Å for symmetry operation -x + 2, y - 1/2, -z + 1/2; Cl2···Cl3 distance = 3.7789 (8) Å for symmetry operation -x + 2, -y + 1, -z] into a network (Fig. 2). In addition, weak intermolecular π–π interactions serve to stabilize the extended structure [Cg···Cg distance = 4.4312 (9) Å for symmetry operation x, -y + 1/2, z - 1/2 and x, -y + 1/2, z + 1/2 (The Cg is the centroid of the phenyl ring)].
Experimental
To a mixture of 1-(3,5-dichloro-2-hydroxy-phenyl)-ethanone (5.1 g, 25 mmol) and chloroethylamine hydrochloride (5.8 g, 50 mmol) in ethanol (150 ml) was added triethylamine (5.1 g, 50 mmol). The mixture was heated to reflux for 10 min. After being cooled to room temperature, the resulting precipitate was filtrated and washed with water to afford the product, E-2,4-dichloro-6-(1-(2-chloroethylimino)ethyl)phenol in 84% yield. Crystals of the title compound suitable for X-ray diffraction were obtained by slow evaporation of a solution of the solid in ethyl acetate at room temperature for 7 d.
Refinement
All H atoms at carbon were placed geometrically and refined using a riding model with C—H = 0.97 Å (for CH2 group), 0.96 Å (for CH3 group) and 0.93 Å (for aryl H atoms). The isotropic atomic displacement parameters of hydrogen atoms were set to 1.5 × Ueq (CH3) and 1.2 × Ueq (CH2, CarH) of the parent atoms. Positions of hydrogen atoms at N1 and O1 were taken from difference Fourier maps and were refined using PART instructions.
Figures
Fig. 1.

Molecular structure of the title compound, showing the atom-numbering scheme. Displacement ellipsoids are drawn at the 30% probability level.
Fig. 2.

Crystal packing of title compound viewed along the b axis. C—H···O and C—H···Cl hydrogen bonds are displayed as red and blue dashed lines, respectively. Cl···Cl interactions are shown as green dashed lines.
Fig. 3.

Crystal packing of title compound viewed along the c axis. The π–π interactions are shown as black dashed lines.
Crystal data
| C10H10Cl3NO | F(000) = 544 |
| Mr = 266.54 | Dx = 1.539 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 2504 reflections |
| a = 14.5710 (2) Å | θ = 2.4–25.9° |
| b = 10.2323 (2) Å | µ = 0.77 mm−1 |
| c = 7.7384 (1) Å | T = 296 K |
| β = 94.376 (1)° | Plate, orange |
| V = 1150.39 (3) Å3 | 0.38 × 0.19 × 0.07 mm |
| Z = 4 |
Data collection
| Bruker APEXII CCD area-detector diffractometer | 2625 independent reflections |
| Radiation source: fine-focus sealed tube | 1940 reflections with I > 2σ(I) |
| graphite | Rint = 0.023 |
| φ and ω scans | θmax = 27.5°, θmin = 2.4° |
| Absorption correction: multi-scan (SADABS; Bruker, 2005) | h = −18→17 |
| Tmin = 0.762, Tmax = 0.951 | k = −10→13 |
| 8253 measured reflections | l = −8→10 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.033 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.091 | H atoms treated by a mixture of independent and constrained refinement |
| S = 1.01 | w = 1/[σ2(Fo2) + (0.0452P)2 + 0.1559P] where P = (Fo2 + 2Fc2)/3 |
| 2625 reflections | (Δ/σ)max = 0.006 |
| 146 parameters | Δρmax = 0.25 e Å−3 |
| 2 restraints | Δρmin = −0.21 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | Occ. (<1) | |
| C1 | 0.57736 (13) | 0.15817 (19) | −0.1505 (2) | 0.0498 (4) | |
| C2 | 0.61146 (13) | 0.0340 (2) | −0.1143 (2) | 0.0516 (5) | |
| H2 | 0.5771 | −0.0395 | −0.1481 | 0.062* | |
| C3 | 0.69659 (12) | 0.02119 (17) | −0.0279 (2) | 0.0455 (4) | |
| C4 | 0.75065 (12) | 0.12988 (16) | 0.0289 (2) | 0.0401 (4) | |
| C5 | 0.71381 (12) | 0.25627 (16) | −0.0117 (2) | 0.0388 (4) | |
| C6 | 0.62665 (12) | 0.26726 (18) | −0.1027 (2) | 0.0458 (4) | |
| H6 | 0.6026 | 0.3495 | −0.1303 | 0.055* | |
| C7 | 0.76729 (12) | 0.37256 (16) | 0.0405 (2) | 0.0403 (4) | |
| C8 | 0.73432 (14) | 0.50736 (18) | −0.0072 (3) | 0.0559 (5) | |
| H8A | 0.6689 | 0.5122 | 0.0002 | 0.084* | |
| H8B | 0.7487 | 0.5268 | −0.1235 | 0.084* | |
| H8C | 0.7642 | 0.5696 | 0.0710 | 0.084* | |
| C9 | 0.90715 (13) | 0.45820 (17) | 0.1910 (2) | 0.0491 (4) | |
| H9A | 0.8749 | 0.5189 | 0.2611 | 0.059* | |
| H9B | 0.9276 | 0.5057 | 0.0925 | 0.059* | |
| C10 | 0.98893 (13) | 0.40380 (19) | 0.2962 (2) | 0.0509 (5) | |
| H10A | 0.9682 | 0.3582 | 0.3961 | 0.061* | |
| H10B | 1.0284 | 0.4752 | 0.3382 | 0.061* | |
| Cl1 | 0.46922 (4) | 0.17442 (7) | −0.26165 (8) | 0.0791 (2) | |
| Cl2 | 0.74236 (4) | −0.13318 (4) | 0.01161 (8) | 0.06643 (18) | |
| Cl3 | 1.05345 (3) | 0.29415 (5) | 0.17400 (7) | 0.05817 (16) | |
| N1 | 0.84464 (10) | 0.35378 (14) | 0.12961 (19) | 0.0418 (3) | |
| H1N | 0.858 (3) | 0.271 (2) | 0.146 (5) | 0.049 (16)* | 0.52 (7) |
| H1O | 0.855 (3) | 0.183 (3) | 0.145 (6) | 0.07 (2)* | 0.48 (7) |
| O1 | 0.83069 (9) | 0.11256 (12) | 0.11379 (18) | 0.0494 (3) |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| C1 | 0.0381 (10) | 0.0583 (11) | 0.0523 (10) | −0.0039 (9) | −0.0005 (8) | 0.0049 (9) |
| C2 | 0.0472 (11) | 0.0491 (10) | 0.0583 (11) | −0.0119 (9) | 0.0041 (9) | −0.0039 (9) |
| C3 | 0.0451 (10) | 0.0370 (9) | 0.0551 (10) | −0.0010 (8) | 0.0077 (8) | −0.0016 (7) |
| C4 | 0.0392 (10) | 0.0368 (8) | 0.0450 (9) | −0.0005 (7) | 0.0069 (7) | −0.0006 (7) |
| C5 | 0.0387 (9) | 0.0364 (8) | 0.0417 (9) | −0.0005 (7) | 0.0051 (7) | 0.0023 (7) |
| C6 | 0.0421 (10) | 0.0449 (10) | 0.0506 (10) | 0.0042 (8) | 0.0041 (8) | 0.0077 (8) |
| C7 | 0.0428 (10) | 0.0360 (8) | 0.0428 (9) | 0.0027 (7) | 0.0081 (7) | 0.0028 (7) |
| C8 | 0.0577 (12) | 0.0382 (9) | 0.0709 (12) | 0.0041 (9) | −0.0014 (10) | 0.0076 (9) |
| C9 | 0.0515 (11) | 0.0348 (8) | 0.0610 (11) | −0.0068 (8) | 0.0040 (9) | −0.0030 (8) |
| C10 | 0.0561 (12) | 0.0511 (10) | 0.0450 (9) | −0.0121 (9) | 0.0002 (8) | −0.0071 (8) |
| Cl1 | 0.0484 (3) | 0.0859 (4) | 0.0987 (4) | −0.0121 (3) | −0.0218 (3) | 0.0179 (3) |
| Cl2 | 0.0629 (4) | 0.0339 (2) | 0.1020 (4) | 0.0003 (2) | 0.0030 (3) | −0.0027 (2) |
| Cl3 | 0.0544 (3) | 0.0519 (3) | 0.0673 (3) | 0.0025 (2) | −0.0016 (2) | −0.0059 (2) |
| N1 | 0.0448 (9) | 0.0310 (7) | 0.0493 (8) | −0.0020 (6) | 0.0019 (7) | −0.0003 (6) |
| O1 | 0.0408 (7) | 0.0350 (7) | 0.0710 (8) | 0.0018 (6) | −0.0055 (6) | 0.0007 (6) |
Geometric parameters (Å, °)
| C1—C6 | 1.363 (3) | C7—C8 | 1.498 (2) |
| C1—C2 | 1.385 (3) | C8—H8A | 0.9600 |
| C1—Cl1 | 1.7446 (19) | C8—H8B | 0.9600 |
| C2—C3 | 1.370 (2) | C8—H8C | 0.9600 |
| C2—H2 | 0.9300 | C9—N1 | 1.460 (2) |
| C3—C4 | 1.413 (2) | C9—C10 | 1.498 (3) |
| C3—Cl2 | 1.7326 (18) | C9—H9A | 0.9700 |
| C4—O1 | 1.306 (2) | C9—H9B | 0.9700 |
| C4—C5 | 1.426 (2) | C10—Cl3 | 1.781 (2) |
| C5—C6 | 1.409 (2) | C10—H10A | 0.9700 |
| C5—C7 | 1.462 (2) | C10—H10B | 0.9700 |
| C6—H6 | 0.9300 | N1—H1N | 0.874 (19) |
| C7—N1 | 1.290 (2) | O1—H1O | 0.827 (19) |
| C6—C1—C2 | 121.53 (17) | C7—C8—H8B | 109.5 |
| C6—C1—Cl1 | 119.55 (15) | H8A—C8—H8B | 109.5 |
| C2—C1—Cl1 | 118.92 (15) | C7—C8—H8C | 109.5 |
| C3—C2—C1 | 118.94 (17) | H8A—C8—H8C | 109.5 |
| C3—C2—H2 | 120.5 | H8B—C8—H8C | 109.5 |
| C1—C2—H2 | 120.5 | N1—C9—C10 | 110.83 (15) |
| C2—C3—C4 | 122.61 (16) | N1—C9—H9A | 109.5 |
| C2—C3—Cl2 | 119.69 (14) | C10—C9—H9A | 109.5 |
| C4—C3—Cl2 | 117.69 (13) | N1—C9—H9B | 109.5 |
| O1—C4—C3 | 120.31 (15) | C10—C9—H9B | 109.5 |
| O1—C4—C5 | 122.71 (15) | H9A—C9—H9B | 108.1 |
| C3—C4—C5 | 116.98 (15) | C9—C10—Cl3 | 112.06 (12) |
| C6—C5—C4 | 119.49 (16) | C9—C10—H10A | 109.2 |
| C6—C5—C7 | 120.94 (15) | Cl3—C10—H10A | 109.2 |
| C4—C5—C7 | 119.56 (15) | C9—C10—H10B | 109.2 |
| C1—C6—C5 | 120.43 (17) | Cl3—C10—H10B | 109.2 |
| C1—C6—H6 | 119.8 | H10A—C10—H10B | 107.9 |
| C5—C6—H6 | 119.8 | C7—N1—C9 | 124.26 (15) |
| N1—C7—C5 | 116.87 (14) | C7—N1—H1N | 113 (3) |
| N1—C7—C8 | 121.32 (16) | C9—N1—H1N | 122 (3) |
| C5—C7—C8 | 121.82 (16) | C4—O1—H1O | 112 (4) |
| C7—C8—H8A | 109.5 | ||
| C6—C1—C2—C3 | 0.2 (3) | C2—C1—C6—C5 | −1.1 (3) |
| Cl1—C1—C2—C3 | 179.64 (14) | Cl1—C1—C6—C5 | 179.53 (13) |
| C1—C2—C3—C4 | 1.1 (3) | C4—C5—C6—C1 | 0.5 (3) |
| C1—C2—C3—Cl2 | −177.60 (14) | C7—C5—C6—C1 | −179.91 (16) |
| C2—C3—C4—O1 | 178.66 (16) | C6—C5—C7—N1 | 177.17 (15) |
| Cl2—C3—C4—O1 | −2.6 (2) | C4—C5—C7—N1 | −3.3 (2) |
| C2—C3—C4—C5 | −1.6 (3) | C6—C5—C7—C8 | −3.0 (3) |
| Cl2—C3—C4—C5 | 177.17 (12) | C4—C5—C7—C8 | 176.56 (16) |
| O1—C4—C5—C6 | −179.54 (15) | N1—C9—C10—Cl3 | 60.56 (18) |
| C3—C4—C5—C6 | 0.7 (2) | C5—C7—N1—C9 | 179.36 (15) |
| O1—C4—C5—C7 | 0.9 (3) | C8—C7—N1—C9 | −0.5 (3) |
| C3—C4—C5—C7 | −178.81 (15) | C10—C9—N1—C7 | 177.78 (16) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1N···O1 | 0.88 (2) | 1.68 (2) | 2.479 (2) | 150 (4) |
| C10—H10B···O1i | 0.97 | 2.48 | 3.416 (2) | 159 |
| C10—H10A···Cl3ii | 0.97 | 2.86 | 3.621 (2) | 136 |
Symmetry codes: (i) −x+2, y+1/2, −z+1/2; (ii) x, −y+1/2, z+1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: IM2238).
References
- Altomare, A., Burla, M. C., Camalli, M., Cascarano, G. L., Giacovazzo, C., Guagliardi, A., Moliterni, A. G. G., Polidori, G. & Spagna, R. (1999). J. Appl. Cryst.32, 115–119.
- Böhme, U. & Günther, B. (2007). Inorg. Chem. Commun.10, 482-484.
- Bruker (2005). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Wang, C.-H., Liu, Y.-C., Lin, C.-H. & Ko, B.-T. (2010). Acta Cryst. E66, o745. [DOI] [PMC free article] [PubMed]
- Westrip, S. P. (2010). J. Appl. Cryst.43, 920–925.
- Yin, H. D., Wang, Q. B. & Xue, S. C. (2004). J. Organomet. Chem.689, 2480–2485.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks I, global. DOI: 10.1107/S160053681004448X/im2238sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S160053681004448X/im2238Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report