

Abstract
The gas phase and aqueous thermochemistry and reactivity of nitroxyl (nitrosyl hydride, HNO) were elucidated with multiconfigurational self-consistent field and hybrid density functional theory calculations and continuum solvation methods. The pKa of HNO is predicted to be 7.2 ± 1.0, considerably different from the value of 4.7 reported from pulse radiolysis experiments. The ground-state triplet nature of NO− affects the rates of acid-base chemistry of the HNO/NO− couple. HNO is highly reactive toward dimerization and addition of soft nucleophiles but is predicted to undergo negligible hydration (Keq = 6.9 × 10−5). HNO is predicted to exist as a discrete species in solution and is a viable participant in the chemical biology of nitric oxide and derivatives.
The discoveries of
nitric oxide (NO) biosynthesis in mammalian cells and the diverse
biological activity associated with NO and NO-derived species (1) have
brought intense interest in the physiological chemistry of nitrogen
oxides. The chemistry of NO and its biologically accessible
oxidized congeners nitrogen dioxide (NO2),
nitrite (NO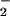 ), peroxynitrite
(ONOO−), dinitrogen trioxide
(N2O3), and nitrate
(NO
), peroxynitrite
(ONOO−), dinitrogen trioxide
(N2O3), and nitrate
(NO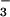 ) is fairly well established (2). By contrast,
the reduced congeners such as nitroxyl (HNO) and its conjugate base
(NO−) are less well understood, and consequently
their role in biology is not clear. The importance of HNO or
NO− in biology has often been neglected or
dismissed, in part because NO metabolism is thought to be primarily
oxidative in nature (3), and because HNO is thought to be only
metastable (3), a strong acid (4), and to dimerize readily (5).
NO− is known to react rapidly and irreversibly
with NO (6), making the examination of NO− in
the presence of NO difficult. Additionally, HNO might be expected to be
electrophilic, and hydration under physiological conditions would serve
to attenuate its aqueous reactivity.
) is fairly well established (2). By contrast,
the reduced congeners such as nitroxyl (HNO) and its conjugate base
(NO−) are less well understood, and consequently
their role in biology is not clear. The importance of HNO or
NO− in biology has often been neglected or
dismissed, in part because NO metabolism is thought to be primarily
oxidative in nature (3), and because HNO is thought to be only
metastable (3), a strong acid (4), and to dimerize readily (5).
NO− is known to react rapidly and irreversibly
with NO (6), making the examination of NO− in
the presence of NO difficult. Additionally, HNO might be expected to be
electrophilic, and hydration under physiological conditions would serve
to attenuate its aqueous reactivity.
Nitroxyl (HNO), or its conjugate base, NO−, is known to be formed under physiological conditions; for example, oxidation of N-hydroxy-l-arginine (an intermediate in NO biosynthesis) (7), reaction of S-nitrosothiols with thiols (8, 9), nitric oxide synthase (10–12), and even direct reduction of NO by mitochondrial cytochrome c (13), may all generate HNO. Nitroxyl has been generated via the interaction of NO with manganese superoxide dismutase (14) and with ubiquinol (15). HNO has biological activity; it can act as a potent cytotoxic agent that causes double-stranded breaks in DNA, depletion of cellular glutathione (16), as well as elicitation of smooth muscle relaxation (17). HNO has been found to be a potent inhibitor of thiol-containing enzymes (18, 19) and attenuates the activity of the NMDA receptor via thiol modification, thus providing neuroprotection (20).
Much of the fundamental biological chemistry associated with HNO is
unknown, aside from the rate constant for dimerization (2–8 ×
109 M−1
s−1) (5). The pKa of HNO
has been reported to be 4.7, as determined by pulse radiolysis studies
(4), indicating that NO− will be the
near-exclusive species present at physiological pH. Subsequent studies
by Seddon et al. demonstrated the temperature and pH
dependence of decay of both NO− and higher-order
adducts N2O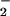 and
N3O
and
N3O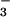 (6).
NO− is a typical nucleophile, yet several
studies find that HNO generated at physiological pH reacts readily
as an electrophile, particularly with thiols, to yield
N-hydroxysulfenamide intermediates (9, 21), implying a
higher pKa.
(6).
NO− is a typical nucleophile, yet several
studies find that HNO generated at physiological pH reacts readily
as an electrophile, particularly with thiols, to yield
N-hydroxysulfenamide intermediates (9, 21), implying a
higher pKa.
NO− and NO react rapidly and irreversibly to
form N2O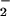 , a reactive radical
anion (6). Nevertheless, at likely physiological concentrations of NO
(<1 μM) (22), the rate of disappearance of
NO− and NO via bimolecular reaction will be slow
compared with that of other biological processes.
, a reactive radical
anion (6). Nevertheless, at likely physiological concentrations of NO
(<1 μM) (22), the rate of disappearance of
NO− and NO via bimolecular reaction will be slow
compared with that of other biological processes.
The generation of HNO has been observed via the decomposition of Piloty's acid (benzenesulfohydroxamic acid) at pH 8 (23). However, the pKa value of 4.7 for HNO would preclude the existence of appreciable concentrations of HNO under these conditions. The unfavorable free energy of protonation at neutral pH (3.2 kcal/mol) would contribute to low reactivity.
We have used quantum mechanical calculations that predict the fundamental chemical properties of HNO in solution and herein report computational results that establish the aqueous thermochemistry of nitroxyl and its reactivity toward species present under physiological conditions. The significant concentration of HNO now predicted at physiological pH, relative to NO−, and the high reactivity toward thiols open new possibilities for the involvement of nitroxyl in biological mechanisms.
Energetics of Singlet and Triplet HNO and NO−
The lowest triplet (3A′′) excited state of HNO has been determined spectroscopically to lie 18 kcal/mol above the singlet (1A′) ground state (24). QCISD(T) and MP2 calculations reported by Brauman et al. (25, 26) agree well (Table 1).
Table 1.
Calculated and experimental singlet-triplet gaps for NO−, O2, and HNO
It is not generally known whether the isomeric HON species is biologically accessible. Spectroscopic and theoretical studies (27–31) and our own calculations place the energy of this triplet ground-state species at 20–23 kcal/mol above the HNO singlet ground state. Therefore, HON is unlikely to be a participant in the physiological chemistry of HNO.
NO− Is a Ground-State Triplet, Isoelectronic with Dioxygen
Experimental measurements place the singlet state of NO− at ≈17 kcal/mol above the ground state (32, 33). We have optimized the singlet and triplet states of NO− with complete active space self-consistent field calculations (34). An (8e, 6o) active space was used, corresponding to full configuration interaction in the 2p valence space. The singlet-triplet (S-T) energy gap, calculated with inclusion of MP2 correction to the CASSCF energy (CASMP2) (35), is provided below in Table 1 and compared with values computed for O2 at the same level of theory. The S-T gap of NO− is predicted to be 21 kcal/mol, suggesting a value slightly higher than the experimental estimates. The calculated value for the isoelectronic O2, 23 kcal/mol, by using this same method, is in excellent agreement with experiment (36).
The pKa of HNO
Is 3NO− protonated at pH 7? Geometry optimizations and harmonic frequency analyses of a series of organic and inorganic acids and their conjugate bases were performed with hybrid density functional theory (B3LYP/6–311+G*) (37). Aqueous solvation energies were determined by using the Polarizable Continuum Model (PCM) of Tomasi and coworkers (38, 39). These values are given in Table 2. The gas-phase deprotonation energies plus aqueous solvation energy differences between acid and conjugate base show a good linear correlation with experimental pKa values (40, 41). The relationship pKa = 0.549 (PAcalc, PCM) − 139.8 was obtained, with a correlation coefficient of 0.95 and standard deviation of 1.0 pKa units. Fig. 1 shows a plot of the predicted versus experimental pKa values.
Table 2.
Calculated gas-phase deprotonation energies (PA), solvation energies, and experimental and calculated pKa values
| Acid | pKa (exp)* | PA (calc, gas),† kcal/mol | ΔΔGsolvation,‡ kcal/mol | pKa, (calc)§ |
|---|---|---|---|---|
| Trifluoroacetic acid | 0.2 | 314.6 | 60.9 | −0.5 |
| Trichloroacetic acid | 0.7 | 312.7 | 59.8 | −1.0 |
| Nitrous acid | 3.3 | 331.5 | 75.8 | 0.6 |
| Dinitromethane | 3.6 | 314.9 | 53.4 | 3.8 |
| Formic acid | 3.8 | 335.6 | 74.8 | 3.4 |
| Benzoic acid | 4.2 | 333.8 | 67.9 | 6.2 |
| Acrylic acid | 4.3 | 337 | 72.2 | 5.6 |
| Hydrazoic acid | 4.6 | 336.2 | 72.6 | 4.9 |
| Acetic acid | 4.8 | 340.6 | 74.3 | 6.4 |
| Dimethyl malonate | 9 | 336.1 | 58.5 | 12.6 |
| Hydrogen cyanide | 9.2 | 346.4 | 79.6 | 6.7 |
| Phenol | 10.0 | 341.5 | 70.6 | 8.9 |
| Nitromethane | 10.2 | 352.2 | 72.1 | 14.0 |
| Methanethiol | 10.3 | 352 | 75.2 | 12.2 |
| Ethanethiol | 10.6 | 350.4 | 71.0 | 13.6 |
| Dicyanomethane | 11.2 | 328 | 55.2 | 10.0 |
| Trichloroethanol | 12.2 | 345.7 | 72.9 | 10.0 |
| Trifluoroethanol | 12.4 | 352 | 71.2 | 14.4 |
| Propargyl alcohol | 13.6 | 362.9 | 84.1 | 13.4 |
| Allyl alcohol | 15.5 | 365.8 | 84.1 | 14.9 |
| Methanol | 15.5 | 373.9 | 92.3 | 14.8 |
| Water | 15.7 | 384.7 | 102.8 | 15.0 |
| Ethanol | 15.9 | 371.0 | 88.6 | 15.2 |
| Isopropanol | 17.1 | 369.2 | 85.6 | 15.9 |
| t-Butanol | 18 | 368.5 | 83.2 | 16.8 |
| Acetone | 19.3 | 366.8 | 73.1 | 21.5 |
| Acetonitrile | 25 | 370.2 | 70.1 | 25.0 |
| Methyl acetate | 25.4 | 370.5 | 66.8 | 27.0 |
| Dimethyl sulfone | 33 | 365.7 | 56.5 | 30.0 |
| Dimethyl sulfoxide | 33.5 | 374.6 | 63.2 | 31.2 |
| HNO | 349.6 | 81.9 | 7.2 |
Figure 1.

Plot of experimental versus calculated pKa. Data are listed in Table 2.
By using this correlation, a calculated pKa
of 7.2 ± 1.0 was determined for the HNO +
H2O/H3O+
+ 3NO− equilibrium,
indicating that about 50% of HNO should exist at equilibrium at
physiological pH. This value is substantially higher than the pulse
radiolytic estimate of 4.7, and more consistent with recent
experimental observations of Wong et al. and of Shoeman
et al., which suggest a substantial concentration of the
nondeprotonated form (9, 19). In the original study by Grätzel
et al. (4), difficulty in obtaining an accurate
NO− concentration measurement directly after the
radiolysis pulse was noted, perhaps because of the generation of both
singlet and triplet nitroxyl, and complicating equilibria between
NO− and its NO adducts,
N2O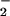 and
N3O
and
N3O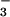 , under the high relative
concentrations of NO used in the experimental conditions.
, under the high relative
concentrations of NO used in the experimental conditions.
Singlet nitroxyl anion (1NO−) is predicted to possess an enormous pKa value of 18.6 (obtained from the CASMP2 singlet-triplet gap of NO− and the corresponding solvation energies). However, given the electronic similarity of NO− and O2, and short lifetime of singlet oxygen in aqueous solution (≈1 × 10−6 s) (36), any NO− present in equilibrium with HNO in aqueous solution should exist in its triplet state. Proton transfer in this system should be slowed substantially relative to typical proton transfers involving conservation of spin. It has been shown by Brauman and coworkers for 3NO− in the gas phase (25, 26) that the process of proton transfer involving spin state interconversion is slowed by as much as 107, as compared with spin-conserving proton transfers.
Donald et al. originally suggested that the triplet forms of HNO should possess enhanced acidity versus the singlet state (42). We predict that the excited triplet state of HNO is an extremely potent acid in aqueous solution, with a pKa of about −2 based on the spectroscopically determined singlet-triplet gap of HNO.
The Hydration Keq of HNO
Because about 50% of HNO is undissociated at physiological pH, the nitroso group might be expected to hydrate, as do many aldehydes (43). We have compared the hydration of HNO with that of aldehydes and ketones. To calibrate our calculations and obtain a quantitative value for the hydration Keq of HNO, the geometries of a series of carbonyl compounds and their respective hydrates were optimized at the B3LYP/6–311 + G* level. Solvation calculations using the PCM formalism were used to model aqueous solvation. Table 3 lists computed hydration energies and experimental values of Keq. A relation of log Keq = −0.56 (ΔErxn, PCM) + 0.32 was obtained, with a correlation coefficient of 0.9 and standard deviation in log Keq of 0.73. A plot of experimental versus calculated hydration equilibria is provided in Fig. 2.
Table 3.
Calculated gas- and solution-phase energetics of hydrate formation [RCHO + H2O → RCH(OH)2] and calculated experimental hydration equilibrium constants
| Compound | Keq (exp)* | ΔErxn, gas phase, kcal/mol† | ΔErxn, PCM, kcal/mol‡ | Keq (calc)§ |
|---|---|---|---|---|
| C6H5COCH3 | 9.3 × 10−6 | +2.3 | +8.5 | 3.6 × 10−5 |
| (CH3)2CO | 1.4 × 10−3 | −0.4 | +4.4 | 7.2 × 10−3 |
| C6H5CHO | 8 × 10−3 | +1.1 | +5.2 | 2.6 × 10−3 |
| (CH3)3CCHO | 2.3 × 10−1 | −2.0 | +1.2 | 4.4 × 10−1 |
| CH3CHO | 1.06 | −3.4 | −0.3 | 3.08 |
| CCl3CHO | 3 × 103 | −5.3 | −1.3 | 1.1 × 102 |
| H2CO | 2.3 × 103 | −7.5 | −5.2 | 1.7 × 103 |
| CF3CHO | 2.9 × 104 | −9.6 | −9.2 | 3.0 × 105 |
| HNO | +7.6 | +8.0 | 6.9 × 10−5 |
Figure 2.

Plot of calculated versus experimental hydration equilibria for aldehydes and ketones in Table 3. PCM-B3LYP/6–311+G*+ zero point energy.
From this correlation, the hydration of HNO is predicted to be highly unfavorable, with a Keq value of 6.9 × 10−5. HNO resembles acetophenone in its reluctance to hydrate, in stark contrast to the analogous parent carbonyl species, formaldehyde (Keq = 2.3 × 103). Essentially all HNO exists in solution as such and does not hydrate to any significant extent. Repulsion between lone pairs on nitrogen and the two adjacent oxygens in the hydrate gives rise to the high energy of this hydrated species.
Dimerization and Reactivity Toward Nucleophiles
The thermodynamics of HNO dimerization and reactions with methanethiol, methylamine, and methanol were predicted at the B3LYP/6–311+G* level of theory. The results are shown in Fig. 3. HNO is predicted to be relatively inert to addition by oxygen-based nucleophiles, but reactions with amines and thiols are highly favorable in either the gas phase or solution. The latter is especially important, because nucleophilic addition to HNO is proposed to occur in the degradation of S-nitrosothiols in the presence of added thiol, to yield the corresponding sulfinamide and ultimately, NH3 (9). The mechanism of aldehyde dehydrogenase inhibition is also thought to involve attack of the active-site sulfhydryl group at nitrogen of HNO (18, 19).
Figure 3.

Energies of reaction of HNO with nucleophiles and for dimerization (kcal/mol), in the gas phase [B3LYP/6–311+G*+ zero point energy (ZPE)] and in solution (PCM-B3LYP/6–311+G*//B3LYP/6–311+G*+ZPE).
At high local concentrations of HNO, dimerization will provide a competing pathway for the HNO degradation, analogous to the dimerization of aliphatic C-nitroso compounds (47). HNO dimerization is rapid (k2 = 2–8 × 109 M−1 s−1) (5) and strongly thermodynamically favored, predicted to occur with an energy of reaction of −37 kcal/mol in the gas phase and −40 kcal/mol in aqueous solution (Fig. 3). This irreversible process leads ultimately to a molecule of N2O and water, the mechanism of which has been studied in detail by ab initio and molecular dynamics methods (48, 49). Our prediction of a pKa of about 7 means that in neutral and slightly acidic aqueous solutions, both HNO and NO− will be present. The reaction between HNO and 3NO− is predicted to be highly thermodynamically favorable (ΔErxn = −40 kcal/mol; Fig. 3). Once again, however, the rate of this spin-forbidden process will be slowed, analogous to the proton transfer reactions discussed above.
Conclusions
HNO is predicted to be stable in aqueous solution and only a weak acid. HNO reacts exothermically with soft nucleophiles such as amines and thiols but is relatively inert to oxygen-based nucleophiles. HNO is a highly reactive but selective electrophile, whereas NO is essentially inert as an electrophile. HNO joins NO and its oxidized congeners as a vital player on the biological stage. In light of the new pKa values, we now provide for singlet and triplet NO−, the redox potentials of these species must now be reassessed. The reduction potentials of −0.35 V and +0.39 V for the NO/1NO− and NO/3NO− couples, as estimated by Stanbury (50), used the pulse radiolysis data of Grätzel et al. (4) and assumed this pKa value corresponded to singlet NO−. These reduction potentials should be reevaluated, and further details of the chemical biology of HNO and its derivatives warrant additional investigation.
Acknowledgments
We thank Professor Dale Margerum and Brent J. Giles for helpful discussions. The support of the National Institute of General Medical Sciences, National Institutes of Health (K.N.H.), and the National Research Service Award, National Institutes of Health (M.D.B.), is gratefully acknowledged.
Abbreviation
- PCM
Polarizable Continuum Model
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Kerwin J F, Jr, Lancaster J R, Feldman P L. J Med Chem. 1995;38:4342–4362. doi: 10.1021/jm00022a001. [DOI] [PubMed] [Google Scholar]
- 2.Bonner F T, Stedman G. In: Methods in Nitric Oxide Research. Freelisch M, Stamler J S, editors. Chichester, U.K.: Wiley; 1996. pp. 3–18. [Google Scholar]
- 3.Fukuto J M, Cho J Y, Switzer C H. In: Nitric Oxide Biology and Pathobiology. Ignarro L J, editor. San Diego: Academic; 2000. pp. 23–40. [Google Scholar]
- 4.Grätzel M, Taniguchi S, Henglein A. Ber Bunsenges Phys Chem. 1970;74:1003–1010. [Google Scholar]
- 5.Bazylinski D A, Hollocher T C. Inorg Chem. 1985;24:4285–4288. [Google Scholar]
- 6.Seddon W A, Fletcher J W, Sopchysyn F C. Can J Chem. 1973;51:1123–1130. [Google Scholar]
- 7.Fukuto J M, Stuehr D J, Feldman P L, Bova M P, Wong P. J Med Chem. 1993;36:2666–2670. doi: 10.1021/jm00070a010. [DOI] [PubMed] [Google Scholar]
- 8.Arnelle D R, Stamler J S. Arch Biochem Biophys. 1995;318:279–285. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- 9.Wong P S-Y, Hyun J, Fukuto J M, Shirota F N, DeMaster E G, Shoeman D W, Nagasawa H T. Biochemistry. 1998;37:5362–5371. doi: 10.1021/bi973153g. [DOI] [PubMed] [Google Scholar]
- 10.Hobbs A J, Fukuto J M, Ignarro L J. Proc Natl Acad Sci USA. 1994;91:10992–10996. doi: 10.1073/pnas.91.23.10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schmidt H W, Hofmann H, Schindler U, Shutenko Z S, Cunningham D D, Feelisch M. Proc Natl Acad Sci USA. 1996;93:14492–14497. doi: 10.1073/pnas.93.25.14492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pufahl R A, Wishnok J S, Marletta M A. Biochemistry. 1995;34:1930–1941. doi: 10.1021/bi00006a014. [DOI] [PubMed] [Google Scholar]
- 13.Sharpe M A, Cooper C E. Biochem J. 1998;332:9–19. doi: 10.1042/bj3320009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Niketic V, Stojanovic S, Nikolic A, Spasic M, Michelson A M. Free Rad Biol Med. 1999;27:992–996. doi: 10.1016/s0891-5849(98)00256-1. [DOI] [PubMed] [Google Scholar]
- 15.Poderoso J J, Carreras M C, Schöpfer F, Lisdero C L, Riobo N A, Giulivi C, Boveris A D, Boveris A, Cadenas E. Free Rad Biol Med. 1999;26:925–935. doi: 10.1016/s0891-5849(98)00277-9. [DOI] [PubMed] [Google Scholar]
- 16.Wink D A, Feelisch M, Fukuto J, Chistodoulou D, Jourd'heuil D, Grisham M B, Vodovotz Y, Cook J A, Krishna M, DeGraff W G, et al. Arch Biochem Biophys. 1998;351:66–74. doi: 10.1006/abbi.1997.0565. [DOI] [PubMed] [Google Scholar]
- 17.Fukuto J M, Chiang K, Hszieh R, Wong P, Chaudhuri G. J Pharmacol Exp Ther. 1992;263:546–551. [PubMed] [Google Scholar]
- 18.Demaster E G, Redfern B, Quast B J, Dahlseid T, Nagasawa H T. Alcohol. 1997;14:181–189. doi: 10.1016/s0741-8329(96)00142-5. [DOI] [PubMed] [Google Scholar]
- 19.Shoeman D W, Shirota F N, DeMaster E G, Nagasawa H T. Alcohol. 2000;20:55–59. doi: 10.1016/s0741-8329(99)00056-7. [DOI] [PubMed] [Google Scholar]
- 20.Kim W K, Choi Y B, Rayudu P V, Das P, Asaad W, Arnelle D R, Stamler J S, Lipton S A. Neuron. 1999;24:461–469. doi: 10.1016/s0896-6273(00)80859-4. [DOI] [PubMed] [Google Scholar]
- 21.Doyle M P, Mahapatro S N, Broene R D, Guy J K. J Am Chem Soc. 1988;110:593–599. [Google Scholar]
- 22.Malinski T, Taha Z. Nature (London) 1992;358:676–678. doi: 10.1038/358676a0. [DOI] [PubMed] [Google Scholar]
- 23.Bonner F T, Ko Y. Inorg Chem. 1992;31:2514–2519. [Google Scholar]
- 24.Ellis H B, Jr, Ellison G B. J Chem Phys. 1983;78:6541–6558. [Google Scholar]
- 25.Janaway G A, Zhong M, Gatev G G, Chabinyc M L, Brauman J I. J Am Chem Soc. 1997;119:11697–11698. [Google Scholar]
- 26.Janaway G A, Brauman J I. J Phys Chem A. 2000;104:1795–1797. [Google Scholar]
- 27.Maier G, Reisenauer P, DeMarco M. Angew Chem Int Ed Engl. 1999;38:108–110. [Google Scholar]
- 28.Gallup G A. Inorg Chem. 1975;14:563–565. [Google Scholar]
- 29.Bruna P J. Chem Phys. 1980;49:39–52. [Google Scholar]
- 30.Bruna P J, Marian C M. Chem Phys Lett. 1979;67:109–114. [Google Scholar]
- 31.Wu A A, Peyerimhoff S D, Buenker R J. Chem Phys Lett. 1975;35:316–322. [Google Scholar]
- 32.Tennyson J, Noble C J. J Phys B. 1986;19:4025–4033. [Google Scholar]
- 33.Szmytkowski C, Maciag K. J Phys B. 1991;24:4273–4279. [Google Scholar]
- 34.Frisch M J, Trucks G W, Schlegel H B, Gill P M W, Johnson B G, Robb M A, Cheeseman J R, Keith T, Petersson G A, Montgomery J A, et al. gaussian 94. Pittsburgh, PA: Gaussian; 1995. [Google Scholar]
- 35.McDouall J J, Peasley K, Robb M A. Chem Phys Lett. 1988;148:183–189. [Google Scholar]
- 36.Wilkinson F, Helman W P, Ross A B. J Phys Chem Ref Data. 1995;24:663–1021. [Google Scholar]
- 37.Becke A D. J Chem Phys. 1996;104:1040–1046. [Google Scholar]
- 38.Miertus S, Tomasi J. Chem Phys. 1982;65:239–245. [Google Scholar]
- 39.Miertus S, Scrocco E, Tomasi J. Chem Phys. 1981;55:117–129. [Google Scholar]
- 40.Serjeant E P, Dempsey B. Ionisation Constants of Organic Liquids in Aqueous Solution. New York: Pergamon; 1979. [Google Scholar]
- 41.Perrin D B. Ionization Constants of Inorganic Acids and Bases in Aqueous Solution. 2nd Ed. Oxford: Pergamon; 1982. [Google Scholar]
- 42.Donald C E, Hughes M N, Thomson J M, Bonner F T. Inorg Chem. 1986;25:2676–2677. [Google Scholar]
- 43.Ogata Y, Kawasaki A. In: The Chemistry of the Carbonyl Group. Patai S, Zabicky J, editors. London: Interscience; 1970. 2, 1–70. [Google Scholar]
- 44.Guthrie J P. Can J Chem. 1975;53:898–906. [Google Scholar]
- 45.Guthrie J P. Can J Chem. 1978;56:962–973. [Google Scholar]
- 46.Wiberg K B, Morgan K M, Maltz H. J Am Chem Soc. 1994;116:11067–11077. [Google Scholar]
- 47.Boyer J H. In: The Chemistry of Nitro and Nitroso Groups. Feuer H, editor. New York: Wiley; 1968. [Google Scholar]
- 48.Ruud K, Helgaker T, Uggerud E. J Mol Struct. 1997;393:59–71. [Google Scholar]
- 49.Lüttke W, Skancke P N, Tratteberg M. Theor Chim Acta. 1994;87:321–323. [Google Scholar]
- 50.Stanbury D M. In: Advances in Inorganic Chemistry. Sykes AG, editor. San Diego: Academic; 1989. 33, 69–138. [Google Scholar]