

Abstract
In the title compound, C14H8F2N2O, the amide plane is inclined at dihedral angles of 28.12 (12) and 32.89 (12)° with respect to the two benzene rings; the dihedral angle between the two rings is 5.58 (5)°. In the crystal, intermolecular N—H⋯O and C—H⋯F hydrogen bonds link adjacent molecules into a double-chain structure along the b axis.
Related literature
For general background to and applications of the title compound, see: Ashwood et al. (1990 ▶); Kees et al. (1989 ▶); Ragavan et al. (2010 ▶); Carmellino et al. (1994 ▶); Rauko et al. (2001 ▶). For a closely related benzamide structure, see: Cronin et al. (2000 ▶). For the stability of the temperature controller used in the data collection, see: Cosier & Glazer (1986 ▶).
Experimental
Crystal data
C14H8F2N2O
M r = 258.22
Monoclinic,
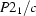
a = 9.3377 (11) Å
b = 5.0793 (6) Å
c = 24.500 (3) Å
β = 100.202 (3)°
V = 1143.6 (2) Å3
Z = 4
Mo Kα radiation
μ = 0.12 mm−1
T = 100 K
0.27 × 0.14 × 0.14 mm
Data collection
Bruker APEXII DUO CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2009 ▶) T min = 0.968, T max = 0.984
27219 measured reflections
4135 independent reflections
3176 reflections with I > 2σ(I)
R int = 0.054
Refinement
R[F 2 > 2σ(F 2)] = 0.043
wR(F 2) = 0.118
S = 1.03
4135 reflections
204 parameters
All H-atom parameters refined
Δρmax = 0.39 e Å−3
Δρmin = −0.23 e Å−3
Data collection: APEX2 (Bruker, 2009 ▶); cell refinement: SAINT (Bruker, 2009 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL and PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810046507/is2630sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810046507/is2630Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1N1⋯O1i | 0.863 (15) | 2.107 (15) | 2.9029 (12) | 153.3 (14) |
| C12—H12⋯F1ii | 0.940 (16) | 2.473 (16) | 3.4066 (14) | 172.4 (13) |
Symmetry codes: (i) 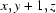 ; (ii)
; (ii) 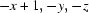 .
.
Acknowledgments
The authors thank Universiti Sains Malaysia (USM) for the Research University Grant (No. 1001/PFIZIK/811160).
supplementary crystallographic information
Comment
A number of benzamide derivatives were reported as anti-hypertensive (Ashwood et al., 1990), anti-diabetic (Kees et al., 1989), anti-bacterial (Ragavan et al., 2010), anti-fungal (Carmellino et al., 1994) and anti-cancer (Rauko et al., 2001) activities. On the basis of these considerations, our particular attention was paid for the synthesis of some benzamide derivatives.
In the title benzamide derivative, the amino moiety (C7/N1/O1) is essentially planar, as indicated by the C7–O1–N1–H1N1 torsion angle of -1.4 (18)°. The mean plane through the amido moiety is inclined at dihedral angles of 32.89 (12) and 28.12 (12)°, respectively, with the C1–C6 and C8–C13 benzene rings. The dihedral angle between the two benzene rings being 5.58 (5)°. All bond lengths and angles are comparable to values observed in a closely related benzamide structure (Cronin et al., 2000). In the crystal packing, adjacent molecules are interconnected into two-molecule-wide infinite chains propagating along the [010] direction (Fig. 2) via intermolecular N1—H1N1···O1 and C12—H12···F1 hydrogen bonds (Table 1).
Experimental
A mixture of 4-amino benzonitrile (4.2 mmol), 2,6-difluorobenzoic acid (4.6 mmol) and triethyl amine (21 mmol) was dissolved in methylene dichloride (5 ml). The resulting solution was cooled to 273 K followed by the drop wise addition of 50 % phosphoric acid cyclic anhydride solution in ethyl acetate (4 ml, 6.3 mmol) and stirred for 12 h. The completion of reaction was checked by TLC. Evaporation of solvent gave 2,6-difluoro-N-(p-cyanophenyl)benzamide as solid mass, which was then stirred with saturated NaHCO3 solution to remove excess of acid. Single crystals suitable for X-ray analysis were obtained by crystallization from acetone under slow evaporation. M.p. 418 K.
Refinement
All H atoms were located from a difference Fourier map and allowed to refine freely [refined distances: N—H = 0.860 (16) Å and C—H = 0.933 (15)–0.984 (15) Å].
Figures
Fig. 1.

The molecular structure of the title compound, showing 50 % probability displacement ellipsoids for non-H atoms and the atom-numbering scheme.
Fig. 2.

The crystal structure of the title compound, viewed along the a axis, showing two-molecule-wide infinite chains along the [010] direction. H atoms not involved in intermolecular hydrogen bonds (dashed lines) have been omitted for clarity.
Crystal data
| C14H8F2N2O | F(000) = 528 |
| Mr = 258.22 | Dx = 1.500 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 5020 reflections |
| a = 9.3377 (11) Å | θ = 2.2–31.3° |
| b = 5.0793 (6) Å | µ = 0.12 mm−1 |
| c = 24.500 (3) Å | T = 100 K |
| β = 100.202 (3)° | Block, yellow |
| V = 1143.6 (2) Å3 | 0.27 × 0.14 × 0.14 mm |
| Z = 4 |
Data collection
| Bruker APEXII DUO CCD area-detector diffractometer | 4135 independent reflections |
| Radiation source: fine-focus sealed tube | 3176 reflections with I > 2σ(I) |
| graphite | Rint = 0.054 |
| φ and ω scans | θmax = 32.6°, θmin = 2.2° |
| Absorption correction: multi-scan (SADABS; Bruker, 2009) | h = −14→14 |
| Tmin = 0.968, Tmax = 0.984 | k = −7→7 |
| 27219 measured reflections | l = −36→37 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.043 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.118 | All H-atom parameters refined |
| S = 1.03 | w = 1/[σ2(Fo2) + (0.0568P)2 + 0.2429P] where P = (Fo2 + 2Fc2)/3 |
| 4135 reflections | (Δ/σ)max < 0.001 |
| 204 parameters | Δρmax = 0.39 e Å−3 |
| 0 restraints | Δρmin = −0.23 e Å−3 |
Special details
| Experimental. The crystal was placed in the cold stream of an Oxford Cryosystems Cobra open-flow nitrogen cryostat (Cosier & Glazer, 1986) operating at 100.0 (1)K. |
| Geometry. All esds (except the esd in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell esds are taken into account individually in the estimation of esds in distances, angles and torsion angles; correlations between esds in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell esds is used for estimating esds involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2sigma(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| F1 | 0.81277 (8) | 0.06726 (14) | 0.14252 (3) | 0.03127 (18) | |
| F2 | 1.02229 (7) | 0.75952 (13) | 0.05423 (3) | 0.02670 (16) | |
| O1 | 0.76102 (9) | 0.12482 (15) | 0.03141 (3) | 0.02539 (18) | |
| N1 | 0.75319 (9) | 0.56187 (17) | 0.01066 (3) | 0.01774 (17) | |
| N2 | 0.33842 (13) | 0.5436 (2) | −0.24586 (5) | 0.0384 (3) | |
| C1 | 0.91364 (12) | 0.2568 (2) | 0.14216 (4) | 0.0207 (2) | |
| C2 | 1.01589 (13) | 0.2889 (2) | 0.18972 (5) | 0.0252 (2) | |
| C3 | 1.11850 (12) | 0.4873 (2) | 0.19138 (5) | 0.0246 (2) | |
| C4 | 1.11804 (11) | 0.6494 (2) | 0.14589 (5) | 0.0228 (2) | |
| C5 | 1.01538 (11) | 0.6060 (2) | 0.09898 (4) | 0.01910 (19) | |
| C6 | 0.90844 (10) | 0.41080 (19) | 0.09449 (4) | 0.01688 (18) | |
| C7 | 0.80070 (11) | 0.35107 (19) | 0.04285 (4) | 0.01768 (19) | |
| C8 | 0.66236 (11) | 0.55108 (19) | −0.04175 (4) | 0.01768 (19) | |
| C9 | 0.67895 (12) | 0.7489 (2) | −0.07969 (4) | 0.0207 (2) | |
| C10 | 0.59456 (12) | 0.7492 (2) | −0.13217 (5) | 0.0224 (2) | |
| C11 | 0.49135 (11) | 0.5511 (2) | −0.14682 (4) | 0.0220 (2) | |
| C12 | 0.47172 (12) | 0.3569 (2) | −0.10873 (5) | 0.0231 (2) | |
| C13 | 0.55706 (11) | 0.3555 (2) | −0.05623 (4) | 0.0209 (2) | |
| C14 | 0.40564 (13) | 0.5474 (2) | −0.20187 (5) | 0.0275 (2) | |
| H1N1 | 0.7848 (16) | 0.716 (3) | 0.0216 (6) | 0.031 (4)* | |
| H2 | 1.0120 (16) | 0.173 (3) | 0.2194 (6) | 0.031 (4)* | |
| H3 | 1.1931 (16) | 0.507 (3) | 0.2249 (6) | 0.031 (4)* | |
| H4 | 1.1877 (17) | 0.787 (3) | 0.1456 (6) | 0.028 (4)* | |
| H9 | 0.7526 (15) | 0.877 (3) | −0.0683 (6) | 0.022 (3)* | |
| H10 | 0.6115 (15) | 0.879 (3) | −0.1573 (6) | 0.026 (4)* | |
| H12 | 0.3999 (17) | 0.227 (3) | −0.1180 (6) | 0.031 (4)* | |
| H13 | 0.5408 (15) | 0.221 (3) | −0.0309 (6) | 0.026 (4)* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| F1 | 0.0336 (4) | 0.0283 (4) | 0.0293 (4) | −0.0134 (3) | −0.0015 (3) | 0.0072 (3) |
| F2 | 0.0242 (3) | 0.0266 (3) | 0.0274 (3) | −0.0063 (3) | −0.0006 (3) | 0.0092 (3) |
| O1 | 0.0327 (4) | 0.0133 (3) | 0.0260 (4) | −0.0007 (3) | −0.0061 (3) | −0.0027 (3) |
| N1 | 0.0212 (4) | 0.0123 (4) | 0.0175 (4) | −0.0004 (3) | −0.0026 (3) | −0.0018 (3) |
| N2 | 0.0415 (6) | 0.0392 (6) | 0.0283 (5) | 0.0086 (5) | −0.0108 (5) | −0.0071 (5) |
| C1 | 0.0227 (5) | 0.0172 (5) | 0.0215 (5) | −0.0029 (4) | 0.0021 (4) | 0.0004 (3) |
| C2 | 0.0283 (5) | 0.0265 (5) | 0.0195 (5) | −0.0012 (4) | 0.0011 (4) | 0.0017 (4) |
| C3 | 0.0235 (5) | 0.0285 (6) | 0.0198 (5) | −0.0004 (4) | −0.0018 (4) | −0.0042 (4) |
| C4 | 0.0190 (5) | 0.0222 (5) | 0.0257 (5) | −0.0025 (4) | −0.0003 (4) | −0.0029 (4) |
| C5 | 0.0187 (4) | 0.0174 (4) | 0.0206 (4) | 0.0002 (3) | 0.0020 (3) | 0.0004 (3) |
| C6 | 0.0173 (4) | 0.0145 (4) | 0.0179 (4) | 0.0004 (3) | 0.0007 (3) | −0.0023 (3) |
| C7 | 0.0188 (4) | 0.0144 (4) | 0.0187 (4) | 0.0009 (3) | 0.0002 (3) | −0.0014 (3) |
| C8 | 0.0186 (4) | 0.0156 (4) | 0.0176 (4) | 0.0020 (3) | −0.0003 (3) | −0.0030 (3) |
| C9 | 0.0240 (5) | 0.0170 (4) | 0.0194 (5) | −0.0008 (4) | −0.0007 (4) | −0.0015 (3) |
| C10 | 0.0264 (5) | 0.0202 (5) | 0.0189 (5) | 0.0021 (4) | −0.0002 (4) | −0.0006 (4) |
| C11 | 0.0214 (5) | 0.0226 (5) | 0.0195 (5) | 0.0049 (4) | −0.0037 (4) | −0.0055 (4) |
| C12 | 0.0196 (5) | 0.0206 (5) | 0.0266 (5) | −0.0003 (4) | −0.0030 (4) | −0.0051 (4) |
| C13 | 0.0206 (5) | 0.0181 (5) | 0.0224 (5) | −0.0004 (4) | −0.0008 (4) | −0.0012 (4) |
| C14 | 0.0279 (5) | 0.0259 (5) | 0.0254 (5) | 0.0056 (4) | −0.0043 (4) | −0.0057 (4) |
Geometric parameters (Å, °)
| F1—C1 | 1.3480 (12) | C4—H4 | 0.956 (15) |
| F2—C5 | 1.3563 (12) | C5—C6 | 1.3975 (14) |
| O1—C7 | 1.2246 (12) | C6—C7 | 1.5014 (13) |
| N1—C7 | 1.3569 (13) | C8—C9 | 1.3961 (14) |
| N1—C8 | 1.4091 (12) | C8—C13 | 1.3985 (14) |
| N1—H1N1 | 0.860 (16) | C9—C10 | 1.3839 (15) |
| N2—C14 | 1.1474 (15) | C9—H9 | 0.953 (14) |
| C1—C2 | 1.3791 (15) | C10—C11 | 1.3955 (15) |
| C1—C6 | 1.3992 (14) | C10—H10 | 0.933 (15) |
| C2—C3 | 1.3861 (16) | C11—C12 | 1.3922 (16) |
| C2—H2 | 0.943 (15) | C11—C14 | 1.4414 (15) |
| C3—C4 | 1.3849 (16) | C12—C13 | 1.3880 (15) |
| C3—H3 | 0.984 (15) | C12—H12 | 0.939 (15) |
| C4—C5 | 1.3777 (14) | C13—H13 | 0.952 (15) |
| C7—N1—C8 | 125.52 (8) | O1—C7—C6 | 120.86 (9) |
| C7—N1—H1N1 | 118.5 (10) | N1—C7—C6 | 115.57 (8) |
| C8—N1—H1N1 | 115.9 (10) | C9—C8—C13 | 119.89 (9) |
| F1—C1—C2 | 117.31 (9) | C9—C8—N1 | 117.32 (9) |
| F1—C1—C6 | 118.94 (9) | C13—C8—N1 | 122.78 (9) |
| C2—C1—C6 | 123.75 (10) | C10—C9—C8 | 120.48 (10) |
| C1—C2—C3 | 118.83 (10) | C10—C9—H9 | 122.3 (8) |
| C1—C2—H2 | 117.6 (9) | C8—C9—H9 | 117.2 (8) |
| C3—C2—H2 | 123.6 (9) | C9—C10—C11 | 119.47 (10) |
| C4—C3—C2 | 120.33 (10) | C9—C10—H10 | 118.6 (9) |
| C4—C3—H3 | 120.8 (9) | C11—C10—H10 | 121.9 (9) |
| C2—C3—H3 | 118.8 (9) | C12—C11—C10 | 120.36 (9) |
| C5—C4—C3 | 118.60 (10) | C12—C11—C14 | 120.06 (10) |
| C5—C4—H4 | 119.0 (9) | C10—C11—C14 | 119.58 (10) |
| C3—C4—H4 | 122.3 (9) | C13—C12—C11 | 120.18 (10) |
| F2—C5—C4 | 117.17 (9) | C13—C12—H12 | 119.3 (9) |
| F2—C5—C6 | 118.68 (9) | C11—C12—H12 | 120.5 (9) |
| C4—C5—C6 | 124.13 (10) | C12—C13—C8 | 119.60 (10) |
| C5—C6—C1 | 114.34 (9) | C12—C13—H13 | 118.5 (9) |
| C5—C6—C7 | 124.92 (9) | C8—C13—H13 | 121.9 (9) |
| C1—C6—C7 | 120.62 (9) | N2—C14—C11 | 179.41 (13) |
| O1—C7—N1 | 123.57 (9) | ||
| F1—C1—C2—C3 | −178.04 (10) | C1—C6—C7—O1 | −31.41 (15) |
| C6—C1—C2—C3 | 1.20 (18) | C5—C6—C7—N1 | −35.04 (14) |
| C1—C2—C3—C4 | −0.17 (18) | C1—C6—C7—N1 | 149.05 (10) |
| C2—C3—C4—C5 | −1.07 (17) | C7—N1—C8—C9 | −149.79 (11) |
| C3—C4—C5—F2 | −176.83 (10) | C7—N1—C8—C13 | 31.27 (16) |
| C3—C4—C5—C6 | 1.43 (17) | C13—C8—C9—C10 | −1.75 (16) |
| F2—C5—C6—C1 | 177.77 (9) | N1—C8—C9—C10 | 179.28 (10) |
| C4—C5—C6—C1 | −0.47 (15) | C8—C9—C10—C11 | 0.62 (16) |
| F2—C5—C6—C7 | 1.63 (15) | C9—C10—C11—C12 | 0.98 (16) |
| C4—C5—C6—C7 | −176.61 (10) | C9—C10—C11—C14 | −178.58 (10) |
| F1—C1—C6—C5 | 178.35 (9) | C10—C11—C12—C13 | −1.44 (16) |
| C2—C1—C6—C5 | −0.88 (16) | C14—C11—C12—C13 | 178.11 (10) |
| F1—C1—C6—C7 | −5.33 (15) | C11—C12—C13—C8 | 0.31 (16) |
| C2—C1—C6—C7 | 175.44 (10) | C9—C8—C13—C12 | 1.28 (16) |
| C8—N1—C7—O1 | −5.34 (17) | N1—C8—C13—C12 | −179.81 (10) |
| C8—N1—C7—C6 | 174.18 (9) | C7—O1—N1—H1N1 | −1.4 (18) |
| C5—C6—C7—O1 | 144.50 (11) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1N1···O1i | 0.863 (15) | 2.107 (15) | 2.9029 (12) | 153.3 (14) |
| C12—H12···F1ii | 0.940 (16) | 2.473 (16) | 3.4066 (14) | 172.4 (13) |
Symmetry codes: (i) x, y+1, z; (ii) −x+1, −y, −z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: IS2630).
References
- Ashwood, V. A., Cassidy, F., Coldwell, M. C., Evans, J. M., Hamilton, T. C., Howlett, D. R., Smith, D. M. & Stemp, G. (1990). J. Med. Chem.33, 2667–2672. [DOI] [PubMed]
- Bruker (2009). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Carmellino, M. L., Pagani, G., Pregnolato, M., Terreni, M. & Pastoni, F. (1994). Eur. J. Med. Chem.29, 743–751.
- Cosier, J. & Glazer, A. M. (1986). J. Appl. Cryst.19, 105–107.
- Cronin, L., Adams, D. A., Nightingale, D. J. & Clark, J. H. (2000). Acta Cryst. C56, 244–245. [DOI] [PubMed]
- Kees, K. L., Cheeseman, R. S., Prozialeck, D. H. & Steiner, K. E. (1989). J. Med. Chem.32, 11–13. [DOI] [PubMed]
- Ragavan, R. V., Vijayakumar, V. & Suchetha Kumari, N. (2010). Eur. J. Med. Chem.43, 1173–1180. [DOI] [PubMed]
- Rauko, P., Novotny, L., Dovinova, I., Hunakova, L., Szekeres, T. & Jayaram, H. N. (2001). Eur. J. Pharm. Sci.12, 387–394. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810046507/is2630sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810046507/is2630Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report