

Abstract
In the title compound, C12H11NO2, the phenolic ring is inclined at an angle of 32.70 (1)° with respect to the pyridine ring. In the crystal, intermolecular O—H⋯N hydrogen bonds link the molecules into C(11) chains along [001].
Related literature
For a related structure, see: Yumoto et al. (2008 ▶).
Experimental
Crystal data
C12H11NO2
M r = 201.22
Monoclinic,
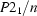
a = 6.6551 (6) Å
b = 9.1160 (8) Å
c = 17.0039 (15) Å
β = 100.501 (1)°
V = 1014.31 (16) Å3
Z = 4
Mo Kα radiation
μ = 0.09 mm−1
T = 293 K
0.28 × 0.24 × 0.22 mm
Data collection
Bruker SMART APEXII CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Sheldrick, 1996 ▶) T min = 0.930, T max = 0.980
5411 measured reflections
1981 independent reflections
1310 reflections with I > 2σ(I)
R int = 0.098
Refinement
R[F 2 > 2σ(F 2)] = 0.041
wR(F 2) = 0.091
S = 0.89
1981 reflections
140 parameters
H atoms treated by a mixture of independent and constrained refinement
Δρmax = 0.14 e Å−3
Δρmin = −0.18 e Å−3
Data collection: APEX2 (Bruker, 2005 ▶); cell refinement: SAINT (Bruker, 2005 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: SHELXTL-Plus (Sheldrick, 2008 ▶); software used to prepare material for publication: SHELXL97.
Supplementary Material
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810045800/ng5062sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810045800/ng5062Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O1—H1A⋯N1i | 0.95 (2) | 1.75 (2) | 2.6991 (17) | 174 (2) |
Symmetry code: (i) 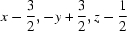 .
.
Acknowledgments
The authors thank The China–Japan Union Hospital of Jilin University for supporting this work.
supplementary crystallographic information
Comment
Pyridine and its derivatives represent one of the most active classes of compounds possessing a wide application of biological activities, such as stent in intestinal or biliary fields. During the past years, considerable efforts have been paid to demonstrate the efficacy of pyridine derivatives including antibacterial, antifungal, herbicidal, insecticidal and other biological activities. A new pyridine derivatives molecule is synthesized, with the aim of studying its single-crystal structure.
The title molecule (Fig. 1) consists of a phenol moiety (O1/C1—C6) and a methoxy moiety (O2/C7) attached to a pyridine ring (N1/C8—C12). The pyridine ring is inclined at an angle of 32.70 (1)° with the phenol ring. Bond lengths and angles are within normal ranges, and comparable to closely related structures (Yumoto et al., 2008). In the crystal structure, the crystal packing is consolidated by intermolecular O1—H1A···N1 hydrogen bonds linking the molecules into one linear structure.
Experimental
A mixture of 1,3-dihydroxybenzene (1.1 g, 10 mmol), 4-chloromethlpyridine hydrochloride (1.64 g, 10 mmol), and NaOH (1.6 g, 40 mmol) in acetonitrile (50 ml) was refluxed under nitrogen with stirring for 24 h. After cooling to room temperature, the reactant was filtered, and the residue was washed with acetonitrile several times. The mixed filtrate was slowly evaporated and colorless crystals were obtained.
Refinement
All H-atoms bound to carbon were refined using a riding model with d(C—H) = 0.93 Å, Uiso = 1.2Ueq (C) for aromatic and 0.97 Å, Uiso = 1.2Ueq (C) for CH2 atoms. H atoms bonded to O atoms were located in a difference Fourier map.
Figures
Fig. 1.

A view of the molecule of (I). Displacement ellipsoids are drawn at the 30% probability level.
Crystal data
| C12H11NO2 | F(000) = 424 |
| Mr = 201.22 | Dx = 1.318 Mg m−3 |
| Monoclinic, P21/n | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2yn | Cell parameters from 1981 reflections |
| a = 6.6551 (6) Å | θ = 1.9–28.3° |
| b = 9.1160 (8) Å | µ = 0.09 mm−1 |
| c = 17.0039 (15) Å | T = 293 K |
| β = 100.501 (1)° | Block, colorless |
| V = 1014.31 (16) Å3 | 0.28 × 0.24 × 0.22 mm |
| Z = 4 |
Data collection
| Bruker APEX CCD area-detector diffractometer | 1981 independent reflections |
| Radiation source: fine-focus sealed tube | 1310 reflections with I > 2σ(I) |
| graphite | Rint = 0.098 |
| φ and ω scans | θmax = 26.0°, θmin = 2.4° |
| Absorption correction: multi-scan (SADABS; Sheldrick, 1996) | h = −8→8 |
| Tmin = 0.930, Tmax = 0.980 | k = −9→11 |
| 5411 measured reflections | l = −15→20 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.041 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.091 | H atoms treated by a mixture of independent and constrained refinement |
| S = 0.89 | w = 1/[σ2(Fo2) + (0.0212P)2] where P = (Fo2 + 2Fc2)/3 |
| 1981 reflections | (Δ/σ)max = 0.001 |
| 140 parameters | Δρmax = 0.14 e Å−3 |
| 0 restraints | Δρmin = −0.18 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| N1 | 0.7272 (2) | 0.82817 (15) | 1.01329 (9) | 0.0343 (4) | |
| O1 | −0.50233 (18) | 0.67160 (13) | 0.65242 (7) | 0.0385 (3) | |
| O2 | 0.13905 (16) | 0.82363 (12) | 0.78584 (7) | 0.0357 (3) | |
| H1A | −0.601 (3) | 0.678 (2) | 0.6047 (13) | 0.073 (7)* | |
| C7 | 0.3149 (2) | 0.91479 (18) | 0.79342 (10) | 0.0317 (4) | |
| H7A | 0.2748 | 1.0170 | 0.7939 | 0.038* | |
| H7B | 0.3829 | 0.8995 | 0.7482 | 0.038* | |
| C6 | −0.1795 (2) | 0.75328 (18) | 0.71613 (9) | 0.0296 (4) | |
| H6 | −0.1743 | 0.6758 | 0.7520 | 0.036* | |
| C5 | −0.0183 (2) | 0.85103 (18) | 0.72303 (9) | 0.0295 (4) | |
| C12 | 0.3904 (2) | 0.81807 (17) | 0.93485 (10) | 0.0320 (4) | |
| H12 | 0.2540 | 0.7921 | 0.9316 | 0.038* | |
| C8 | 0.4572 (2) | 0.87800 (17) | 0.86953 (10) | 0.0271 (4) | |
| C11 | 0.5288 (3) | 0.79727 (18) | 1.00500 (11) | 0.0345 (4) | |
| H11 | 0.4809 | 0.7596 | 1.0489 | 0.041* | |
| C1 | −0.3484 (2) | 0.77025 (19) | 0.65603 (10) | 0.0305 (4) | |
| C2 | −0.3563 (3) | 0.88767 (19) | 0.60343 (10) | 0.0356 (4) | |
| H2 | −0.4703 | 0.9013 | 0.5635 | 0.043* | |
| C9 | 0.6626 (2) | 0.90938 (18) | 0.87728 (10) | 0.0313 (4) | |
| H9 | 0.7143 | 0.9480 | 0.8344 | 0.038* | |
| C4 | −0.0223 (2) | 0.96653 (18) | 0.67026 (10) | 0.0348 (4) | |
| H4 | 0.0871 | 1.0313 | 0.6743 | 0.042* | |
| C3 | −0.1947 (2) | 0.98324 (19) | 0.61087 (10) | 0.0384 (5) | |
| H3 | −0.2005 | 1.0613 | 0.5753 | 0.046* | |
| C10 | 0.7902 (2) | 0.88286 (18) | 0.94908 (10) | 0.0346 (4) | |
| H10 | 0.9283 | 0.9043 | 0.9532 | 0.041* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| N1 | 0.0290 (8) | 0.0365 (9) | 0.0345 (9) | 0.0046 (6) | −0.0018 (7) | −0.0058 (7) |
| O1 | 0.0276 (7) | 0.0540 (8) | 0.0305 (7) | −0.0094 (6) | −0.0038 (6) | 0.0043 (6) |
| O2 | 0.0296 (7) | 0.0405 (7) | 0.0315 (7) | −0.0063 (5) | −0.0091 (5) | 0.0046 (5) |
| C7 | 0.0266 (9) | 0.0371 (10) | 0.0304 (10) | −0.0015 (8) | 0.0025 (8) | −0.0030 (8) |
| C6 | 0.0292 (9) | 0.0352 (10) | 0.0231 (9) | 0.0006 (8) | 0.0014 (8) | 0.0026 (8) |
| C5 | 0.0266 (9) | 0.0365 (10) | 0.0231 (9) | 0.0017 (8) | −0.0012 (7) | −0.0033 (8) |
| C12 | 0.0228 (9) | 0.0375 (11) | 0.0345 (10) | 0.0018 (7) | 0.0020 (8) | −0.0024 (8) |
| C8 | 0.0237 (9) | 0.0280 (9) | 0.0283 (9) | 0.0033 (7) | 0.0012 (7) | −0.0063 (7) |
| C11 | 0.0329 (10) | 0.0388 (11) | 0.0315 (10) | 0.0025 (8) | 0.0050 (8) | −0.0018 (8) |
| C1 | 0.0252 (9) | 0.0401 (11) | 0.0255 (9) | −0.0006 (8) | 0.0029 (7) | −0.0043 (8) |
| C2 | 0.0312 (10) | 0.0406 (11) | 0.0307 (10) | 0.0036 (8) | −0.0061 (8) | 0.0026 (8) |
| C9 | 0.0287 (10) | 0.0328 (10) | 0.0323 (10) | 0.0001 (7) | 0.0055 (8) | −0.0063 (8) |
| C4 | 0.0325 (10) | 0.0324 (10) | 0.0364 (11) | −0.0041 (8) | −0.0016 (8) | 0.0018 (8) |
| C3 | 0.0422 (11) | 0.0319 (10) | 0.0367 (11) | −0.0006 (8) | −0.0046 (9) | 0.0057 (8) |
| C10 | 0.0243 (9) | 0.0358 (10) | 0.0419 (12) | 0.0003 (8) | 0.0015 (8) | −0.0112 (9) |
Geometric parameters (Å, °)
| N1—C11 | 1.332 (2) | C12—C8 | 1.382 (2) |
| N1—C10 | 1.335 (2) | C12—H12 | 0.9300 |
| O1—C1 | 1.3560 (19) | C8—C9 | 1.379 (2) |
| O1—H1A | 0.95 (2) | C11—H11 | 0.9300 |
| O2—C5 | 1.3756 (17) | C1—C2 | 1.390 (2) |
| O2—C7 | 1.4218 (18) | C2—C3 | 1.372 (2) |
| C7—C8 | 1.496 (2) | C2—H2 | 0.9300 |
| C7—H7A | 0.9700 | C9—C10 | 1.375 (2) |
| C7—H7B | 0.9700 | C9—H9 | 0.9300 |
| C6—C5 | 1.383 (2) | C4—C3 | 1.392 (2) |
| C6—C1 | 1.383 (2) | C4—H4 | 0.9300 |
| C6—H6 | 0.9300 | C3—H3 | 0.9300 |
| C5—C4 | 1.381 (2) | C10—H10 | 0.9300 |
| C12—C11 | 1.381 (2) | ||
| C11—N1—C10 | 116.44 (15) | N1—C11—C12 | 123.70 (17) |
| C1—O1—H1A | 113.5 (12) | N1—C11—H11 | 118.2 |
| C5—O2—C7 | 117.50 (13) | C12—C11—H11 | 118.2 |
| O2—C7—C8 | 109.17 (13) | O1—C1—C6 | 117.70 (16) |
| O2—C7—H7A | 109.8 | O1—C1—C2 | 122.82 (15) |
| C8—C7—H7A | 109.8 | C6—C1—C2 | 119.46 (16) |
| O2—C7—H7B | 109.8 | C3—C2—C1 | 119.52 (16) |
| C8—C7—H7B | 109.8 | C3—C2—H2 | 120.2 |
| H7A—C7—H7B | 108.3 | C1—C2—H2 | 120.2 |
| C5—C6—C1 | 120.27 (16) | C10—C9—C8 | 119.27 (16) |
| C5—C6—H6 | 119.9 | C10—C9—H9 | 120.4 |
| C1—C6—H6 | 119.9 | C8—C9—H9 | 120.4 |
| O2—C5—C4 | 124.39 (15) | C5—C4—C3 | 118.06 (16) |
| O2—C5—C6 | 114.70 (15) | C5—C4—H4 | 121.0 |
| C4—C5—C6 | 120.91 (15) | C3—C4—H4 | 121.0 |
| C11—C12—C8 | 119.11 (15) | C2—C3—C4 | 121.76 (17) |
| C11—C12—H12 | 120.4 | C2—C3—H3 | 119.1 |
| C8—C12—H12 | 120.4 | C4—C3—H3 | 119.1 |
| C9—C8—C12 | 117.64 (15) | N1—C10—C9 | 123.82 (16) |
| C9—C8—C7 | 119.78 (15) | N1—C10—H10 | 118.1 |
| C12—C8—C7 | 122.54 (14) | C9—C10—H10 | 118.1 |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| O1—H1A···N1i | 0.95 (2) | 1.75 (2) | 2.6991 (17) | 174 (2) |
Symmetry codes: (i) x−3/2, −y+3/2, z−1/2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: NG5062).
References
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablocks global, I. DOI: 10.1107/S1600536810045800/ng5062sup1.cif
Structure factors: contains datablocks I. DOI: 10.1107/S1600536810045800/ng5062Isup2.hkl
Additional supplementary materials: crystallographic information; 3D view; checkCIF report