

Abstract
Abundant abnormal aggregates of cytoskeletal proteins are neuropathological signatures of many neurodegenerative diseases that are broadly classified by filamentous aggregates of neuronal intermediate filament (IF) proteins, or by inclusions containing the microtubule-associated protein (MAP) tau. The discovery of mutations in neuronal IF and tau genes firmly establishes the importance of neuronal IF proteins and tau in the pathogenesis of neurodegenerative diseases. Multiple IF gene mutations are pathogenic for Charcot–Marie–Tooth (CMT) disease and amyotrophic lateral sclerosis (ALS) — in addition to those in the copper/zinc superoxide dismutase-1 (SOD1) gene. Tau gene mutations are pathogenic for frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17), and tau polymorphisms are genetic risk factors for sporadic progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD). Thus, IF and tau abnormalities are linked directly to the aetiology and pathogenesis of neurodegenerative diseases. In vitro and transgenic animal models are being used to demonstrate that different mutations impair protein function, promote tau fibrilization, or perturb tau gene splicing, leading to aberrant and distinct tau aggregates. For recognition of these disorders at neuropathological examination, immunohistochemistry is needed, and this may be combined with biochemistry and molecular genetics to properly determine the nosology of a particular case. As reviewed here, the identification of molecular genetic defects and biochemical alterations in cytoskeletal proteins of human neurodegenerative diseases has facilitated experimental studies and will promote the development of assays of molecules which inhibit abnormal neuronal IF and tau protein inclusions.
Keywords: neuronal intermediate filament, tau, cytoskeleton, mutation, neurodegenerative disease, peripheral neuropathy
Introduction
Many chronic progressive neurodegenerative disorders are characterized by the presence of abnormal protein aggregates in neurons and glia of the central nervous system (CNS) [1–6]. The identification of disease-specific abnormal protein inclusions has illuminated mechanisms of pathogenesis as well as facilitating the molecular classification of the neurodegenerative diseases. Several sporadic and familial neurodegenerative diseases are characterized by the formation of filamentous deposits of abnormal brain proteins. Thus, a heterogeneous group of movement disorders and dementias is linked by the presence of pathological intra-cellular inclusions of neuronal intermediate filament (IF) proteins or the microtubule-associated protein (MAP) tau; each appears to share common mechanisms of disease [6]. These disorders are called, respectively, neuronal intermediate filamentopathies and tauopathies (Table 1). Despite the diverse phenotypic expression, brain dysfunction and neurodegeneration in both classes of disease are linked to the progressive accumulation of abnormal filamentous protein; and this, together with the absence of other disease-specific neuropathological abnormalities, provides evidence implicating neuronal IF and tau in disease onset and progression. The discovery of multiple mutations in neuronal IF genes in the hereditary neuropathy Charcot–Marie–Tooth disease (CMT) and amyotrophic lateral sclerosis (ALS) (Table 2) [7–17] and in the tau gene in frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) (Table 3) [18–47] has led to the unequivocal evidence that neuronal IF and tau abnormalities alone are sufficient to cause neurodegenerative disease. These discoveries have opened up new avenues of research into the roles of neuronal IF proteins and tau in mechanisms of brain dysfunction and neurodegeneration.
Table 1.
Neurodegenerative diseases with neuronal intermediate filament- and tau-positive filamentous inclusions
| Neuronal intermediate filaments | Tau |
|---|---|
| Alzheimer’s disease* Amyotrophic lateral sclerosis Charcot–Marie–Tooth disease Diabetic neuropathy Dementia with Lewy bodies* Giant axonal neuropathy Neuronal intermediate filament Inclusion disease Parkinson’s disease* |
Alzheimer’s disease Amyotrophic lateral sclerosis/parkinsonism–dementia complex of Guam (ALS/PDC)† Argyrophilic grain disease† Corticobasal degeneration† Dementia pugilistica† Diffuse neurofibrillary tangles with calcification† Down’s syndrome Frontotemporal dementia with parkinsonism linked to chromosome 17† Gerstmann–Sträussler–Scheinker disease Myotonic dystrophy Niemann–Pick disease, type C Non-Guamanian motor neuron disease with neurofibrillary tangles Pick’s disease† Post-encephalitic parkinsonism Prion disease with neurofibrillary tangles Progressive supranuclear palsy† Subacute sclerosing panencephalitis Tangle only dementia† |
Neuronal intermediate filaments are chaperone proteins and a minor component of inclusions.
Diseases in which neurofibrillary pathology is the most predominant neuropathological feature.
Table 2.
Neuronal intermediate filament mutations in human diseases
| IF | Mutation | Domain | Phenotype | Reference |
|---|---|---|---|---|
| Peripherin | ND | — | — | — |
| α-Internexin | ND | — | — | — |
| NF-L | E7L + P8R | Head | CMT-unspecified | 7 |
| NF-L | P8R | Head | CMT-2 | 8 |
| NF-L | P8Q | Head | CMT-1 | 7 |
| NF-L | P8A | Head | CMT-1 | 7 |
| NF-L | P8L | Head | CMT-1 | 7 |
| NF-L | P22T | Head | CMT-1 | 9 |
| NF-L | P22S | Head | CMT-2 | 10,11 |
| NF-L | E89K | Head | CMT-1 | 7 |
| NF-L | N97S | Rod | CMT-1 | 9 |
| NF-L | N148V | Rod | CMT-unspecified | 9 |
| NF-L | Q333P | Rod | CMT-2 | 12 |
| NF-L | E393K | Rod | CMT-2 | 13 |
| NF-L | ΔE528 | Tail | CMT-unspecified | 7 |
| NF-M | G336S | Rod | PD | 14 |
| NF-H | Δ34 aa 528–561 | KSP | ALS | 15 |
| NF-H | Δ8 aa 655–662 | KSP | ALS | 16 |
| NF-H | Δ6 aa 663–668 | KSP | ALS | 16 |
| NF-H | Δ14 aa 663–677 | KSP | ALS | 16 |
| NF-H | 28 aa insert 708 | KSP | ALS | 17 |
| NF-H | Δ6 aa 743–748 | KSP | ALS | 16 |
| NF-H | ΔK790 | KSP | ALS | 15 |
ND = none detected; PD = Parkinson’s disease; CMT-1 = Charcot–Marie–Tooth disease, type 1; ALS = amyotrophic lateral sclerosis; Δaa = amino acid deletion.
Table 3.
Tau mutations in FTDP-17
| Mutation | Location | Exon 10 splicing | MT binding | Phenotype | Reference |
|---|---|---|---|---|---|
| R5H | Exon 1 | No change | Reduced | FTDP-17 | 18 |
| R5L | Exon 1 | No change | Reduced | PSP-like | 19 |
| K257T | E9, R1 | No change | Reduced | PiD-like | 20,21 |
| I260V | E9, R1 | ND | ND | NA | 22 |
| L266V | E9, R1 | Decreased* | Reduced | PiD-like | 23 |
| G272V | E9, R1 | No change | Reduced | FTDP-17 | 24 |
| E9 + 33 | I9 | ND | NA | NA | 25 |
| N279K | E10, IR1–2 | Increased† | Variable | PSP-like | 26 |
| ΔK280 | E10, IR1–2 | Decreased | Reduced | FTDP-17 | 25 |
| L284L | E10, IR1–2 | Increased | NA | AD-like | 27 |
| N296N | E10, R2 | Increased | NA | CBD-like | 28 |
| N296H | E10, R2 | Increased | Decreased | FTDP-17 | 29 |
| ΔN296 | E10, R2 | No change | Decreased | PSP-like | 30 |
| P301L | E10, R2 | No change | Reduced | FTDP-17 | 24 |
| P301S | E10, R2 | No change | Reduced | CBD-like, FTDP-17 | 31,32 |
| S305N | E10, IR2–3 | Increased | No effect | CBD-like | 33,34 |
| S305S | E10, IR2–3 | Increased | NA | PSP-like | 35 |
| E10 + 3 | I10 | Increased | NA | FTDP-17 | 36 |
| E10 + 11 | I10 | Increased | NA | FTDP-17 | 37 |
| E10 + 12 | I10 | Increased | NA | FTDP-17 | 38 |
| E10 + 13 | I10 | Increased | NA | NA | 24 |
| E10 + 14 | I10 | Increased | NA | PSP-like, FTDP-17 | 24 |
| E10 + 16 | I10 | Increased | NA | AD-, PiD-, PSP-, CBD-like, FTDP-17 | 24,39 |
| L315R | E11 | No change | Reduced | PiD-like | 40 |
| S320F | E11 | No change | Reduced | PiD-like | 41 |
| Q336R | E12 | No change | Increased | PiD-like | 42 |
| V337M | E12, IR3–4 | No change | Reduced | FTDP-17 | 43 |
| E342V | E12, IR3–4 | Increased | ND | FTDP-17 | 44 |
| S352L | E12, IR3–4 | ND | No effect | Atypical | 45 |
| K369I | E12, IR3–4 | ND | Reduced | PiD-like | 46 |
| G389R | E13 | No change | Reduced | PiD-like | 47 |
| R406W | E13 | No change | Reduced | PSP-like | 24 |
E = exon; I = intron; R = MT binding repeat; IR = inter-repeat regions; ND = not determined; NA = not applicable.
Decreased indicates reduced exon 10 utilization.
Increased indicates enhanced exon 10 utilization.
This review is designed to integrate and interpret the remarkable recent advances that have led to new insights into the nosology and mechanisms of action of neuronal IF proteins and tau in neurodegenerative diseases. It starts with brief summaries of the human neuronal IF and tau genes; the functions of neuronal IFs and the six alternatively spliced tau isoforms are reviewed; the role of neuronal IF proteins and tau abnormalities in neurodegenerative diseases is discussed; and data from transgenic (TG) models of neuronal intermediate filamentopathies and tauopathies are considered.
Structure, function, and molecular genetics of neuronal intermediate filaments
There are six types of IF proteins classified by gene structure and sequence homology. The name ‘intermediate’ derives from their diameter (10–12 nm), being intermediate between microtubules (25 nm) and microfilaments (7–10 nm). Five major neuronal IF proteins are expressed in the adult human CNS: three neurofilament (NF) proteins: light (NF-L), medium (NF-M), and heavy (NF-H) subunits of approximately 68, 145, and 200 kD, respectively; peripherin of 57 kD; and α-internexin of 66 kD (Figure 1). NFs and α-internexin genes have homologous intron–exon organization and are type IV, while peripherin encodes a type III IF protein resembling vimentin [6,48]. The IF proteins have a tripartite structure: a central rod domain of about 300 amino acids formed from a highly conserved α-helix, and amino- and carboxy-terminal regions called head and tail domains, respectively, which are less conserved [6].
Figure 1.

Structure of neuronal IF proteins. All proteins share a conserved structure of a head, rod with coils forming an α-helix, and tail domains containing glutamic acid-rich sequences and repeat phosphorylation motifs of lysine–serine–proline (KSP). Figures refer to the initial and terminal amino acids of each protein
The assembly and transport of neuronal IF proteins are probably regulated by post-translational modification of the head region by phosphorylation and O-glycosylation [5]. All subunits are constitutively phosphorylated and most of the phosphorylation sites containing lysine–serine–proline (KSP) motifs are located in the tail domains of NF-M and NF-H [5,6,49]. Most of the serines in these motifs may be phosphorylated, which makes NF-M and NF-H highly phosphorylated proteins. The phosphorylation state of these proteins relates to their function, ie phosphorylation of the tail domain modifies the axonal diameter, which is important for controlling axonal conductivity, an important function of motor neurons. After synthesis in the perikaryon, neuronal IFs are rapidly assembled into filaments and actively transported along microtubules (MTs) in axons by motors like kinesin and dynein, where they move at a net slow velocity of 0.2–1 mm/day [6].
Assemblies of IFs form 10 nm filaments. NFs copolymerize requiring NF-L with either NF-M or NF-H for proper filament formation [5,6]. Peripherin and α-internexin, in contrast, can self-assemble and co-assemble with NFs [6,50]. α-Internexin is widely expressed during development and throughout the adult CNS, but at lower levels than NFs. Peripherin is found predominantly in the peripheral nervous system (PNS), although it is also expressed in specific populations of neurons including spinal motor neurons, cranial nerve nuclei of sensory origin, and small interneurons.
Neuronal IFs and disease
The use of phosphorylation-dependent and -independent antibodies to NF epitopes has enabled the immunohistochemical dissection of these proteins and has revealed that NFs within the perikaryon and proximal segments of axons and dendrites are normally hypophosphorylated, while NFs in axons are heavily phosphorylated [51,52]. In neurodegenerative diseases including Alzheimer’s disease (AD), neuronal IF proteins are present either as innocent bystanders or as chaperone-like proteins together with tau in neurofibrillary tangles (NFTs), dystrophic neurites of neuritic plaques (NPs), and neuropil threads. In Parkinson’s disease (PD) and dementia with Lewy bodies (DLB), α-internexin and NF triplet proteins are found in a subset of α-synuclein-positive Lewy bodies, although their role in lesion formation or neurodegeneration is uncertain [53,54]. In ALS, abnormal accumulations of phosphorylated NF proteins are present in the perikaryon of affected neurons, swollen axons, and spheroids, although the significance of the phosphorylation of NF proteins within the cytoplasm is unclear [49,55,56]. However, abnormal phosphorylation may impede axonal transport and contribute to neuronal dysfunction, while constitutive phosphorylation of NFs may protect them against proteolysis [57].
α-Internexin is expressed by most, if not all, neurons as they commence differentiation and precedes the expression of the NF triplet proteins. In the adult brain, α-internexin is expressed at relatively low levels in comparison to the NF proteins and there is selective anatomical expression with greater immunoreactivity being seen in the cerebellar granule cells, the source of thin-calibre parallel fibres, and in the neuron cell bodies and processes of cortical layer II neurons [58]. α-Internexin has recently been identified as a major component of the pathological inclusions of the frontotemporal dementia, neuronal intermediate filament inclusion disease (NIFID) (Figure 2). The signature lesion of this disease is the neuronal cytoplasmic inclusion, which is tau- and α-synuclein negative, variably ubiquitinated, and contains epitopes of all type IV IF proteins [59–61]. In addition to NFs in swollen axons and spheroids in ALS, peripherin has also been demonstrated by immunohistochemistry (IHC) in the ubiquitinated inclusions of ALS [62]. However, protein chemistry has not revealed any change in mobility on western blots of NFs, α-internexin or peripherin in ALS, PD, DLB, or NIFID when compared with normal controls [59,63]. Thus, other factors may play a role in the formation of abnormal neuronal IF aggregates including dysregulation of protein synthesis, failure of axonal transport, abnormal phosphorylation, and proteolysis.
Figure 2.
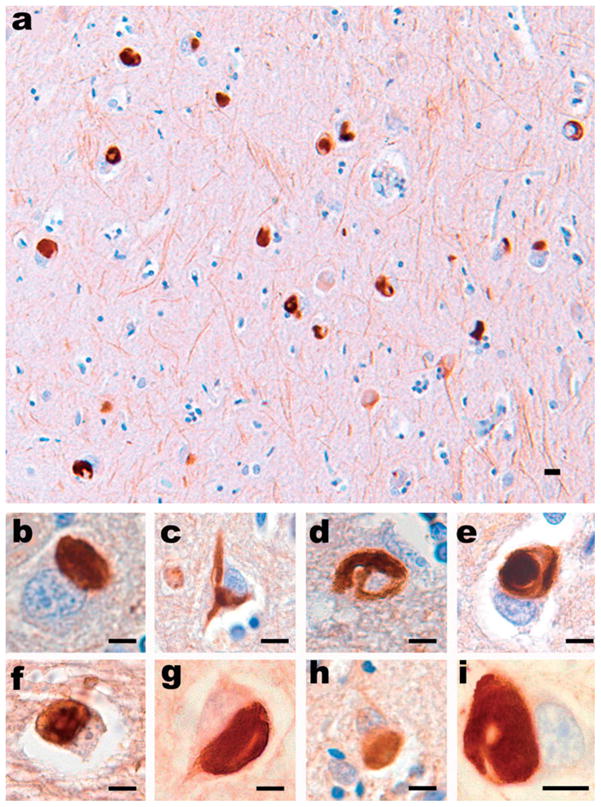
All type IV neuronal IF proteins are present in the pathological inclusions of NIFID. (a) Neuronal inclusions in the subiculum of a case of NIFID contain α-internexin. α-Internexin immunohistochemistry. Neuronal inclusions in NIFID are pleomorphic. (b) Pick body-like inclusions are the most common morphological type. (c) A flame-shaped, NFT-like inclusion. (d) A filamentous serpiginous inclusion. (e) A globose NFT-like inclusion. α-Internexin immunohistochemistry. Epitopes of NF triplet proteins are present in inclusions of NIFID and are recognized by (f) phosphorylation-dependent NF-H; (g) non-phosphorylation-dependent NF-H; (h) phosphorylation-independent NF-M; and (i) phosphorylation-independent NF-L antibodies. NF immunohistochemistry. Scale bars = 10 μm
Strong evidence for the role of neuronal IFs in pathogenesis has come from the discovery of mutations in IF genes in ALS and CMT in particular (Table 2). For example, both the Q333P mutation in the rod domain and to a lesser degree the P8R mutation in the head domain of NF-L disrupt the self-assembly of NF-L and the formation of NF-L/NF-M heteropolymers in a transient transfection system [64]. Codon deletions and insertions in the phosphorylation domain (KSP) of the tail region of NF-H have been reported in sporadic cases of ALS, and mutations in the NF-L gene located on chromosome 8 have been reported in several cases of CMT with neuroaxonal degeneration [7–17]. Thus, mutations in neuronal IF genes can directly cause selective motor neuron degeneration, axonal disorganization, and death.
Experimental animal models of neuronal intermediate filamentopathies
Although no gross developmental abnormalities have been reported in mouse knock-out experiments, the differential expression of neuronal IF proteins during development indicates a role for these proteins in axon formation and maintenance [4–6]. The absence of IFs in these TG models is, however, associated with measurable functional deficits in axogenesis. Models over-expressing single or multiple neuronal IF genes have replicated some of the features of human diseases. For example, overexpression of NF-M and NF-H has been shown to produce perykaryal inclusions of IFs resembling those seen in human disease [65], and a TG mouse model with overexpression of rat α-internexin has been shown to cause abnormal neurofilamentous accumulations and motor co-ordination deficits [66]. A model of CMT, type 2 (CMT-2) has been associated with NF assembly disruption and transport, mechanisms probably underlying neurodegeneration in this disease [5]. In a mouse model with a leucine to proline mutation at residue 394, selective motor neuron degeneration developed probably as a result of NF assembly disruption [67]. Ribonucleic acid (RNA) processing has also been implicated in the pathogenesis of neurodegenerative diseases: the expression of an NF-L transgene with a mutant messenger RNA (mRNA) stability determinant disrupts enteric and motor neurons in a TG mouse, indicating that motor neuron degeneration may be attributable to expression of mutant mRNA rather than mutant protein by the NF-L transgene [68]. The expression of neurotoxic splice variants of peripherin may also contribute to the neurodegenerative mechanism in ALS [69]. In a TG model, mice overexpressing peripherin developed a late-onset, and progressive, motor neuron disease with neuronal IF inclusions comparable to the spheroids seen in ALS [70]. However, these mice also developed motor neuron death during ageing. The mechanism leading to peripherin-induced neurodegeneration is unclear. Although familial PD has been linked to mutations in α-synuclein and parkin genes, a point mutation has been reported in the NF-M gene causing a substitution of serine for glycine at residue 336 in an affected woman at age 16 years [14]. Thus, mutations in NF genes can generate heterogeneous clinical and neuropathological phenotypes and although TG models recapitulate features of these human diseases, additional models are required to elucidate the mechanisms of action of each genetic defect.
Structure, function, and molecular genetics of tau
Several sporadic and familial neurodegenerative disorders that are characterized clinically by dementia and/or motor dysfunction are characterized pathologically by abnormal intracellular accumulations of the MAP tau, collectively called tauopathies (Table 1). The progressive accumulation of filamentous tau inclusions in the absence of other disease-specific neuropathological abnormalities provides evidence implicating tau dysfunction in disease onset and progression. However, the discovery of pathogenic tau gene mutations in the heterogeneous group of disorders known as FTDP-17 provided confirmation of the central role of tau abnormalities in the aetiology of neurodegenerative disorders [24,43,71,72]. These findings have opened novel areas of investigation into the mechanisms of tau dysfunction and the relationship of tau abnormalities to brain degeneration.
Tau proteins are low-molecular-weight MAPs that are abundant in the CNS, where they are expressed predominantly in axons [73,74], and at low levels in astrocytes and oligodendrocytes [75,76]. They are also expressed in axons of PNS neurons [77]. Human tau proteins are encoded by a single copy gene on chromosome 17q21 of 16 exons, with CNS isoforms generated by alternative mRNA splicing of 11 of these exons (Figure 3) [78–80]. In adult human brain, alternative splicing of exons 2, 3, and 10 generates six tau isoforms ranging from 352 to 441 amino acids in length which differ by the presence of either three or four MT binding repeats (3R tau or 4R tau, respectively) consisting of carboxy-terminal tandem repeat sequences of 31 or 32 amino acids each that are encoded by exons 9–12 [81,82]. Additionally, alternative splicing of exons 2 and 3 leads to the absence (0N) or presence of inserted sequences of 29 (1N) or 58 (2N) amino acids in the amino-terminal third of the molecule. In the adult human brain, the ratio of 3R : 4R tau isoforms is approximately 1 : 1, while the 0N, 1N, and 2N tau isoforms comprise about 37%, 54%, and 9%, respectively, of total tau [83,84].
Figure 3.

Schematic representation of the human tau gene and six human CNS tau isoforms generated by alternative splicing. The human tau gene contains 16 exons, including exon 0 that is part of the promoter. Exons 1, 4, 5, 7, 9, and 11–13 are constitutively expressed. Alternative splicing of exons 2 (E2), 3 (E3), and 10 produces the six alternative tau isoforms. Exons 6 and 8 are not transcribed in the human CNS. Exon 4a, which is also not transcribed in the human CNS, is expressed in the PNS leading to the larger tau isoforms, termed ‘big tau’. The black bars depict the 18 amino acid MT binding repeats and are designated R1 to R4. The relative sizes of the exons and introns are not drawn to scale
Tau binds to and stabilizes MTs and promotes MT polymerization [73,85]. The MT binding domains of tau are localized within the four MT binding motifs (Figures 3 and 4). These motifs are composed of highly conserved binding elements [85–87]. The function of tau as an MT binding protein is regulated by phosphorylation [87–92]. Phosphorylation at approximately 30 of these sites has been reported in normal tau proteins [93–95]. Several Ser/Thr protein kinases and Ser/Thr protein phosphatases have been implicated in regulating the phosphorylation state and thus the function of tau. The phosphorylation sites are clustered in regions flanking the MT binding repeats, and increasing tau phosphorylation at multiple sites negatively regulates MT binding [89–92]. However, in both sporadic and familial tauopathies including AD and FTDP-17, tau is hyperphosphorylated and it is this ‘abnormal’ tau that is the principal component of the filamentous aggregates in neurons and glia that are the pathological hallmarks of these disorders [96–98].
Figure 4.

Schematic representation of mutations in the tau gene in FTDP-17. The structure of the largest tau isoform is shown, with known coding region mutations indicated above. The grey boxes near the amino terminus represent the alternatively spliced inserts encoded for by exons 2 and 3, while the black boxes represent each of the four MT binding repeats (not drawn to scale). The second MT binding repeat is encoded by exon 10. Part of the mRNA sequence encoding exon 10 and the intron following exon 10 is enlarged to visualize the 5′ splice site as well as the mutations both in exon 10 and within the 5′ splice site. Nucleotides that are part of intron 10 are shown in lower case
Although there is clinical and neuropathological overlap between the neurodegenerative tauopathies, each can be distinguished with variable probability by the distribution, severity, and morphology of tau-positive inclusions. In cases with a tau gene mutation, in addition to extensive neuronal loss and astrocytosis, tau-positive neuronal and glial inclusions may resemble those seen in AD, PSP, CBD, and Pick’s disease. This neuropathological heterogeneity is a striking feature of FTDP-17 and it is complemented by biochemical heterogeneity where there is variation in the proportions of tau isoforms, not only with different mutations, but also within the same brain. Nevertheless, cases with tau gene mutations may be broadly grouped according to the pattern of tau immunostaining and tau isoform ratios as demonstrated by western blotting (Figure 5).
Figure 5.

Schematic representation of western blot banding patterns of soluble and insoluble tau from different tauopathies. The drawing depicts the typical banding pattern of soluble tau (top panels) and insoluble/filamentous tau (bottom panels) from the brains of patients with FTDP-17 as well as sporadic tauopathies following resolution with SDS-PAGE and immunoblotting with anti-tau antibodies. The FTDP-17 mutations show several different western blot banding patterns of soluble and insoluble tau protein that are depicted as groups A to D. The soluble fraction from the brains of unaffected (normal) individuals, sporadic tauopathies, and FTDP-17 with mutations that do not affect tau splicing (groups A, B, and C) shows expression of all six tau isoforms. Insoluble tau from the brains of patients with FTDP-17, group A (S320F, V337M, K369I, G389R, and R406W), resolves as three major proteins of 68, 64 and 60 kD; and a minor band of 72 kD similar to that observed in AD. When dephosphorylated, they resolve into six proteins that correspond to all six tau isoforms similar to the soluble fraction. In FTDP-17 group B (R5H, P301L, and G342V), two prominent 68- and 64-kD protein bands are detected (the 72 kD minor band is variably detected) that align with 4R tau following dephosphorylation similar to that observed in PSP and CBD, indicating the selective aggregation of 4R tau. In FTDP-17 group C (K257T) and Pick’s disease, the 64 and 60 kD insoluble tau protein isoforms predominate and align with 3R tau isoforms following dephosphorylation, indicating selective aggregation of 3R tau. In contrast, in FTDP-17 mutations that affect mRNA splicing (group D: N279K, L284L, N296N, N296H, S305S, S305N, and intron 10 mutations), there is expression of predominantly 4R tau throughout the entire brain, which is reflected in the insoluble tau aggregates
Tau gene mutations cause tau dysfunction by several distinct mechanisms. Intronic and some exonic mutations affect the alternative splicing of exon 10 and consequently alter the relative proportions of 3R and 4R tau. Other exonic mutations impair the ability of tau to bind MTs and to promote MT assembly. Some mutations also promote the assembly of tau into pathological amyloid filaments. Moreover, additional mechanisms may play a role in the case of some coding region mutations [1]. The intronic mutations clustered around the 5′ splice site of exon 10, as well as several mutations within exon 10 (Figure 4), increase the ratio of 4R : 3R tau by altering exon splicing [24,27,99–105]. As a result of these mutations, there is a relative increase in mRNA containing exon 10. Biochemical analysis of insoluble tau extracted from FTDP-17 brain tissue reveals predominantly 4R tau isoforms (Figure 5) [72,106–108]. Furthermore, 4R tau protein levels are increased in both affected and unaffected regions of FTDP-17 brains [72,84,107].
Mutations in the tau gene may alter exon 10 splicing by affecting several of the regulatory elements described above. For example, the intronic mutations as well as the exonic mutations at codon 305 (S305N and S305S) may destabilize the inhibitory stem-loop structure and alter the ratio of 3R : 4R tau [24,27,102]. The mechanisms by which changes in the ratio of 3R : 4R tau lead to neuronal and glial dysfunction and cell death remain unclear. However, 3R and 4R tau may bind to distinct sites on MTs [109] and it is possible that a specific ratio of tau isoforms is necessary for normal MT function [110]. Thus, the altered ratio of 3R : 4R tau may directly affect MT function. In addition, overproduction of 4R tau isoforms may lead to an excess of free tau in the cytoplasm that is prone to aggregate and polymerize into filaments over time.
Another subset of the tau mutations has no effect on tau splicing, but instead alters the ability of tau to interact with MTs: missense mutations K257T, G272V, ΔK280, ΔN296, P301L, P301S, V337M, G389R, and R406W reduce the binding of tau to MTs and decrease its ability to promote MT stability and assembly in vitro [20,25,31,83,111,112]. In contrast to mutations that affect the splicing of tau, these mutations do not alter the expression pattern of 3R and 4R tau [84]. The P301L mutation causes a moderate (25%) decrease in soluble 4R tau due to the selective aggregation of mutant 4R tau isoforms [84,113,114]. Biochemical analysis of insoluble tau extracted from brain tissue of patients with these mutations reveals a variety of patterns.
A subset of missense tau gene mutations may cause FTDP-17, at least in part, by promoting tau aggregation. In vitro studies demonstrated that mutations, including K257T, G272V, ΔK280, ΔN296, P301L, P301S, V337M, and R406W, promote heparin- or arachidonic acid-induced tau filament formation relative to wild type tau [114–118]. The missense tau gene mutations may also affect tau function by altering its phosphorylation state, and several mutations decrease the binding affinity of tau for protein phosphatase 2A, a major phosphatase implicated in the regulation of the MT-binding activity of tau [119].
Experimental animal models of tauopathies
Several TG models of tau pathology have been generated by overexpressing human tau proteins in mice [120,121]. However, these mice either were asymptomatic or developed pathology that was localized to the spinal cord and/or lacked many of the key features of tau-based disorders. In contrast, the introduction of the P301L mutation led to the development of TG mice that develop age- and gene dose-dependent accumulation of tau tangles in the brain and spinal cord with associated nerve cell loss and gliosis, as well as behavioural abnormalities [120,122]. Similar to human disease, the tau aggregates were composed of only mutant human tau, further implicating the P301L change in promoting the selective aggregation of mutant tau. Other systems were also developed to model various aspects of human tauopathies including a transgenic mouse overexpressing the shortest human tau isoform which acquired age-dependent tau pathology similar to that seen in FTDP-17 and ALS/PDC [123]. Overexpression of either wild-type or mutant tau (R406W and V337M) in Drosophila melanogaster demonstrated features of tauopathy including adult-onset progressive neurodegeneration with accumulation of abnormal tau [124]. However, the neurodegeneration occurred in the absence of NFT formation. More recent studies demonstrated NFT-like pathology when tau was co-expressed with shaggy, a homologue of glycogen synthase 3-kinase, an enzyme implicated in tau phosphorylation [125]. Neurodegeneration and defective neurotransmission have also been demonstrated in a tau TG Caenorhabditis elegans. In this model, pan-neuronal expression of normal and mutant tau resulted in altered behaviour, accumulation of insoluble phosphorylated tau, age-dependent loss of axons and neurons, and structural damage to axonal tracts [126]. Clearly, these models recapitulate various features of the tauopathies that will facilitate understanding of the molecular mechanisms underlying tau neurodegeneration.
Conclusions
The accumulation of filamentous neuronal IF and tau proteins are common features of a wide variety of sporadic and familial neurodegenerative disorders. These diseases are distinguished by the distinct topographic and cell type-specific distribution of inclusions. The biochemical and ultrastructural characteristics of the inclusions also reveal a significant phenotypic overlap. The discovery that multiple mutations in neuronal IF and tau genes lead to the abnormal protein aggregation demonstrates that neuronal IF and tau dysfunction are sufficient to produce neurodegenerative disease. Experimental evidence indicates that mutations lead to specific alterations in expression, function, and biochemistry of neuronal IF and tau proteins. The identification of additional gene mutations or polymorphisms at distinct genetic loci that either cause or are risk factors for disease will provide additional insights into disease pathogenesis. Taken together, these new insights will lead to the development of novel strategies for treatment and prevention.
Acknowledgments
Support for this work was provided by grants from the National Institute on Aging of the National Institutes of Health (AG-09215, AG-10124, AG-17586 to VM-YL and JQT), and from the Wellcome Trust (GR066166AIA) to NJC. VM-YL is the John H Ware, Third Professor of Alzheimer’s Research and JQT is the William Maul Measey–Truman G Schnabel, Jr, MD, Professor of Geriatric Medicine and Gerontology. We would also like to thank the members of the Center for Neurodegenerative Disease Research who contributed to the work, and the many patients studied and their families, for making the research reviewed here possible.
References
- 1.Lee K, Cleveland DW. Neuronal intermediate filaments. Annu Rev Neurosci. 1996;19:187–217. doi: 10.1146/annurev.ne.19.030196.001155. [DOI] [PubMed] [Google Scholar]
- 2.Lee VM-Y, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 3.Goedert M, Spillantini MG, Serpell LC, et al. From genetics to pathology: tau and alpha-synuclein assemblies in neurodegenerative diseases. Philos Trans R Soc London Ser B Biol Sci. 2001;356:213–227. doi: 10.1098/rstb.2000.0767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3:55–56. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 5.Al-Chalabi A, Miller CCJ. Neurofilaments and neurological disease. Bioessays. 2003;25:346–365. doi: 10.1002/bies.10251. [DOI] [PubMed] [Google Scholar]
- 6.Lariviere RC, Julien JP. Functions of intermediate filaments in neuronal development and disease. J Neurobiol. 2004;58:131–148. doi: 10.1002/neu.10270. [DOI] [PubMed] [Google Scholar]
- 7.Jordanova A, De Jonghe P, Boerkoel CF, et al. Mutations in the neurofilament light chain gene (NEFL) cause early onset severe Charcot–Marie–Tooth disease. Brain. 2003;126:590–597. doi: 10.1093/brain/awg059. [DOI] [PubMed] [Google Scholar]
- 8.De Jonghe P, Mersivanova I, Nelis E, et al. Further evidence that neurofilament light chain gene mutations can cause Charcot–Marie–Tooth disease type 2E. Ann Neurol. 2001;49:245–249. doi: 10.1002/1531-8249(20010201)49:2<245::aid-ana45>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 9.Yoshihara T, Yamamoto M, Hattori N, et al. Identification of novel sequence variants in the neurofilament-light gene in a Japanese population: analysis of Charcot–Marie–Tooth disease patients and normal individuals. J Peripher Nerv Syst. 2002;7:221–224. doi: 10.1046/j.1529-8027.2002.02028.x. [DOI] [PubMed] [Google Scholar]
- 10.Georgiou DM, Zidar J, Korosec M, Middleton LT, Kyriakides T, Christodoulou K. A novel NF-L mutation Pro22Ser is associated with CMT2 in a large Slovenian family. Neurogenetics. 2002;4:93–96. doi: 10.1007/s10048-002-0138-4. [DOI] [PubMed] [Google Scholar]
- 11.Fabrizi GM, Cavallaro T, Angiari C, et al. Giant axon and neurofilament accumulation in Charcot–Marie–Tooth disease type 2E. Neurology. 2004;62:1429–1431. doi: 10.1212/01.wnl.0000120664.07186.3c. [DOI] [PubMed] [Google Scholar]
- 12.Mersiyanova IV, Perepelov AV, Polyakov AV, et al. A new variant of Charcot–Marie–Tooth disease type 2 is probably the result of a mutation in the neurofilament-light gene. Am J Hum Genet. 2000;67:37–46. doi: 10.1086/302962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zuchner S, Vorgerd M, Sindern E, Schroder JM. The novel neurofilament light (NEFL) mutation Glu397Lys is associated with a clinically and morphologically heterogeneous type of Charcot–Marie–Tooth neuropathy. Neuromuscul Disord. 2004;14:147–157. doi: 10.1016/j.nmd.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 14.Lavedan C, Buchholtz S, Nussbaum RL, Albin RL, Polymeropoulos MH. A mutation in the human neurofilament M gene in Parkinson’s disease that suggests a role for the cytoskeleton in neuronal degeneration. Neurosci Lett. 2002;322:57–61. doi: 10.1016/s0304-3940(01)02513-7. [DOI] [PubMed] [Google Scholar]
- 15.Figlewicz DA, Krizus A, Martinoli MG, et al. Variants of the heavy neurofilament subunit are associated with the development of amyotrophic lateral sclerosis. Hum Mol Genet. 1994;3:1757–1761. doi: 10.1093/hmg/3.10.1757. [DOI] [PubMed] [Google Scholar]
- 16.Al-Chalabi A, Andersen PM, Nilsson P, et al. Deletions of the heavy neurofilament subunit tail in amyotrophic lateral sclerosis. Hum Mol Genet. 1999;8:157–164. doi: 10.1093/hmg/8.2.157. [DOI] [PubMed] [Google Scholar]
- 17.Tomkins J, Usher P, Slade JY, et al. Novel insertion in the KSP region of the neurofilament heavy gene in amyotrophic lateral sclerosis (ALS) Neuroreport. 1998;9:3967–3970. doi: 10.1097/00001756-199812010-00036. [DOI] [PubMed] [Google Scholar]
- 18.Hayashi S, Toyoshima Y, Hasegawa M, et al. Late-onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann Neurol. 2002;51:525–530. doi: 10.1002/ana.10163. [DOI] [PubMed] [Google Scholar]
- 19.Poorkaj P, Muma NA, Zhukareva V, et al. An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol. 2002;52:511–516. doi: 10.1002/ana.10340. [DOI] [PubMed] [Google Scholar]
- 20.Pickering-Brown S, Baker M, Yen SH, et al. Pick’s disease is associated with mutations in the tau gene. Ann Neurol. 2000;48:859–867. [PubMed] [Google Scholar]
- 21.Rizzini C, Goedert M, Hodges JR, et al. Tau gene mutation K257T causes a tauopathy similar to Pick’s disease. J Neuropathol Exp Neurol. 2000;59:990–1001. doi: 10.1093/jnen/59.11.990. [DOI] [PubMed] [Google Scholar]
- 22.Reed LA, Wszolek ZK, Hutton M. Phenotypic correlations in FTDP-17. Neurobiol Aging. 2001;22:89–107. doi: 10.1016/s0197-4580(00)00202-5. [DOI] [PubMed] [Google Scholar]
- 23.Kobayashi T, Ota S, Tanaka K, et al. A novel L266V mutation of the tau gene causes frontotemporal dementia with a unique tau pathology. Ann Neurol. 2003;53:133–137. doi: 10.1002/ana.10447. [DOI] [PubMed] [Google Scholar]
- 24.Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 25.Rizzu P, Van Swieten JC, Joosse M, et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in The Netherlands. Am J Hum Genet. 1999;64:414–421. doi: 10.1086/302256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clark LN, Poorkaj P, Wszolek Z, et al. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci U S A. 1998;95(13):13 103–13 107. doi: 10.1073/pnas.95.22.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D’Souza I, Poorkaj P, Hong M, et al. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism–chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci U S A. 1999;96:5598–5603. doi: 10.1073/pnas.96.10.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spillantini MG, Yoshida H, Rizzini C, et al. A novel tau mutation (N296N) in familial dementia with swollen achromatic neurons and corticobasal inclusion bodies. Ann Neurol. 2000;48:939–943. doi: 10.1002/1531-8249(200012)48:6<939::aid-ana17>3.3.co;2-t. [DOI] [PubMed] [Google Scholar]
- 29.Iseki E, Matsumura T, Marui W, et al. Familial frontotemporal dementia and parkinsonism with a novel N296H mutation in exon 10 of the tau gene and a widespread tau accumulation in glial cells. Acta Neuropathol. 2001;102:285–292. doi: 10.1007/s004010000333. [DOI] [PubMed] [Google Scholar]
- 30.Pastor P, Pastor E, Carnero C, et al. Familial atypical progressive supranuclear palsy associated with homozygosity for the delN296 mutation in the tau gene. Ann Neurol. 2001;49:263–267. doi: 10.1002/1531-8249(20010201)49:2<263::aid-ana50>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 31.Bugiani O, Murrell JR, Giaccone G, et al. Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J Neuropathol Exp Neurol. 1999;58:667–677. doi: 10.1097/00005072-199906000-00011. [DOI] [PubMed] [Google Scholar]
- 32.Sperfeld AD, Collatz MB, Baier H, et al. FTDP-17: an early-onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann Neurol. 1999;46:708–715. doi: 10.1002/1531-8249(199911)46:5<708::aid-ana5>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 33.Hasegawa M, Smith MJ, Iijima M, Tabira T, Goedert M. FTDP-17 mutations N279K and S305N in tau produce increased splicing of exon 10. FEBS Lett. 1999;443:93–96. doi: 10.1016/s0014-5793(98)01696-2. [DOI] [PubMed] [Google Scholar]
- 34.Iijima M, Tabira T, Poorkaj P, et al. A distinct familial presenile dementia with a novel missense mutation in the tau gene. Neuroreport. 1999;10:497–501. doi: 10.1097/00001756-199902250-00010. [DOI] [PubMed] [Google Scholar]
- 35.Stanford PM, Halliday GM, Brooks WS, et al. Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: expansion of the disease phenotype caused by tau gene mutations. Brain. 2000;123:880–893. doi: 10.1093/brain/123.5.880. [DOI] [PubMed] [Google Scholar]
- 36.Spillantini MG, Bird TD, Ghetti B. Frontotemporal dementia and parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol. 1998;8:387–402. doi: 10.1111/j.1750-3639.1998.tb00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyamoto K, Kowalska A, Hasegawa M, et al. Familial frontotemporal dementia and parkinsonism with a novel mutation at an intron 10 + 11-splice site in the tau gene. Ann Neurol. 2001;50:117–120. doi: 10.1002/ana.1083. [DOI] [PubMed] [Google Scholar]
- 38.Yasuda M, Takamatsu J, D’Souza I, et al. A novel mutation at position +12 in the intron following exon 10 of the tau gene in familial frontotemporal dementia (FTD-Kumamoto) Ann Neurol. 2000;47:422–429. [PubMed] [Google Scholar]
- 39.Lantos PL, Cairns NJ, Khan MN, et al. Neuropathologic variation in frontotemporal dementia due to the intronic tau 10 (+16) mutation. Neurology. 2002;58:1169–1175. doi: 10.1212/wnl.58.8.1169. [DOI] [PubMed] [Google Scholar]
- 40.van Herpen E, Rosso SM, Serverijnen LA, et al. Variable phenotypic expression and extensive tau pathology in two families with the novel tau mutation L315R. Ann Neurol. 2003;54:573–581. doi: 10.1002/ana.10721. [DOI] [PubMed] [Google Scholar]
- 41.Rosso SM, van Herpen E, Deelen W, et al. A novel tau mutation, S320F, causes a tauopathy with inclusions similar to those in Pick’s disease. Ann Neurol. 2002;51:373–376. doi: 10.1002/ana.10140. [DOI] [PubMed] [Google Scholar]
- 42.Pickering-Brown SM, Baker M, Nonaka T, et al. Frontotemporal dementia with Pick-type histology associated with Q336R mutation in the tau gene. Brain. 2004;127:1415–1426. doi: 10.1093/brain/awh147. [DOI] [PubMed] [Google Scholar]
- 43.Poorkaj P, Bird TD, Wijsman E, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 44.Lippa CF, Zhukareva V, Kawarai T, et al. Frontotemporal dementia with novel tau pathology and a Glu342Val tau mutation. Ann Neurol. 2000;48:850–858. [PubMed] [Google Scholar]
- 45.Nicholl DJ, Greenstone MA, Clarke CE, et al. An English kindred with a novel recessive tauopathy and respiratory failure. Ann Neurol. 2003;54:682–686. doi: 10.1002/ana.10747. [DOI] [PubMed] [Google Scholar]
- 46.Neumann M, Schulz-Schaeffer W, Crowther RA, et al. Pick’s disease associated with the novel tau gene mutation K369I. Ann Neurol. 2001;50:503–513. doi: 10.1002/ana.1223. [DOI] [PubMed] [Google Scholar]
- 47.Murrell JR, Spillantini MG, Zolo P, et al. Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J Neuropathol Exp Neurol. 1999;58:1207–1226. doi: 10.1097/00005072-199912000-00002. [DOI] [PubMed] [Google Scholar]
- 48.Ching GY, Liem RKH. Structure of the gene for the neuronal intermediate filament protein α-internexin and functional analysis of its promoter. J Biol Chem. 1991;266:19 459–19 468. [PubMed] [Google Scholar]
- 49.Schmidt ML, Carden MJ, Lee VM-Y, Trojanowski JQ. Phosphate dependent and independent neurofilament epitopes in the axonal swellings of patients with motor neuron disease and controls. Lab Invest. 1987;56:282–294. [PubMed] [Google Scholar]
- 50.Ching GY, Liem RKH. Roles of head and tail domains in α-internexin’s self-assembly and coassembly with the neurofilament triplet proteins. J Cell Science. 1998;111:321–333. doi: 10.1242/jcs.111.3.321. [DOI] [PubMed] [Google Scholar]
- 51.Lee VM-Y, Carden MJ, Schlaepfer WW, Trojanowski JQ. Monoclonal antibodies distinguish several differentially phosphorylated states of the two largest rat neurofilament subunits (NF-H and NF-M) and demonstrate their existence in the normal nervous system of adult rats. J Neurosci. 1987;7:3478–3488. doi: 10.1523/JNEUROSCI.07-11-03474.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Carden MJ, Trojanowski JQ, Schlaepfer WW, Lee VM-Y. Two-stage expression of neurofilament polypeptides during rat neurogenesis with early establishment of adult phosphorylation patterns. J Neurosci. 1987;7:3499–3504. doi: 10.1523/JNEUROSCI.07-11-03489.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hill WD, Lee VM-Y, Hurtig HI, Murray JM, Trojanowski JQ. Epitopes located in spatially separate domains of each neurofilament subunit are present in Parkinson’s disease Lewy body. J Comp Neurol. 1991;309:150–160. doi: 10.1002/cne.903090111. [DOI] [PubMed] [Google Scholar]
- 54.Schmidt ML, Murray J, Lee VM-Y, Hill WD, Wertkin A, Trojanowski JQ. Epitope map of neurofilament protein domains in cortical and peripheral nervous system Lewy bodies. Am J Pathol. 1991;139:53–65. [PMC free article] [PubMed] [Google Scholar]
- 55.Manetto V, Sternberger NH, Perry G, Sternberger LA, Gambetti P. Phosphorylation of neurofilaments is altered in amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 1988;47:642–653. doi: 10.1097/00005072-198811000-00007. [DOI] [PubMed] [Google Scholar]
- 56.Leigh PN, Dodson A, Swash M, Brion JP, Anderton BH. Cytoskeletal abnormalities in motor neuron disease: an immunohistochemical study. Brain. 1989;112:521–535. doi: 10.1093/brain/112.2.521. [DOI] [PubMed] [Google Scholar]
- 57.Goldstein E, Sternberger NM, Sternberger LA. Phosphorylation protects neurofilaments against proteolysis. J Neuroimmunol. 1987;14:149–160. doi: 10.1016/0165-5728(87)90049-x. [DOI] [PubMed] [Google Scholar]
- 58.Fliegner KH, Kaplan MP, Wood TL, Pintar JE, Liem RK. Expression of the gene for the neuronal intermediate filament protein α-internexin coincides with the onset of neuronal differentiation in the developing rat nervous system. J Comp Neurol. 1994;342:161–173. doi: 10.1002/cne.903420202. [DOI] [PubMed] [Google Scholar]
- 59.Cairns NJ, Zhukareva V, Uryu K, et al. α-Internexin is present in the pathological inclusions of neuronal intermediate filament inclusion disease. Am J Pathol. 2004;164:2153–2161. doi: 10.1016/s0002-9440(10)63773-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cairns NJ, Uryu K, Bigio E, et al. α-Internexin aggregates are abundant in neuronal intermediate filament inclusion disease (NIFID) but rare in other neurodegenerative diseases. Acta Neuropathol. 2004;108:213–223. doi: 10.1007/s00401-004-0882-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Josephs KA, Holton JL, Rossor MN, et al. Neurofilament inclusion body disease: a new proteinopathy? Brain. 2003;126:2291–2303. doi: 10.1093/brain/awg231. [DOI] [PubMed] [Google Scholar]
- 62.He CZ, Hays AP. Expression of peripherin in ubiquinated inclusions of amyotrophic lateral sclerosis. J Neurol Sci. 2004;217:47–54. doi: 10.1016/j.jns.2003.08.016. [DOI] [PubMed] [Google Scholar]
- 63.Strong MJ, Strong WL, Jaffe H, Traggert B, Sopper MM, Pant HC. Phosphorylation state of the native high-molecular-weight neurofilament subunit protein from cervical spinal cord in sporadic amyotrophic lateral sclerosis. J Neurochem. 2001;76:1315–1325. doi: 10.1046/j.1471-4159.2001.00094.x. [DOI] [PubMed] [Google Scholar]
- 64.Perez-Olle R, Leung CL, Liem RK. Effects of Charcot–Marie–Tooth-linked mutations of the neurofilament light subunit on intermediate filament formation. J Cell Sci. 2002;115:4937–4946. doi: 10.1242/jcs.00148. [DOI] [PubMed] [Google Scholar]
- 65.Galvin JE, Nakamura M, McIntosh TK, et al. Neurofilament-rich intraneuronal inclusions exacerbate neurodegenerative sequelae of brain trauma in NFH/LacZ transgenic mice. Exp Neurol. 2000;165:77–89. doi: 10.1006/exnr.2000.7461. [DOI] [PubMed] [Google Scholar]
- 66.Ching GY, Chien C-L, Flores R, Liem RKH. Overexpression of α-internexin causes abnormal neurofilamentous accumulations and motor coordination deficits in transgenic mice. J Neurosci. 1999;19:2974–2986. doi: 10.1523/JNEUROSCI.19-08-02974.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee MK, Marszalek JR, Cleveland DW. A mutant neurofilament subunit causes massive, selective motor neuron death: implications for the pathogenesis of human motor neuron disease. Neuron. 1994;13:975–988. doi: 10.1016/0896-6273(94)90263-1. [DOI] [PubMed] [Google Scholar]
- 68.Canete-Soler R, Silberg DG, Gershon MD, Schlaepfer WW. Mutation in neurofilament transgene implicates RNA processing in the pathogenesis of neurodegenerative disease. J Neurosci. 1999;19:1273–1283. doi: 10.1523/JNEUROSCI.19-04-01273.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beaulieu JM, Julien JP. Peripherin-mediated death of motor neurons rescued by overexpression of neurofilament NF-H proteins. J Neurochem. 2003;85:248–256. doi: 10.1046/j.1471-4159.2003.01653.x. [DOI] [PubMed] [Google Scholar]
- 70.Robertson J, Doroudchi MM, Nguyen MD, et al. A neurotoxic peripherin splice variant in a mouse model of ALS. J Cell Biol. 2003;160:939–949. doi: 10.1083/jcb.200205027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Foster NL, Wilhelmsen K, Sima AA, Jones MZ, D’Amato CJ, Gilman S. Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Ann Neurol. 1997;41:706–715. doi: 10.1002/ana.410410606. [DOI] [PubMed] [Google Scholar]
- 72.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cleveland DW, Hwo SY, Kirschner MW. Purification of tau, a microtubule-associated protein that induces assembly of microtubules from purified tubulin. J Mol Biol. 1977;116:207–225. doi: 10.1016/0022-2836(77)90213-3. [DOI] [PubMed] [Google Scholar]
- 74.Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101:1371–1378. doi: 10.1083/jcb.101.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shin RW, Iwaki T, Kitamoto T, Tateishi J. Hydrated autoclave pretreatment enhances tau immunoreactivity in formalin-fixed normal and Alzheimer’s disease brain tissues. Lab Invest. 1991;64:693–702. [PubMed] [Google Scholar]
- 76.LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP, Binder LI. Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. Proc Natl Acad Sci U S A. 1995;92:10 369–10 373. doi: 10.1073/pnas.92.22.10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Couchie D, Mavilia C, Georgieff IS, Liem RK, Shelanski ML, Nunez J. Primary structure of high molecular weight tau present in the peripheral nervous system. Proc Natl Acad Sci U S A. 1992;89:4378–4381. doi: 10.1073/pnas.89.10.4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Neve RL, Harris P, Kosik KS, Kurnit DM, Donlon TA. Identification of cDNA clones for the human microtubule-associated protein tau and chromosomal localization of the genes for tau and microtubule-associated protein 2. Brain Res. 1986;387:271–280. doi: 10.1016/0169-328x(86)90033-1. [DOI] [PubMed] [Google Scholar]
- 79.Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A. 1988;85:4051–4055. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Andreadis A, Brown WM, Kosik KS. Structure and novel exons of the human tau gene. Biochemistry. 1992;31:10 626–10 633. doi: 10.1021/bi00158a027. [DOI] [PubMed] [Google Scholar]
- 81.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 82.Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 1989;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9:4225–4230. doi: 10.1002/j.1460-2075.1990.tb07870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 85.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Himmler A, Drechsel D, Kirschner MW, Martin DW., Jr Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol Cell Biol. 1989;9:1381–1388. doi: 10.1128/mcb.9.4.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lee G, Neve RL, Kosik KS. The microtubule binding domain of tau protein. Neuron. 1989;2:1615–1624. doi: 10.1016/0896-6273(89)90050-0. [DOI] [PubMed] [Google Scholar]
- 88.Butner KA, Kirschner MW. Tau protein binds to microtubules through a flexible array of distributed weak sites. J Cell Biol. 1991;115:717–730. doi: 10.1083/jcb.115.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Drechsel DN, Hyman AA, Cobb MH, Kirschner MW. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol Biol Cell. 1992;3:1141–1154. doi: 10.1091/mbc.3.10.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yoshida H, Ihara Y. Tau in paired helical filaments is functionally distinct from fetal tau: assembly incompetence of paired helical filament-tau. J Neurochem. 1993;61:1183–1186. doi: 10.1111/j.1471-4159.1993.tb03642.x. [DOI] [PubMed] [Google Scholar]
- 91.Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron. 1993;10:1089–1099. doi: 10.1016/0896-6273(93)90057-x. [DOI] [PubMed] [Google Scholar]
- 92.Biernat J, Gustke N, Drewes G, Mandelkow EM, Mandelkow E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: distinction between PHF-like immunoreactivity and microtubule binding. Neuron. 1993;11:153–163. doi: 10.1016/0896-6273(93)90279-z. [DOI] [PubMed] [Google Scholar]
- 93.Billingsley ML, Kincaid RL. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J. 1997;323:577–591. doi: 10.1042/bj3230577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:1–36. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 95.Hong M, Trojanowski JQ, Lee VM-Y. Tau-based neurofibrillary lesions. In: Clark CM, Trojanowski JQ, editors. Neurodegenerative Dementias. McGraw-Hill; New York: 2000. pp. 161–175. [Google Scholar]
- 96.Hasegawa M, Jakes R, Crowther RA, Lee VM, Ihara Y, Goedert M. Characterization of mAb AP422, a novel phosphorylation-dependent monoclonal antibody against tau protein. FEBS Lett. 1996;384:25–30. doi: 10.1016/0014-5793(96)00271-2. [DOI] [PubMed] [Google Scholar]
- 97.Hoffmann R, Lee VM-Y, Leight S, Varga I, Otvos L., Jr Unique Alzheimer’s disease paired helical filament specific epitopes involve double phosphorylation at specific sites. Biochemistry. 1997;36:8114–8124. doi: 10.1021/bi970380+. [DOI] [PubMed] [Google Scholar]
- 98.Zheng-Fischhofer Q, Biernat J, Mandelkow EM, Illenberger S, Godemann R, Mandelkow E. Sequential phosphorylation of tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur J Biochem. 1998;252:542–552. doi: 10.1046/j.1432-1327.1998.2520542.x. [DOI] [PubMed] [Google Scholar]
- 99.Yasuda M, Kawamata T, Komure O, et al. A mutation in the microtubule-associated protein tau in pallido-nigro-luysian degeneration. Neurology. 1999;53:864–868. doi: 10.1212/wnl.53.4.864. [DOI] [PubMed] [Google Scholar]
- 100.Delisle MB, Murrell JR, Richardson R, et al. A mutation at codon 279 (N279K) in exon 10 of the Tau gene causes a tauopathy with dementia and supranuclear palsy. Acta Neuropathol. 1999;98:62–77. doi: 10.1007/s004010051052. [DOI] [PubMed] [Google Scholar]
- 101.Varani L, Hasegawa M, Spillantini MG, et al. Structure of tau exon 10 splicing regulatory element RNA and destabilization by mutations of frontotemporal dementia and parkinsonism linked to chromosome 17. Proc Natl Acad Sci U S A. 1999;96:8229–8234. doi: 10.1073/pnas.96.14.8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Grover A, Houlden H, Baker M, et al. 5′ splice site mutations in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure that regulates alternative splicing of exon 10. J Biol Chem. 1999;274:15 134–15 143. doi: 10.1074/jbc.274.21.15134. [DOI] [PubMed] [Google Scholar]
- 103.D’Souza I, Schellenberg D. Determinants of 4 repeat tau expression: coordination between enhancing and inhibitory splicing sequences for exon 10 inclusion. J Biol Chem. 2000;275:17 700–17 709. doi: 10.1074/jbc.M909470199. [DOI] [PubMed] [Google Scholar]
- 104.Gao QS, Memmott J, Lafyatis R, Stamm S, Screaton G, Andreadis A. Complex regulation of tau exon 10, whose missplicing causes frontotemporal dementia. J Neurochem. 2000;74:490–500. doi: 10.1046/j.1471-4159.2000.740490.x. [DOI] [PubMed] [Google Scholar]
- 105.Grover A, DeTure M, Yen SH, Hutton M. Effects on splicing and protein function of three mutations in codon N296 of tau in vitro. Neurosci Lett. 2002;323:33–36. doi: 10.1016/s0304-3940(02)00124-6. [DOI] [PubMed] [Google Scholar]
- 106.Hulette CM, Pericak-Vance MA, Roses AD, et al. Neuropathological features of frontotemporal dementia and parkinsonism linked to chromosome 17q21–22 (FTDP-17): Duke family 1684. J Neuropathol Exp Neurol. 1999;58:859–866. doi: 10.1097/00005072-199908000-00008. [DOI] [PubMed] [Google Scholar]
- 107.Goedert M, Spillantini MG, Crowther RA, et al. Tau gene mutation in familial progressive subcortical gliosis. Nature Med. 1999;5:454–457. doi: 10.1038/7454. [DOI] [PubMed] [Google Scholar]
- 108.Arima K, Kowalska A, Hasegawa M, et al. Two brothers with frontotemporal dementia and parkinsonism with an N279K mutation of the tau gene. Neurology. 2000;54:1787–1795. doi: 10.1212/wnl.54.9.1787. [DOI] [PubMed] [Google Scholar]
- 109.Goode BL, Feinstein SC. Identification of a novel microtubule binding and assembly domain in the developmentally regulated inter-repeat region of tau. J Cell Biol. 1994;124:769–782. doi: 10.1083/jcb.124.5.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Goode BL, Denis PE, Panda D, et al. Functional interactions between the proline-rich and repeat regions of tau enhance microtubule binding and assembly. Mol Biol Cell. 1997;8:353–365. doi: 10.1091/mbc.8.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hasegawa M, Smith MJ, Goedert M. Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett. 1998;437:207–210. doi: 10.1016/s0014-5793(98)01217-4. [DOI] [PubMed] [Google Scholar]
- 112.Barghorn S, Zheng-Fischhofer Q, Ackmann M, et al. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry. 2000;39:11 714–11 721. doi: 10.1021/bi000850r. [DOI] [PubMed] [Google Scholar]
- 113.Rizzu P, Joosse M, Ravid R, et al. Mutation-dependent aggregation of tau protein and its selective depletion from the soluble fraction in brain of P301L FTDP-17 patients. Hum Mol Genet. 2000;9:3075–3082. doi: 10.1093/hmg/9.20.3075. [DOI] [PubMed] [Google Scholar]
- 114.Miyasaka T, Morishima-Kawashima M, Ravid R, et al. Selective deposition of mutant tau in the FTDP-17 brain affected by the P301L mutation. J Neuropathol Exp Neurol. 2001;60:872–884. doi: 10.1093/jnen/60.9.872. [DOI] [PubMed] [Google Scholar]
- 115.Arrasate M, Perez M, Armas-Portela R, Avila J. Polymerization of tau peptides into fibrillar structures. The effect of FTDP-17 mutations. FEBS Lett. 1999;446:199–202. doi: 10.1016/s0014-5793(99)00210-0. [DOI] [PubMed] [Google Scholar]
- 116.Nacharaju P, Lewis J, Easson C, et al. Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. FEBS Lett. 1999;447:195–199. doi: 10.1016/s0014-5793(99)00294-x. [DOI] [PubMed] [Google Scholar]
- 117.Goedert M, Jakes R, Crowther RA. Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of tau filaments. FEBS Lett. 1999;450:306–311. doi: 10.1016/s0014-5793(99)00508-6. [DOI] [PubMed] [Google Scholar]
- 118.Gamblin TC, King ME, Dawson H, et al. In vitro polymerization of tau protein monitored by laser light scattering: method and application to the study of FTDP-17 mutants. Biochemistry. 2000;39:6136–6144. doi: 10.1021/bi000201f. [DOI] [PubMed] [Google Scholar]
- 119.Goedert M, Satumtira S, Jakes R, et al. Reduced binding of protein phosphatase 2A to tau protein with frontotemporal dementia and parkinsonism linked to chromosome 17 mutations. J Neurochem. 2000;75:2155–2162. doi: 10.1046/j.1471-4159.2000.0752155.x. [DOI] [PubMed] [Google Scholar]
- 120.Götz J, Chen F, Barmettler R, Nitsch RM. Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem. 2001;276:529–534. doi: 10.1074/jbc.M006531200. [DOI] [PubMed] [Google Scholar]
- 121.Götz J. Tau and transgenic animal models. Brain Res Rev. 2001;35:266–286. doi: 10.1016/s0165-0173(01)00055-8. [DOI] [PubMed] [Google Scholar]
- 122.Lewis J, McGowan E, Rockwood J, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nature Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 123.Ishihara T, Hong M, Zhang B, et al. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- 124.Wittmann CW, Wszolek MF, Shulman JM, et al. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 125.Jackson GR, Wiedau-Pazos M, Sang TK, et al. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 2002;34:509–519. doi: 10.1016/s0896-6273(02)00706-7. [DOI] [PubMed] [Google Scholar]
- 126.Kraemer BC, Zhang B, Leverenz JB, Thomas JH, Trojanowski JQ, Schellenberg GD. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci U S A. 2003;100:9980–9985. doi: 10.1073/pnas.1533448100. [DOI] [PMC free article] [PubMed] [Google Scholar]