

Abstract
Rationale
Autophagy, a bulk degradation process of cytosolic proteins and organelles, is protective during nutrient starvation in cardiomyocytes. However, the underlying signaling mechanism mediating autophagy is not well understood.
Objective
We investigated the role of FoxOs and its posttranslational modification in mediating starvation-induced autophagy.
Methods and Results
Glucose deprivation (GD) increased autophagic flux in cultured CMs, as evidenced by increased mRFP-GFP-LC3 puncta and decreases in p62, which was accompanied by upregulation of Sirt1 and FoxO1. Overexpression of either Sirt1 or FoxO1 was sufficient for inducing autophagic flux, whereas both Sirt1 and FoxO1 were required for GD-induced autophagy. GD increased deacetylation of FoxO1, and Sirt1 was required for GD-induced deacetylation of FoxO1. Overexpression of FoxO1(3A/LXXAA), which cannot interact with Sirt1, or p300, a histone acetylase, increased acetylation of FoxO1 and inhibited GD-induced autophagy. FoxO1 increased expression of Rab7, a small GTP- binding protein that mediates late autophagosome-lysosome fusion, which was both necessary and sufficient for mediating FoxO1-induced increases in autophagic flux. Although cardiac function was maintained in control mice after 48 hours of food starvation, it was significantly deteriorated in mice with cardiac specific overexpression of FoxO1(3A/LXXAA), those with cardiac specific homozygous deletion of FoxO1 (c-FoxO1−/−), and beclin1+/− mice, in which autophagy is significantly inhibited.
Conclusions
These results suggest that Sirt1-mediated deacetylation of FoxO1 and upregulation of Rab7 play an important role in mediating starvation-induced increases in autophagic flux, which in turn plays an essential role in maintaining left ventricular function during starvation.
Keywords: Autophagy, starvation, FoxO, Sirt1, Rab7, deacetylation
Introduction
Macroautophagy (hereafter autophagy) is a dynamic process of intracellular bulk degradation in which cytosolic proteins and organelles are sequestered into double membrane vesicles called autophagosomes, to be fused with lysosomes for degradation. 1 In the heart, autophagy maintains protein quality control, adapts to nutrient and oxygen deprivation during myocardial ischemia, and mediates cell death during reperfusion injury. 2, 3 Autophagy during nutrient deprivation is an adaptive mechanism which allows the cells to survive by degrading the intracellular protein and lipid cargo and recycling the amino and fatty acids to generate ATP. 2 The nutrient status has a profound effect upon cardiac contractility, and activation of autophagy during starvation is protective for the heart. The intracellular signaling mechanism by which nutrient starvation activates autophagy in cardiomyocytes (CMs) is not well understood, however.
The forkhead box, class O (FoxO) family of transcription factors are present as four distinct isoforms (FoxO1, FoxO3, FoxO4 and FoxO6) in mammals. FoxO proteins play an important role in several intracellular functions, such as metabolism, stress resistance, longevity, tumor suppression and cell size regulation. 4 The key to the myriad functions of FoxO proteins lies in the complex post-translational modifications they undergo. They are phosphorylated in response to insulin and growth factors, dephosphorylated by protein phosphatases, ubiquitinated in response to oxidative stress, acetylated by p300/CBP and deacetylated by Sirt1.4 FoxO3 and FoxO1 regulate autophagy in skeletal and cardiac muscles by activating genes that are involved in autophagosome formation. 5–7 However, the specific role of FoxO post-translational modification in mediating autophagy is not yet known.
Sirt1, the mammalian ortholog of yeast Sir2 (silent information regulator), is a class III histone deacetylase. Sirt1 extends lifespan in lower organisms, whereas it suppresses aging-induced cardiomyopathy and protects against oxidative stress in the mouse heart. 8 Sirt1 regulates autophagy by interacting with autophagy related genes Atg5, Atg7, and Atg8 and deacetylating them. 9 We have shown previously that endogenous Sirt1 positively regulates autophagy in CMs. 10 Sirt1 deacetylates FoxO1, FoxO3 and FoxO4, and regulates FoxO dependent gene transcription either positively or negatively. 11, 12 FoxO proteins bear a conserved coactivator-interacting LXXLL motif that is important for their interaction with Sirt1. 13 However, whether or not the effect of Sirt1 upon autophagy is mediated through FoxO proteins is unknown.
In this study, we used activation of autophagy by glucose deprivation (GD) in cultured CMs as a model, and investigated the signaling mechanism mediating autophagy. Goals of this study were to 1) elucidate the role of FoxO1 in mediating starvation-induced increases in autophagy, 2) evaluate the role of FoxO1 deacetylation by Sirt1 in mediating starvation-induced autophagy, and 3) clarify the molecular mechanism by which FoxO1 can enhance autophagic flux.
Methods
An expanded Methods section is available in the Online Data Supplement at http://circres.ahajournals.org.
Adeno- and Lentiviruses
Adenoviruses harboring murine wildtype (WT) FoxO1 (Ad-FoxO1-WT), FoxO1(3A/LXXAA) (Ad-3A/LXXAA) 13, Sirt1 (Ad-Sirt1) 8, shRNA Sirt1 (Ad-sh-Sirt1) 10, shRNA Scramble (Ad-sh-Scr)10, GFP-LC3 (Ad-GFP-LC3) 2, tandem fluorescent mRFP-GFP-LC3 14 (Ad-tf-LC3) 15, tTA (Ad-tTA) 16, and LacZ (Ad-LacZ) 16 have been described. Adenovirus harboring inducible p300 (Ad-p300) was purchased from Cell Biolabs. The plasmid constructs of hemagglutinin (HA) tagged human Rab7 (obtained from Missouri S&T cDNA Resource Center) and HA tagged WT FoxO3a (from Dr. Michael Greenberg) 17 were used to generate adenoviruses Ad-HA-Rab7 and Ad-FoxO3-WT, respectively, using the Admax system (Microbix). Adenovirus harboring shRNA for FoxO1 (Ad-sh-FoxO1) was generated as previously described 2 using the following hairpin forming oligo 5’ – CGCCAAACTCACTACACCATTTCAAGAGAATGGTGTAGTGAGTTTGGCTTTTTA – 3’. The hairpin loop sequence is underlined. Adenovirus harboring control scramble shRNA (Ad-sh-Scr) has been described. 10 Lentivirus (Lt) harboring sh-Rab7 (Lt-sh-Rab7) was purchased from Open Biosystems. For knockdown studies, adenoviral and lentiviral transduction was carried out for 96 hours, while overexpression models involved 24 hours of adenoviral transduction.
Transgenic Mice
Transgenic mice with cardiac specific over-expression of FoxO1(3A/LXXAA) (Tg-FoxO1m) were generated on an FVB background with the α-MHC promoter (courtesy, Dr. J Robbins, Children’s Hospital, Cincinnati). FoxO1 homozygous floxed mice (flox) 18 were bred with transgenic mice with cardiac specific overexpression of Cre recombinase (αMHC-Cre-Tg) (courtesy, Dr. M.D. Schneider, Imperial College, London) to generate cardiac specific FoxO1 homozygous knockout mice (c-FoxO1−/−). Beclin1 heterozygous knockout mice (beclin1+/−) have been described. 2
Results
GD increases autophagic flux in cultured CMs
Cultured CMs were subjected to GD for 2 hours. As previously shown 2, GD induced a significant increase in the expression of LC3-II (Fig. 1AB). When CMs were transduced with Ad-GFP-LC3, GD significantly increased the number of CMs with numerous GFP-LC3 dots (Online Figure I-A). Expression of p62, a protein sequestered in autophagosomes for lysosomal degradation was significantly reduced (Fig. 1AB). To separately evaluate the extent of autophagosome and autolysosome accumulation, we generated an adenovirus harboring tandem fluorescent mRFP-GFP-LC3 (Ad-tf-LC3).14, 15 mRFP retains its fluorescence even in the acidic environment of lysosomes where GFP loses its fluorescence. Thus, green LC3 puncta primarily indicate autophagosomes, while red LC3 puncta indicate both autophagosomes and autolysosomes. The red puncta that overlay with the green ones and appear yellow in merged images are indicators of autophagosomes, while the free red puncta that do not overlay with the green ones and appear red in merged images are indicative of autolysosomes. 14, 15 The numbers of green and red puncta were both significantly higher after GD (Fig. 1CD). Furthermore, yellow and red puncta were both significantly increased after GD in merged images, indicating increased autophagosomes and autolysosomes (Fig. 1E). These results suggest that autophagic flux is enhanced in response to GD in CMs.
Figure 1. GD induces autophagy in cultured CMs with enhanced FoxO1 expression.

Neonatal rat CMs were transduced with Ad-LacZ and incubated in serum free media or treated with serum free-glucose free media. A) Immunoblot analyses.B) Densitometric analyses. C-E) CMs were transduced with Ad-tf-LC3 and Ad-LacZ and treated with glucose free media. C) Representative images of fluorescent LC3 puncta. Arrows indicate red puncta that overlay with green puncta indicating autophagosomes. D) Mean number of green and red puncta per cell. E) Mean number of autophagosomes (puncta with both red and green colors, i.e puncta with yellow color in merged images) and autolysosomes (puncta with only red but not green color, i.e puncta with red color in merged images) per cell. Graph with error bars is shown in Online Figure XI-A. Results represent the means from at least 4 independent experiments. * p<0.05, ** p<0.01.
FoxO1 is required for GD-induced autophagy
FoxO1, a major isoform of the FoxO family in CMs, was significantly up-regulated following GD (Fig. 1AB). Overexpression of FoxO1 by Ad-FoxO1-WT caused a significant increase in LC3-II and a decrease in p62, indicating increased autophagy (Fig. 2AB), whereas knockdown of FoxO1 by Ad-sh-FoxO1 caused significant accumulation of both LC3-II and p62 indicating inhibition of autophagic flux in CMs (Fig. 2AB). To further evaluate the effect of FoxO1 upon autophagic flux, CMs were transduced with Ad-tf-LC3. Expression of FoxO1 significantly increased both green and red puncta compared to that of LacZ (Fig. 2C and Online Figure I-B). Merged images showed that the increase in red puncta was significantly greater than that in yellow puncta (Fig. 2D). These results suggest that FoxO1 increases autolysosomes more than autophagosomes, and, thus, stimulates autophagic flux. In addition, there was a significant increase in the FoxO1-induced accumulation of LC3-II in the presence of lysosomal protease inhibitors (PI), including phenyl methane sulfonyl fluoride and leupeptin, further supporting the idea that FoxO1 stimulates autophagic flux in CMs (Online Figure I-CD). FoxO1 also increased the number of yellow puncta in merged images in the presence of Bafilomycin A1, an inhibitor of autophagosome-lysosome fusion 19 (Online Figure I-E-G), suggesting that FoxO1 increases autophagosome formation as well. In Ad-sh-FoxO1 transduced CMs, although the number of green and red puncta was robustly increased compared to Ad-FoxO1-WT (Online Figure I-B), most of the red puncta overlaid with the green ones (Fig. 2CD), suggesting that knockdown of FoxO1 inhibits autophagic flux. Similar to FoxO1, overexpression of FoxO3 also increased autophagic flux (Online Figure II).
Figure 2. FoxO1 is required for GD-induced autophagy in CMs.


CMs were transduced with Ad-FoxO1-WT or Ad-LacZ, Ad-sh-FoxO1 or Ad-sh-Scr. A) Immunoblot analyses showing FoxO1, LC3, p62 and Tubulin expressions. B) Densitometric analyses. C) Representative images of fluorescent LC3 puncta after Ad-tf-LC3 transduction. Insets in Ad-FoxO1-WT transduced myocytes show higher magnification. D) Mean number of autophagosomes represented by yellow puncta in merged images and autolysosomes represented by red puncta in merged images per cell. E–H) CMs were transduced with Ad-sh-FoxO1 or Ad-sh-Scr and treated with glucose free media. E) Immunoblots showing p62, FoxO1 and Tubulin expressions. F) Densitometric analyses. G) Representative images of fluorescent LC3 puncta after Ad-tf-LC3 transduction. H) Mean number of autophagosomes represented by yellow puncta in merged images and autolysosomes represented by red puncta in merged images per cell. In D and H, graph with error bars is shown in Online Figure XI-BC. Results represent means from at least 4 independent experiments. * p<0.05, ** p<0.01, N.S Not Significant.
To evaluate whether FoxO1 is required for GD-induced autophagy, CMs were transduced with Ad-sh-FoxO1 (Fig. 2E). Knockdown of FoxO1 with Ad-sh-FoxO1 induced p62 accumulation in the presence and absence of GD, indicating that endogenous FoxO1 is required for GD-induced autophagy (Fig. 2EF). Analyses with Ad-tf-LC3 showed that upon knockdown of FoxO1, even in the presence of GD, most of the red puncta overlaid with green puncta, indicating inhibition of autophagic flux (Fig. 2GH). These results suggest that FoxO1 is required for GD-induced autophagy.
Sirt1 deacetylates FoxO1 during GD
Sirt1 is activated upon starvation in cancer cell lines. 20 We hypothesized that Sirt1 deacetylates FoxO1 during GD, thereby inducing autophagy in CMs. An acetylated form of FoxO1, evaluated with anti-acetylated FoxO1 antibody, was significantly decreased following GD (Fig. 3AB). Conversely, expression of Sirt1 was increased following GD (Fig. 3AB). Since the activity of Sirt1 is NAD+-dependent, we evaluated NAD+ content in starved CMs. The NAD+ content was significantly increased following GD (Fig. 3C). GD-induced deacetylation of FoxO1 was abolished when Sirt1 was downregulated by Ad-sh-Sirt1 (Fig. 3DE). Taken together, these results suggest that GD upregulates and activates Sirt1, which in turn induces deacetylation of FoxO1.
Figure 3. Sirt1 deacetylates FoxO1 during GD in CMs.
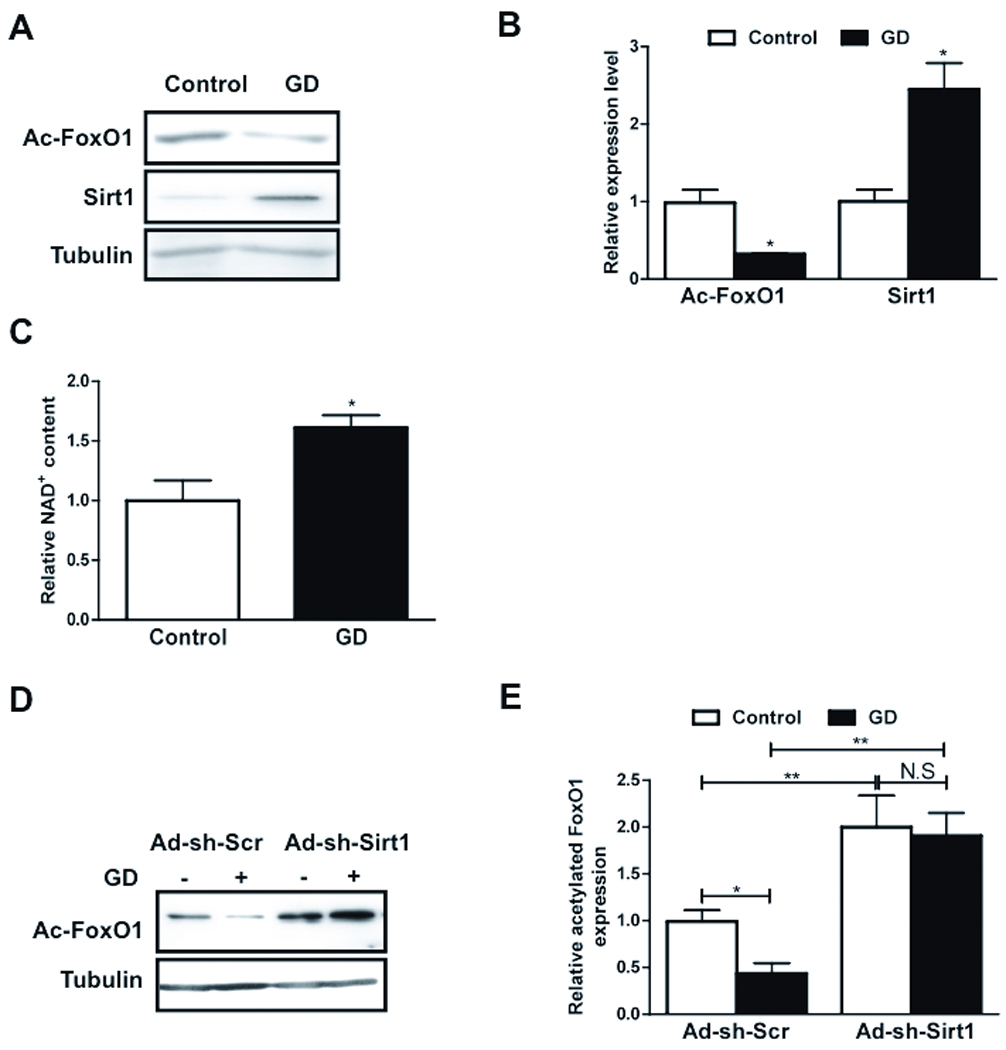
A–C) CMs were transduced with Ad-LacZ and treated with glucose free media. A) Immunoblots showing acetylated FoxO1 (Ac-FoxO1), Sirt1 and Tubulin expressions. B) Densitometric analyses. C) Relative NAD+ content is shown. D–E) Myocytes were transduced with Ad-sh-Sirt1 or Ad-sh-Scr and were glucose starved. D) Immunoblots showing Ac-FoxO1 and Tubulin expressions. E) Densitometric analyses. Data represent means from at least 4 independent experiments. * p<0.05, ** p<0.01, N.S Not significant.
Sirt1 is required for GD-induced autophagy
To evaluate the role of Sirt1 in mediating autophagy, CMs were transduced with Ad-Sirt1 and Ad-sh-Sirt1. Overexpression of Sirt1 increased LC3-II and decreased p62 accumulation, suggesting that Sirt1 stimulates autophagy (Fig. 4AB). Knockdown of Sirt1 inhibited autophagic flux, as evidenced by increased accumulation of both LC3-II and p62 at baseline, consistent with our previous results 10 (Fig. 4AB). Analyses with tf-LC3 showed that the number of green and red puncta was higher in Ad-Sirt1 transduced CMs than in Ad-LacZ transduced CMs (Fig. 4C and Online Figure III-A). Furthermore, there were more red puncta than yellow puncta, indicating that Sirt1 increases autolysosome formation more strongly than autophagosome formation (Fig. 4D). In Ad-Sirt1 transduced CMs, LC3-II accumulation was significantly enhanced in the presence of PI, indicating that Sirt1 enhances autophagic flux in CMs (Online Figure III-BC). Ad-Sirt1 significantly enhanced accumulation of yellow puncta in the presence of Bafilomycin A1 (Online Figure I-E-G), suggesting that Sirt1 increases autophagosome formation as well. Although the number of green and red puncta were significantly greater in Ad-sh-Sirt1 transduced CMs than in Ad-LacZ or Ad-Sirt1 transduced CMs (Fig. 4C and Online Figure III-A), most of the red puncta overlaid with green puncta in Ad-sh-Sirt1 transduced CMs (Fig. 4D), suggesting that autophagic flux was inhibited in the absence of Sirt1.
Figure 4. Sirt1 is required to mediate GD-induced autophagy in CMs.

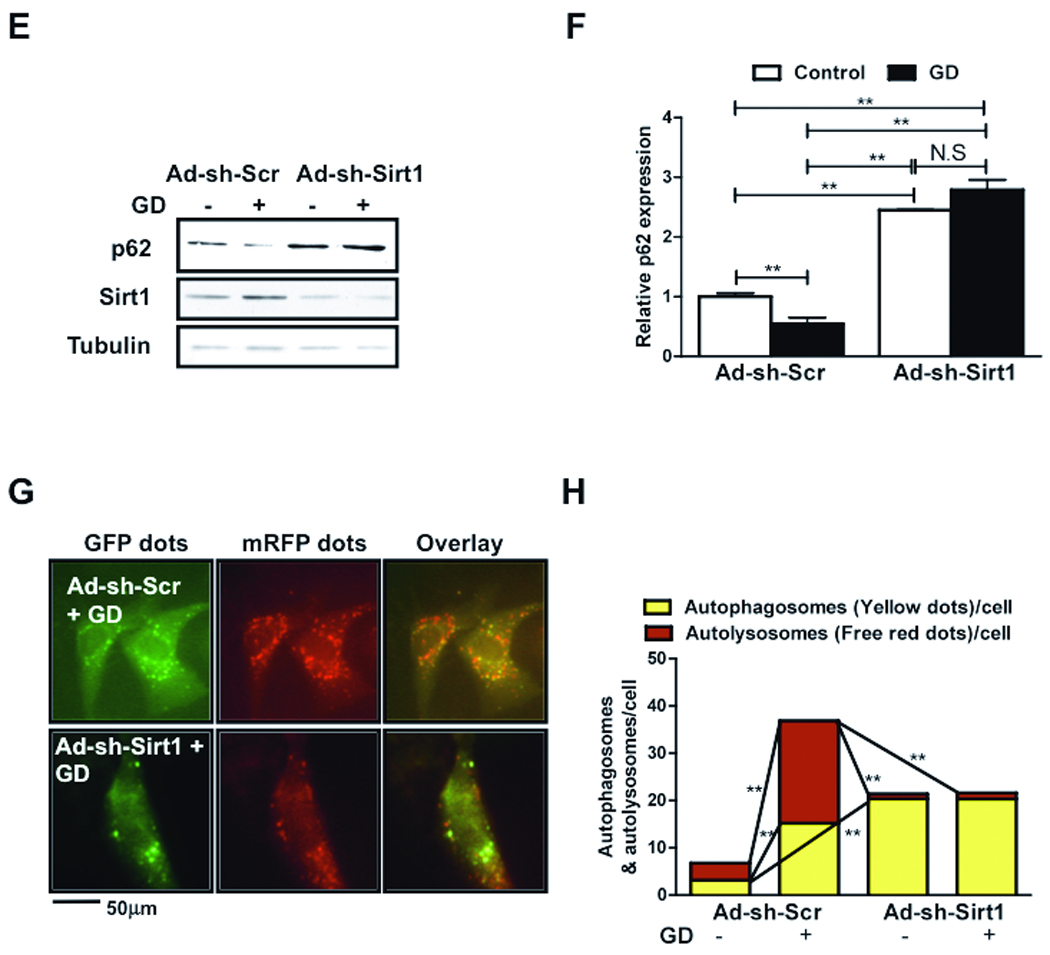
CMs were transduced with Ad-LacZ or Ad-Sirt1, Ad-sh-Sirt1 or Ad-sh-Scr. A) Immunoblot analyses showing expressions of Sirt1, LC3 (at two different exposure times), p62 and Tubulin. B) Densitometric analyses. C) Representative images of fluorescent LC3 puncta after transduction with Ad-tf-LC3. D) Mean number of autophagosomes represented by yellow puncta in merged images and autolysosomes represented by red puncta in merged images per cell. E-H) CMs were transduced with Ad-sh-FoxO1 or Ad-sh-Scr and treated with glucose free media. E) Immunoblots showing Sirt1, p62 and Tubulin expressions. F) Densitometric analyses. G) Representative images of fluorescent LC3 puncta after Ad-tf-LC3 transduction. H) Mean number of autophagosomes represented by yellow puncta in merged images and autolysosomes represented by red puncta in merged images per cell. In D and H, graph with error bars is shown in Online Figure XI-DE. Data represent means from at least 4 independent experiments. * p<0.05, ** p<0.01, N.S Not Significant.
To evaluate whether Sirt1 is required for GD-induced autophagy, Ad-sh-Sirt1 transduced myocytes were subjected to GD. Knockdown of Sirt1 inhibited GD-induced stimulation of autophagic flux, as evidenced by increased accumulation of p62 (Fig. 4EF). Furthermore, Ad-sh-Sirt1 inhibited GD-induced increases in autolysosome formation (Fig. 4GH), indicating that Sirt1 is required for GD induced increases in autophagic flux in CMs.
Deacetylation of FoxO1 and the LXXLL motif are required for GD induced autophagy
The LXXLL motif in FoxOs is important for interaction with Sirt1. Mutations in this motif are introduced in FoxO1(3SA), a mutant which cannot be phosphorylated by Akt and which is localized constitutively in the nucleus (FoxO1(3A/LXXAA)). Expression of FoxO1(3A/LXXAA) mutant abolishes interaction between Sirt1 and FoxO1 and Sirt1-induced stimulation of FoxO1-mediated transcription. 13 FoxO1(3A/LXXAA) is a useful tool for investigating the role of Sirt1 in regulating the function of FoxO1. We confirmed that FoxO1(3A/LXXAA) is localized in the nucleus (Online Figure IV). We hypothesized that FoxO1-Sirt1 interaction is required for FoxO1 to induce autophagy in response to GD. We transduced CMs with Ad-3A/LXXAA or Ad-LacZ and subjected them to GD for 2 hours. In Ad-3A/LXXAA transduced myocytes, acetylation of FoxO1 was increased at baseline and even after GD treatment (Fig. 5A). In these myocytes, GD-induced stimulation of autophagy was inhibited, as indicated by reduced LC3-II and increased p62 accumulation (Fig. 5A).
Figure 5. Interaction with Sirt1 and deacetylation of FoxO1 are required for autophagy in CMs.
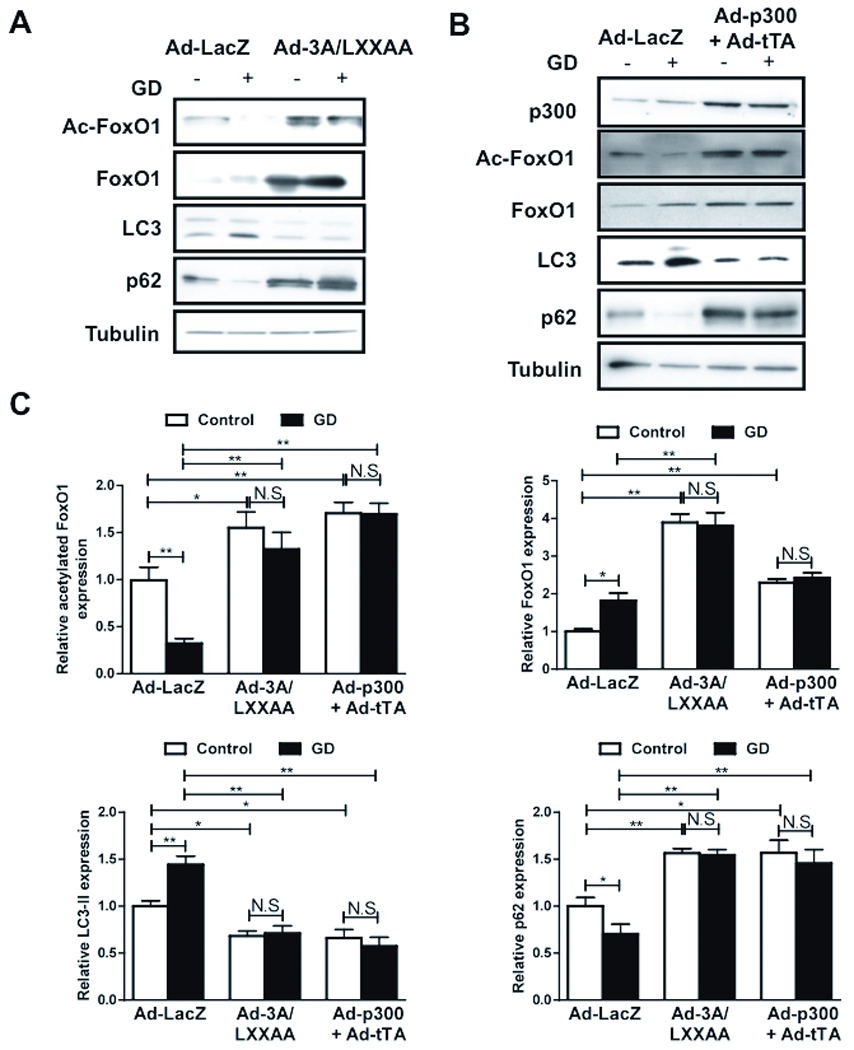
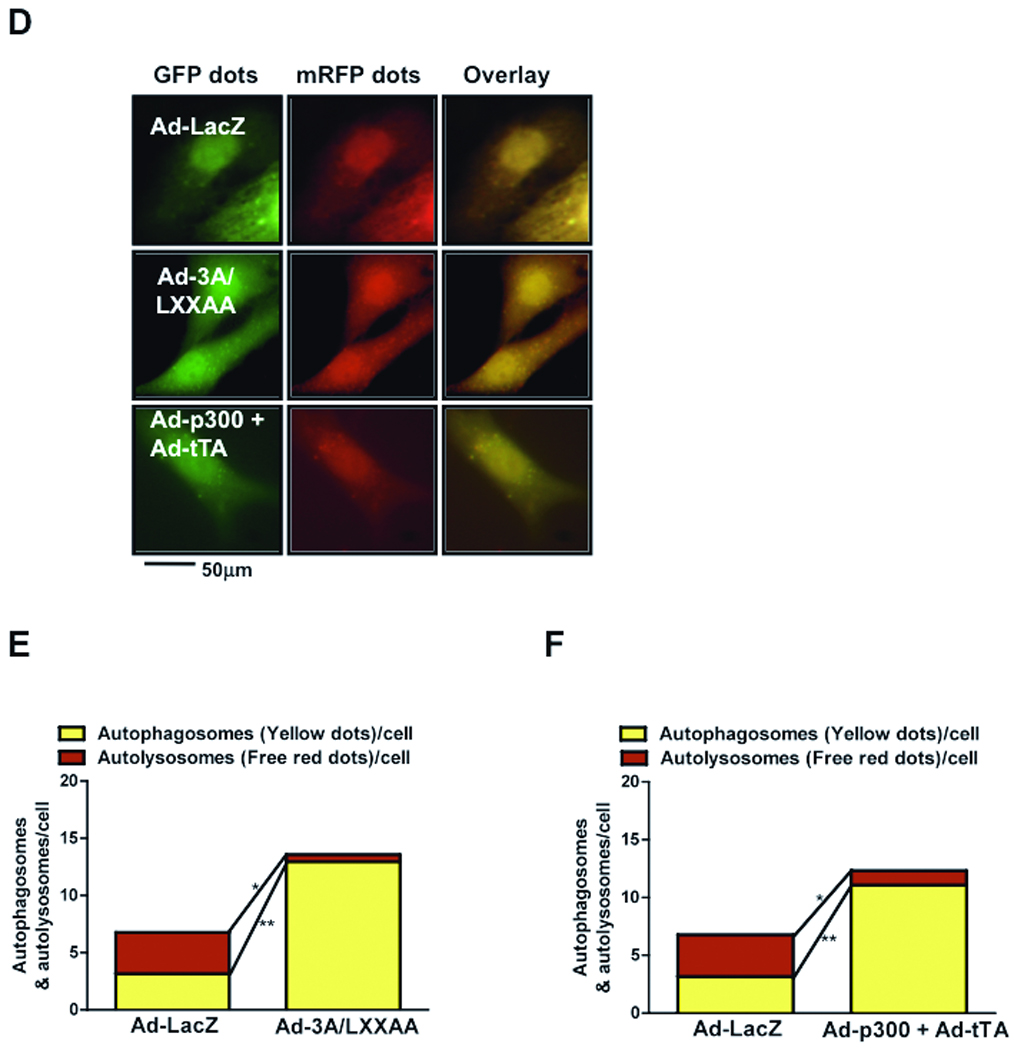
A) CMs were transduced with Ad-3A/LXXAA or Ad-LacZ and treated with glucose free media. Immunoblots for acetylated FoxO1, total FoxO1, LC3, p62 and Tubulin. B) CMs were transduced with Ad-p300 and Ad-tTA. Immunoblots for p300, acetylated FoxO1, total FoxO1, LC3, p62 and Tubulin. C) Densitometric analyses. D) Representative images of fluorescent LC3 puncta after transduction with Ad-tf-LC3. E–F) Mean number of autophagosomes represented by yellow puncta in merged images and autolysosomes represented by red puncta in merged images per cell. Graph with error bars is shown in Online Figure XI-FG. Data represent means from at least 4 independent experiments. * p<0.05, ** p<0.01, N.S Not Significant.
To show that deacetylation is required for autophagy, CMs were transduced with adenoviruses harboring inducible p300 (Ad-p300+Ad-tTA), a histone acetyl transferase. Overexpression of p300 significantly increased FoxO1 acetylation and inhibited autophagy, as indicated by decreased LC3-II and increased p62 accumulation, at baseline and upon GD (Fig. 5BC). Increases in FoxO1 acetylation and suppression of autophagy were not observed when myocytes were transduced with Ad-p300 or Ad-tTA alone. Ad-3A/LXXAA and Ad-p300+Ad-tTA both increased accumulation of autophagosomes in Ad-tf-LC3 transduced myocytes, as indicated by increases in yellow puncta in merged images, but not autolysosomes, as indicated by significant decreases in red puncta (Fig. 5DEF). These results suggest that deacetylation of FoxO1 by Sirt1, which is dependent on an intact LXXLL motif, is required for GD-induced autophagy.
FoxO1 and Sirt1 act synergistically to induce autophagy
Since deacetylation of FoxO1 mediated by Sirt1 is required for GD-induced autophagy, we hypothesized that FoxO1 is required for Sirt1-induced autophagy. To this end, the effect of FoxO1 knockdown upon Sirt1-mediated autophagy was evaluated with tf-LC3 (Online Figure V-A-C). Although the number of autophagosomes was significantly greater, the number of autolysosomes was significantly smaller in CMs transduced with both Ad-Sirt1 and Ad-sh-FoxO1 than in those with Ad-Sirt1 alone (Online Figure V-C). These results suggest that FoxO1 mediates Sirt1-induced increases in autophagic flux. Conversely, in CMs transduced with Ad-FoxO1 and Ad-sh-Sirt1, autophagosome formation was significantly increased while autolysosome formation was robustly decreased compared to in those with Ad-FoxO1 alone (Online Figure V-D-F). These results suggest that Sirt1 also plays an important role in mediating FoxO1-induced increases in autophagic flux in CMs and that Sirt1 and FoxO1 act in concert to mediate autophagy in CMs (Online Figure VI).
FoxO1 increases expression of Rab7
Since FoxO1 not only stimulates autophagosome formation but also prominently increases autolysosomes, we hypothesized that FoxO1 increases autophagic flux by increasing the expression of Rab7, a small GTPase protein which stimulates lysosomal biogenesis and maturation of autophagic vacuoles by promoting their fusion with endosomes and lysosomes.21 Both mRNA (Online Figure VII-A) and protein expression of Rab7 (Fig. 6A) were significantly increased in Ad-FoxO1-WT transduced myocytes, while they were reduced significantly by knockdown of FoxO1 or transduction with Ad-3A/LXXAA (Fig. 6A). Rab7 expression was also increased significantly upon GD, but this increase was abolished when FoxO1 was knocked down (Fig. 6B), indicating that the GD-induced increase in Rab7 is critically mediated through FoxO1. Rab7 is accumulated as puncta in the peri-nuclear region in response to GD (Online Figure VII-B). Rab7 co-stained predominantly with Cathepsin L, a lysosomal cysteine protease (Online Figure VII-B), but less so with GFP-LC3 (Online Figure VII-C). These results suggest that, in cultured CMs, Rab7 is localized to autolysosomes and late autophagic vacuoles, but not autophagosomes. Overexpression of Sirt1 upregulates Rab7, while knockdown of Sirt1 downregulates it (Online Figure VII-D). Taken together, these results indicate that the Sirt1-FoxO1 pathway regulates expression of Rab7.
Figure 6. FoxO1 regulates Rab7 expression and FoxO1-induced autophagy is attenuated by Rab7 knockdown.
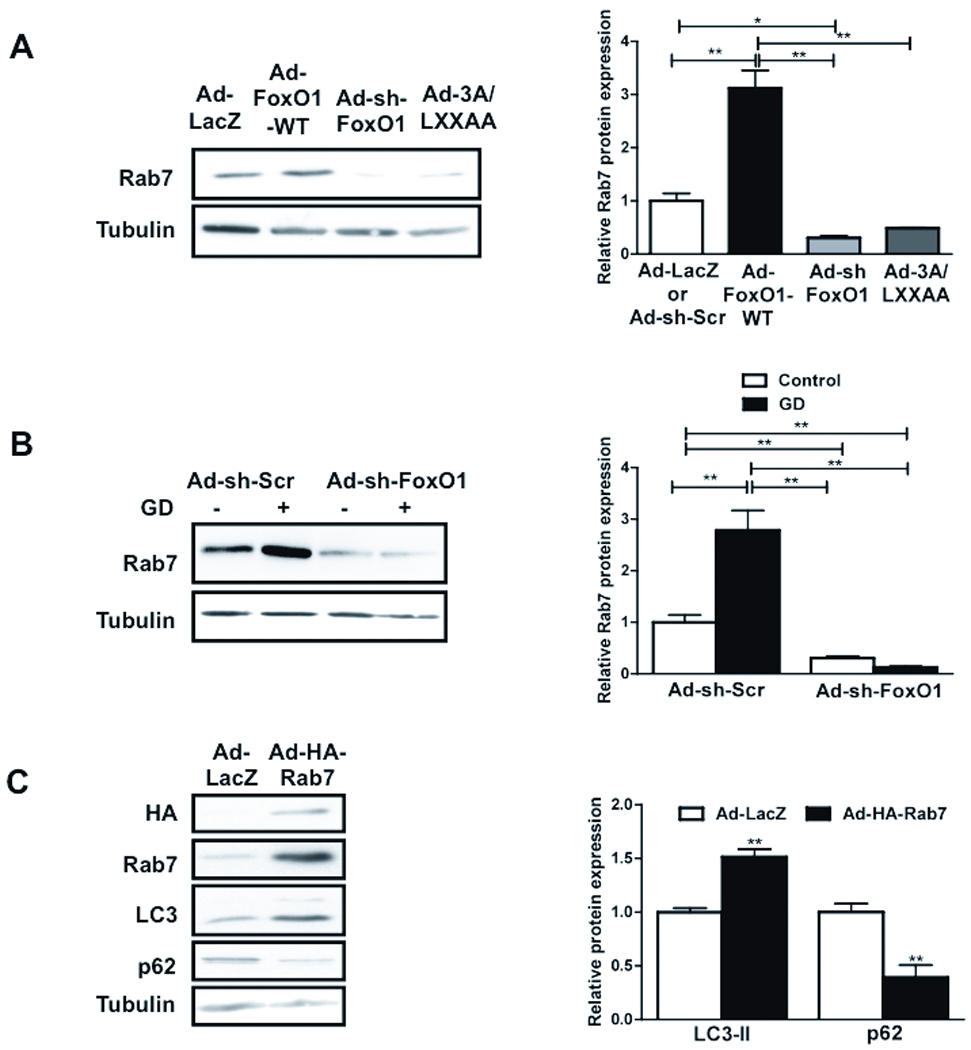

A) CMs were transduced with Ad-LacZ, Ad-FoxO1-WT, Ad-3A/LXXAA or Ad-sh-FoxO1. Immunoblots and densitometric analyses showing Rab7 expression. B) CMs were transduced with Ad-sh-FoxO1 or Ad-sh-Scr and treated with glucose free media. Immunoblot and densitometric analyses showing Rab7 expression. C) CMs were transduced with Ad-HA-Rab7 or Ad-LacZ. Immunoblots and densitometric analyses. D) CMs were transduced with Ad-LacZ, Ad-FoxO1-WT, Ad-sh-Scr or lentivirus harboring sh-Rab7 (Lt-sh-Rab7) for 96 hours. Immunoblots for LC3 (at two different exposure times) and p62 and densitometric analyses are shown. Data represent means from at least 4 independent experiments. * p<0.05, ** p<0.01.
FoxO1-induced autophagy is inhibited by Rab7 knockdown
In order to evaluate the role of Rab7 in mediating autophagic flux, we generated an adenovirus harboring HA-Rab7 (Ad-HA-Rab7). Overexpression of Rab7 in CMs increased LC3-II and decreased p62 (Fig. 6C), indicating that Rab7 stimulates autophagy. To evaluate the extent of autophagosome and autolysosome formation, Rab7-overexpressing CMs were transduced with Ad-tf-LC3 (Online Figure VIII-A). Rab7 increased both yellow and red puncta, but it increased red puncta significantly more (Online Figure VIII-BC), suggesting that Rab7 enhances autophagic flux in CMs.
To evaluate the role of endogenous Rab7 in mediating FoxO1-induced autophagy, we treated CMs with a lentivirus harboring sh-Rab7 (Lt-sh-Rab7). Knockdown of Rab7 was confirmed by immunoblots (Online Figure VIII-D). Expression of LC3-II was reduced and p62 was accumulated at baseline in Lt-sh-Rab7-transduced myocytes (Fig. 6D). Furthermore, the FoxO1-induced increase in LC3-II expression and p62 degradation was significantly attenuated upon knockdown of Rab7 (Fig. 6D). These results suggest that stimulation of autophagic flux by FoxO1 is mediated through upregulation of Rab7.
FoxO1 and its interaction with Sirt1 are required for starvation-induced autophagy in mouse hearts
We hypothesized that FoxO1 and its modulation by Sirt1 are required for starvation-induced autophagy in the heart in vivo. We used transgenic mice with cardiac specific overexpression of FoxO1(3A/LXXAA) (Tg-FoxO1m) and cardiac specific FoxO1 homozygous knockout mice (c-FoxO1−/−). These mice were subjected to starvation for 48 hours. LC3-II expression and p62 degradation were increased in control mice (non-transgenic mice and FoxO1flox/fox without Cre mice) after starvation, indicating increased autophagy (Fig. 7). These changes were abolished in Tg-FoxO1m and c-FoxO1−/− mice, where LC3-II expression was reduced and p62 accumulation was increased at baseline and in response to starvation, indicating inhibition of autophagic flux (Fig. 7). These results suggest that FoxO1 and its interaction with Sirt1 are required for starvation-induced autophagy in the heart in vivo.
Figure 7. FoxO1 is required for starvation-induced autophagy in vivo.
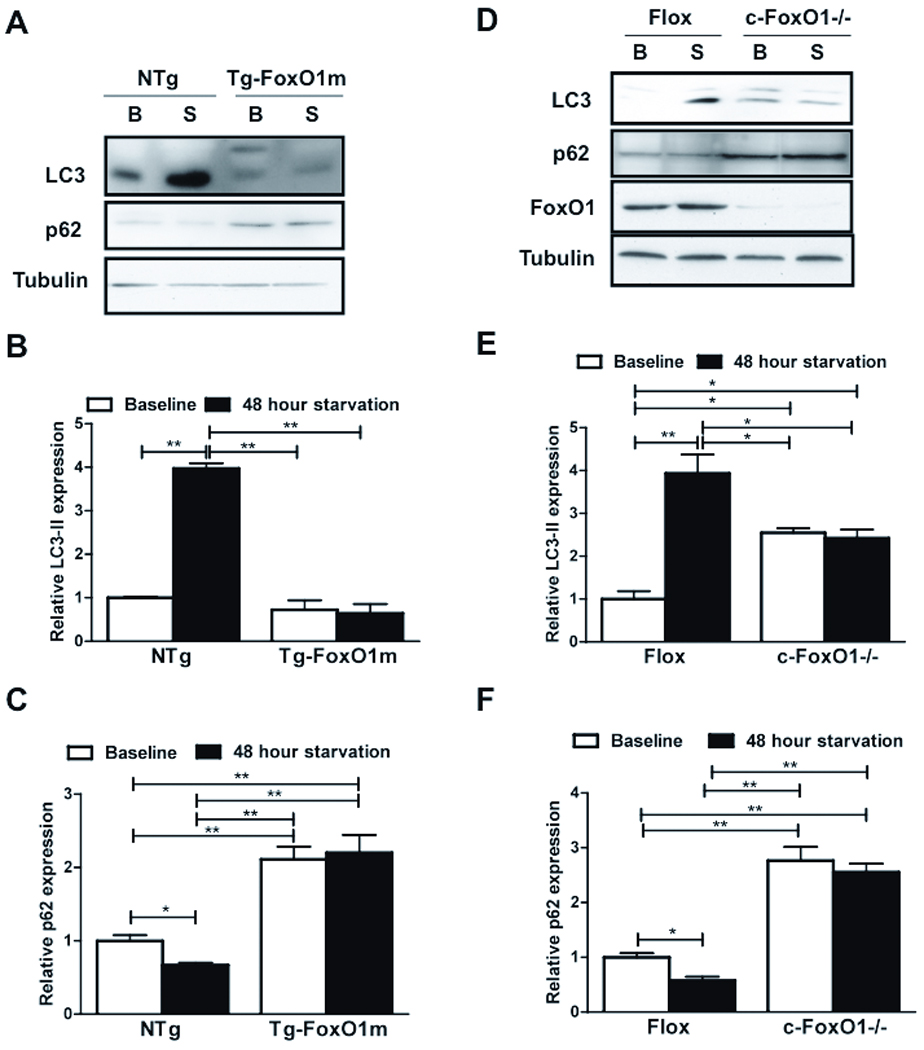
Tg-FoxO1m, c-FoxO1−/− and NTg or floxed control mice were starved for 48 hours. A) Immunoblots showing expression of LC3, p62 and Tubulin in Tg-FoxO1m mice, both at baseline (B) and after starvation (S). B-C) Densitometric analyses. D) Immunoblots at baseline and after starvation of c-FoxO1−/− mice. E-F) Densitometric analyses. Data represent means from at least 3 individual mice. * p<0.05, ** p<0.01.
To evaluate the role of FoxO1 in regulating cardiac function during starvation, echocardiographic analyses were conducted before and after starvation. In control mice, ejection fraction and fractional shortening were maintained after starvation (Fig. 8AB and Online Figure IX). However, in both Tg-FoxO1m and c-FoxO1−/− mice, cardiac function deteriorated significantly after starvation (Fig. 8AB, Online Table I). A decrease in cardiac function caused by starvation was also observed in beclin1+/− mice (Fig. 8CD), consistent with the notion that suppression of autophagy may contribute to starvation-induced cardiac dysfunction. These results suggest that FoxO1 is required for maintaining cardiac function after starvation, possibly through stimulation of autophagy.
Figure 8. FoxO1-induced autophagy is required to maintain cardiac function after starvation.
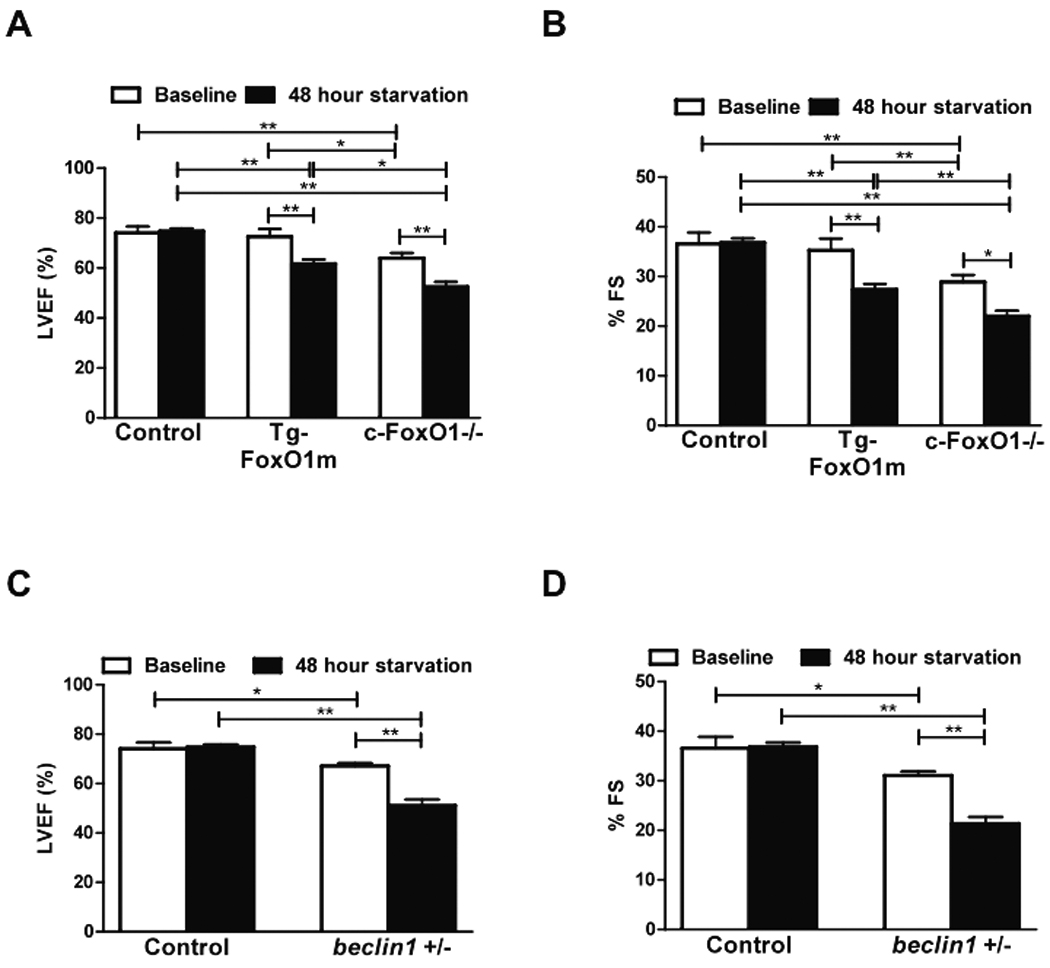
Cardiac function was evaluated in the Tg-FoxO1m, c-FoxO1−/−, beclin1+/− and control mice with echocardiographic analyses both at baseline and after 48 hours of starvation. A–B) Left ventricular ejection fraction (LVEF) and fractional shortening (%FS) in control, Tg-FoxO1m, c-FoxO1−/− mice. C–D) LVEF and FS in control and beclin1+/− mice. * p<0.05, **p<0.01. N=6 in each group.
Discussion
We have demonstrated that 1) deacetylation of FoxO1 by Sirt1 is required for starvation induced autophagy in the heart, 2) Sirt1 and FoxO1 cooperatively mediate starvation-induced autophagy, 3) FoxO1 enhances autophagic flux by activating Rab7, and 4) FoxO1 is required to maintain cardiac function after starvation. Overall, deacetylation of FoxO1 and activation of autophagy form an essential adaptive mechanism for maintaining LV function under starvation conditions.
Experiments with Ad-tf-LC3 allowed us to demonstrate that both GD and FoxO1 not only stimulate autophagosome formation but also enhance formation of autolysosomes, and, thus, they both stimulate autophagic flux. FoxO1 is upregulated by GD and upregulation of FoxO1 is necessary for stimulating starvation-induced autophagy. FoxO1 is a key molecule which is important for maintaining energy homeostasis and regulating metabolism in the liver, adipose tissue and skeletal muscle. 22 During fasting and exercise, FoxO1 can activate gluconeogenic enzymes in the liver and turn on lipid metabolism in order to cope with conditions of nutrient and energy depletion. 4, 22 FoxO1 induced autophagy could be a part of such an adaptive mechanism, providing a source of energy by recycling amino and fatty acids and mediating cell survival during the energy crisis.
Multiple FoxO family transcription factors have many overlapping functions. Since FoxO3 also stimulates autophagy (Online Figure II), FoxO3 may be involved in GD-induced autophagy in CMs. However, since specific downregulation of FoxO1 potently suppresses GD-induced autophagy, FoxO1 may have non-overlapping mechanisms to stimulate autophagy in CMs. Whether or not FoxO isoforms differentially contribute to starvation-induced autophagy remains to be clarified.
Different post-translational modifications regulate the function of FoxO proteins. In the presence of insulin, FoxO is phosphorylated by Akt and translocates from the nucleus to the cytosol, and FoxO-mediated transcription is attenuated 17. In response to GD, FoxO proteins are dephosphorylated (Online Figure X) and localized in the nucleus (Online Figure IV), which in turn could stimulate transcription of autophagy genes. 22 Besides phosphorylation, the acetylation status of FoxO proteins also affects their transcriptional activity. 22 Here we show that FoxO1 is deacetylated upon GD in a Sirt1-dependent manner. FoxO1(3A/LXXAA), which cannot interact with Sirt1, remains acetylated, indicating that interaction with Sirt1 is required for FoxO1 to be deacetylated in the presence of GD. Importantly, FoxO1 cannot stimulate autophagy when it is acetylated in the presence of p300, a histone acetyltransferase known to acetylate Sirt1, and FoxO1(3A/LXXAA) inhibits GD-induced autophagy despite being localized in the nucleus, suggesting that interaction with Sirt1 and consequent deacetylation are required for FoxO1 to mediate GD-induced autophagy. Furthermore, nuclear translocation of FoxO1 caused by dephosphorylation alone may not be sufficient for inducing autophagy in response to GD. Interestingly, nuclear localization of FoxO1 is enhanced in the presence of Sirt1 overexpression (Online Figure IV), raising the possibility that Sirt1-mediated deacetylation may also contribute to the nuclear localization of FoxO1 during GD. Taken together, our results suggest that Sirt1-mediated deacetylation of FoxO1 plays an important role in mediating GD-induced stimulation of autophagy in CMs. Our results do not exclude the involvement of phosphorylation-dependent regulation of FoxO1 in modulation of autophagy.
Although both sh-FoxO1 and FoxO1 (3A/LXXAA) inhibit autophagic flux, as evidenced by accumulation of p62 and decreases in autolysosomes, Ad-sh-FoxO1 increased LC3-II, whereas FoxO1 (3A/LXXAA) decreased LC3-I and LC3-II, suggesting that sh-FoxO1 inhibits autophagic flux primarily at autophagosome-lysosome fusion, whereas FoxO1 (3A/LXXAA) either inhibits expression of LC3 or interferes with autophagosome formation as well as autophagosome-lysosome fusion. It is possible that sh-FoxO1 induced incomplete knockdown of FoxO1 or that FoxO3 may compensate for downregulation of FoxO1. On the other hand, FoxO1 (3A/LXXAA), as a dominant negative (against FoxO1 and/or FoxO3), may be a stronger inhibitor of FoxO, so that not only genes involved in autophagosome-lysosome fusion but also those involved in autophagosome formation are affected.
Both expression of Sirt1 and the cellular amount of NAD+ are significantly increased during GD in CMs, which would enhance the total activity of Sirt1, favoring deacetylation of FoxO1. The facts that upregulation of Sirt1 is sufficient to increase both autophagosomes and autolysosomes and stimulate autophagy, an effect very similar to that of FoxO1, and that stimulation of autophagy by Sirt1 is abolished in the absence of FoxO1 are consistent with the notion that Sirt1 is an upstream regulator of FoxO1 for induction of autophagy (Online Figure VI). Interestingly, however, FoxO1-induced autophagy also requires Sirt1. These results suggest that FoxO1 and Sirt1 in concert mediate autophagy under GD by making a functional complex in CMs.
Previous studies have shown that FoxO3 regulates autophagy in skeletal muscle cells by transcriptional activation of genes that are involved in autophagosome formation, including LC3, Gabarapl1, Atg12l, Atg4, Beclin1, Ulk2, Vps34 and Bnip3. 5, 7 It should be noted that the causative role of autophagy related molecules in mediating FoxO-mediated autophagy remains to be elucidated. Our results suggest that Rab7 plays an important role in mediating FoxO1-induced stimulation of autophagy. Since Rab7 plays an important role in the fusion of the matured autophagic vacuole with the lysosome, we speculate that Rab7 may primarily contribute to the late stages of autophagy and the overall increase in autophagic flux. Since FoxO1 upregulates other autophagy-related genes specifically involved in different steps of autophagy, and because autophagy is executed through multiple steps, including nucleation, autophagosome formation, autophagosome-lysosome fusion and lysosomal degradation, other FoxO1 targets may also contribute to FoxO1-induced autophagy by modulating other processes of autophagy.
Starvation of newborn mice lacking atg5 increases perinatal death due to heart failure 23, suggesting that the heart function critically relies on autophagy during nutrient starvation. Our results show that downregulation of FoxO1 or expression of a FoxO1 mutant which cannot interact with Sirt1 not only inhibits autophagy but also results in LV dysfunction under starvation conditions. These results are consistent with the notion that the Sirt1-FoxO1 pathway is a compensatory signaling mechanism essential for maintaining LV function under starvation conditions. Since the suppression of autophagy in beclin1+/− mice also leads to LV dysfunction during food starvation, suppression of autophagy may in part mediate LV dysfunction in Tg-FoxO1m and c-FoxO1−/− mice under starvation conditions. When Sirt1 and FoxO proteins interact, transcription of genes mediating stress resistance, cell survival and longevity is turned on, while transcription of those mediating apoptosis and cell death is turned off. 4 We therefore speculate that FoxO1-induced transcription of autophagy genes is an important adaptive mechanism during starvation in the heart.
Both FoxOs and Sirt1 affect glucose metabolism as well as insulin signaling. 22 Thus, we should point out the possibility that manipulation of FoxOs and Sirt1 secondarily affects the extent of autophagy through changes in metabolism and insulin signaling in the heart and the myocytes therein.
In summary, Sirt1-mediated deacetylation of FoxO1 plays an essential role in mediating starvation-induced autophagy. Deacetylation of FoxO1 induces expression of genes involved in autophagy, including Rab7, which in turn stimulate autophagy. We propose that autophagy activated by the Sirt1-FoxO1 pathway is beneficial for the heart during nutrient and energy deficiency.
Novelty and Significance
What is known?
The FoxO family transcription factors stimulate autophagy in cardiac myocytes
Autophagy is activated by nutrient deprivation.
What new information does this article contribute?
In cardiac myocytes, deacetylation of FoxO1 by Sirt1, a histone deacetylase, is required for mediating starvation-induced autophagy.
FoxO1 upregulates Rab7, which in turn mediates autophagosome-lysosome fusion, thereby enhancing autophagic flux.
FoxO1-induced autophagy maintains cardiac function during starvation.
Summary
Activation of autophagy preserves ATP and increases survival during nutrient deprivation. The signaling mechanism by which nutrient deprivation activates autophagy in the heart is not well understood. Here we demonstrate that in cardiac myocytes FoxO1 and its deacetylation by Sirt1 mediates autophagy in response to glucose deprivation. FoxO1 upregulates Rab7, a small GTP binding protein, which in turn enhances fusion of autophagosomes and lysosomes and stimulates autophagic flux. Deacetylation of FoxO1 and subsequent activation of autophagy are essential for the heart to maintain left ventricular contractility during nutrient starvation in adult mice. Our findings suggest a critical role of Sirt1-mediated deacetylation of FoxO1 in mediating starvation-induced autophagy and indicate that activation of autophagy through the Sirt1-FoxO1 pathway protects the heart during nutrient/energy starvation.
Supplementary Material
Acknowledgements
The authors thank Drs. Tamotsu Yoshimori and Beth Levine for tf-LC3 and beclin1+/− mice, respectively. We thank Daniela Zablocki for critical reading of the manuscript.
Sources of Funding
This work was supported in part by U.S. Public Health Service Grants HL 59139, HL67724, HL69020, HL91469, HL102738, and AG27211, and Foundation of Leducq Transatlantic Network of Excellence.
Non-standard Abbreviations and Acronyms
- Ad
adenovirus
- BW
body weight
- CM
neonatal rat cardiac myocytes
- EF
ejection fraction
- FS
fractional shortening
- FoxO1m
FoxO1 mutant
- GD
glucose deprivation
- GFP
green fluorescent protein
- HA
hemagglutinin
- Lt
lentivirus
- LV
left ventricular
- MOI
multiplicities of infection
- mRFP
monomeric red fluorescent protein
- MHC
myosin heavy chain
- NAD+
nicotinamide adenine dinucleotide
- NTg
non-transgenic
- PI
protease inhibitors
- sh
short hairpin
- tf-LC3
tandem fluorescent mRFP-GFP-LC3
- Tg
transgenic
- tTA
tetracycline trans-activator
- WT
wildtype
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
None
Subject Codes:
[138] Cell signaling/signal transduction
[151] Ischemic biology – basic studies
References
- 1.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 2.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 3.Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K. The role of autophagy in the heart. Cell Death Differ. 2009;16:31–38. doi: 10.1038/cdd.2008.163. [DOI] [PubMed] [Google Scholar]
- 4.Greer EL, Brunet A. FOXO transcription factors at the interface between longevity and tumor suppression. Oncogene. 2005;24:7410–7425. doi: 10.1038/sj.onc.1209086. [DOI] [PubMed] [Google Scholar]
- 5.Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S, Sandri M. FoxO3 Controls Autophagy in Skeletal Muscle In Vivo. Cell Metab. 2007;6:458–471. doi: 10.1016/j.cmet.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Sengupta A, Molkentin JD, Yutzey KE. FoxO transcription factors promote autophagy in cardiomyocytes. J Biol Chem. 2009;284:28319–28331. doi: 10.1074/jbc.M109.024406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 Coordinately Activates Protein Degradation by the Autophagic/Lysosomal and Proteasomal Pathways in Atrophying Muscle Cells. Cell Metab. 2007;6:472–483. doi: 10.1016/j.cmet.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 8.Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF, Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 9.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu CP, Oka S, Shao D, Hariharan N, Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res. 2009;105:481–491. doi: 10.1161/CIRCRESAHA.109.203703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 12.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 13.Nakae J, Cao Y, Daitoku H, Fukamizu A, Ogawa W, Yano Y, Hayashi Y. The LXXLL motif of murine forkhead transcription factor FoxO1 mediates Sirt1-dependent transcriptional activity. J Clin Invest. 2006;116:2473–2483. doi: 10.1172/JCI25518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 15.Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal. 2010 doi: 10.1089/ars.2010.3488. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho J, Rameshwar P, Sadoshima J. Distinct roles of glycogen synthase kinase (GSK)-3alpha and GSK-3beta in mediating cardiomyocyte differentiation in murine bone marrow-derived mesenchymal stem cells. J Biol Chem. 2009;284:36647–36658. doi: 10.1074/jbc.M109.019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 18.Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, Jiang S, Gilliland DG, Chin L, Wong WH, Castrillon DH, DePinho RA. FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell. 2007;128:309–323. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perry CN, Kyoi S, Hariharan N, Takagi H, Sadoshima J, Gottlieb RA. Novel methods for measuring cardiac autophagy in vivo. Methods Enzymol. 2009;453:325–342. doi: 10.1016/S0076-6879(08)04016-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004;306:2105–2108. doi: 10.1126/science.1101731. [DOI] [PubMed] [Google Scholar]
- 21.Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117:4837–4848. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- 22.Gross DN, van den Heuvel AP, Birnbaum MJ. The role of FoxO in the regulation of metabolism. Oncogene. 2008;27:2320–2336. doi: 10.1038/onc.2008.25. [DOI] [PubMed] [Google Scholar]
- 23.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.