

Abstract
OBJECTIVE
Hypoglycemia-associated autonomic failure (HAAF) constitutes one of the main clinical obstacles to optimum treatment of type 1 diabetes. Neurons in the ventromedial hypothalamus are thought to mediate counterregulatory responses to hypoglycemia. We have previously hypothesized that hypoglycemia-induced hypothalamic angiotensin might contribute to HAAF, suggesting that the angiotensin blocker valsartan might prevent HAAF. On the other hand, clinical studies have demonstrated that the opioid receptor blocker naloxone ameliorates HAAF. The goal of this study was to generate novel hypothalamic markers of hypoglycemia and use them to assess mechanisms mediating HAAF and its reversal.
RESEARCH DESIGN AND METHODS
Quantitative PCR was used to validate a novel panel of hypothalamic genes regulated by hypoglycemia. Mice were exposed to one or five episodes of insulin-induced hypoglycemia, with or without concurrent exposure to valsartan or naloxone. Corticosterone, glucagon, epinephrine, and hypothalamic gene expression were assessed after the final episode of hypoglycemia.
RESULTS
A subset of hypothalamic genes regulated acutely by hypoglycemia failed to respond after repetitive hypoglycemia. Responsiveness of a subset of these genes was preserved by naloxone but not valsartan. Notably, hypothalamic expression of four genes, including pyruvate dehydrogenase kinase 4 and glycerol 3-phosphate dehydrogenase 1, was acutely induced by a single episode of hypoglycemia, but not after antecedent hypoglycemia; naloxone treatment prevented this failure. Similarly, carnitine palmitoyltransferase-1 was inhibited after repetitive hypoglycemia, and this inhibition was prevented by naloxone. Repetitive hypoglycemia also caused a loss of hypoglycemia-induced elevation of glucocorticoid secretion, a failure prevented by naloxone but not valsartan.
CONCLUSIONS
Based on these observations we speculate that acute hypoglycemia induces reprogramming of hypothalamic metabolism away from glycolysis toward β-oxidation, HAAF is associated with a reversal of this reprogramming, and naloxone preserves some responses to hypoglycemia by preventing this reversal.
Hypoglycemia-associated autonomic failure (HAAF) is thought to constitute one of the main obstacles to optimum treatment of type 1 diabetes (1). The causes of HAAF are not known, but counterregulatory responses appear to be mediated by glucose-sensing neurons in the ventromedial hypothalamus (2–6). We have reported that acute hypoglycemia induces hypothalamic expression of angiotensinogen (7). Furthermore, inhibition of ACE activity appears to reduce the risk of hypoglycemia episodes (8,9), suggesting that angiotensin receptor blockers might prevent or even reverse HAAF. On the other hand, the angiotensin receptor blocker losartan is reported to attenuate counterregulatory responses to hypoglycemia (10), though it is not clear if losartan crosses the blood-brain barrier. In contrast, clinical studies have demonstrated that naloxone improves counterregulatory responses (11) and prevents HAAF (12) in humans. In this study we therefore assessed the effects of treatment with valsartan, an angiotensin receptor blocker that crosses the blood-brain barrier, or naloxone on counterregulatory and molecular responses to hypoglycemia after antecedent hypoglycemia.
RESEARCH DESIGN AND METHODS
All studies were approved by the appropriate institutional animal review board (Institutional Animal Care and Use Committee). Twelve-week-old male C57BL/6J mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and housed five per cage with free access to food and water under 12:12-h light-dark cycle (lights on at 7:00 a.m.).
Drugs.
This study was designed to assess if blocking angiotensin receptors would prevent counterregulatory failure based on our observation that hypoglycemia induces expression of angiotensin (7). Acute inhibition of angiotensin II production by an ACE inhibitor attenuates acute sympathetic responses to hypoglycemia in humans (13), but specific blockade of the angiotensin receptor A1 subtype does not block responses to hypoglycemia in humans (14), suggesting that the AT2 receptor may mediate acute counterregulatory responses. Furthermore, in a wide variety of circumstances, the AT1 receptor antagonizes effects of the AT2 receptor. For example, the AT1 receptor inhibits effects of the AT2 receptor on vasodilation (15), which is consistent with opposing effects of these receptors in a variety of systems (16). Furthermore, activation of the AT1 receptor increases glucose uptake (17), suggesting that activation of the AT1 receptor would reduce sensitivity to hypoglycemia and thus lead to counterregulatory failure. Therefore, valsartan was chosen for the present studies because it primarily blocks AT1 receptors, and it is the optimum protocol for oral delivery to produce protective effects in the mouse brain without producing hypotension, which has been exhaustively characterized by Wang et al. (18). We therefore administered valsartan orally as described in that article.
Naloxone hydrochloride dihydrate (Sigma Chemical, St. Louis, MO) was dissolved in sterile saline and injected intraperitoneally at a dose of 2 mg/kg in a volume of 0.1 ml/10 g of body weight.
Insulin-induced hypoglycemia.
Hypoglycemia was produced by insulin (2.5 units/kg body weight) injected intraperitoneally into mice previously fasted for 3 h, a protocol that produced blood glucose <40 mg/dl when measured at 90 min after injection and without producing unconsciousness, seizures, or death. We previously used a similar protocol to study hypothalamic gene expression following hypoglycemia (7). The insulin dose was tested and optimized prior to the experiments on age-matched C57Bl6/J mice. Blood glucose was measured before insulin injection and after 30, 90, and 180 min via tail prick. For antecedent hypoglycemia episodes, mice were placed in cages without food for 3 h after the insulin injection and then moved back into home cages with food 3 h after the insulin injection. The euglycemic experimental group was similarly denied food access after the saline injection concurrently with the hypoglycemia groups. On the final day of hypoglycemia, blood glucose was additionally measured over 240 min, at which time the animals were killed.
Experimental design.
Animals were randomly assigned to one of seven groups (n = 10), designated in Figure 1: Eu (saline-injected euglycemic), 1XH (acute insulin-induced hypoglycemia without antecedent hypoglycemia), 5XH (acute hypoglycemia with four antecedent days of hypoglycemia), Eu-V (euglycemic group with oral valsartan 40 mg/kg/day), 1XH-V (acute hypoglycemia with oral valsartan), 5XH-V (acute hypoglycemia with four antecedent days of hypoglycemia maintained on oral valsartan), 5XH-N (acute hypoglycemia with four antecedent days of hypoglycemia with 2 mg/kg naloxone injected intraperitoneally 15 min before every insulin injection). Antecedent hypoglycemia (5XH) consisted of four consecutive days of insulin-induced hypoglycemia (3 h), followed on the 5th day with the final episode of acute hypoglycemia and the animals killed 4 h after insulin injection. The group exposed to acute insulin–induced hypoglycemia without antecedent hypoglycemia (1XH) received physiological saline injections for 4 days, followed by acute hypoglycemia on the 5th day. The euglycemic group (Eu) received saline injections for five consecutive days. The valsartan groups (1XH-V, 5XH-V) ingested 40 mg/kg/day oral valsartan starting 5 days prior to commencing the 5-day injection protocol and continued on oral valsartan throughout the study. Before the 40-mg/kg/day dose, the valsartan groups received a 20-mg/kg/day dose for a 2-day adjustment period. The naloxone group (5XH-N) received an injection of naloxone (2 mg/kg, intraperitoneally) 15 min before every insulin injection. Therefore, the 5XH-N group received a total of 5 days of insulin injections preceded on each day by an injection of naloxone.
FIG. 1.

Blood glucose concentration throughout the study for all experimental groups: Eu, 1XH, 5XH, Eu-V, 1XH-V, 5XH-V, 5XH-N. The x-axis indicates the day and time points of the study (with insulin or saline injected at time 0, and for the 5XH-N group, naloxone was injected 15 min prior). Data are means ± SE (n = 10 for all groups).
In all cases, mice were killed 4 h after the injection of insulin (or saline for euglycemic mice). Mice were killed following a balanced design at the start of the light period (10:00 a.m. to 2:00 p.m.). Mice were killed by decapitation after a brief exposure to carbon dioxide. Hypothalamic and cortical areas, along with peripheral tissues, were quickly removed, frozen on dry ice, and stored at −70°C until extraction of RNA. Trunk blood was collected for analysis of corticosterone levels.
Blood chemistry.
Blood chemistry was carried out in all mice (n = 10, 7 groups). Blood glucose was measured by a Contour glucose meter (Bayer, Mountain View, CA). Blood corticosterone levels were measured using an enzyme-linked immunosorbent assay (ELISA) from Assay Designs (Ann Arbor, MI). Blood glucagon was measured using an ELISA from Wako Chemicals USA (Richmond, VA), and epinephrine was measured using an ELISA from Rocky Mountain Diagnostics (Colorado Springs, CO).
Extraction of hypothalamic RNA and cDNA synthesis.
Gene expression was assessed for six mice per group based on the quality of the RNA. To obtain the RNA for gene expression analysis by real-time RT-PCR, hypothalamic tissue was homogenized in tubes containing RLT buffer (Qiagen, Valencia, CA) supplemented with 2-ME, and total RNA was extracted using an RNeasy Mini Kit (Qiagen). The quality of the total RNA was assessed using the Biophotometer (Eppendorf, Hauppauge, NY). Due to the capacity limitations of the PCR array plates, six out of ten samples from each experimental group were selected (based on superior RNA quality) and were subjected to reverse transcription. One μg of high-quality total RNA was used for cDNA synthesis using RT2 First Strand Kit (SABiosciences, Frederick, MD). All procedures were performed according to the manufacturers' protocols.
RT-PCR with custom RT2 profiler PCR arrays.
RT2 Profiler PCR Array (SABiosciences) technology for gene expression analysis entails a synthesis between the profiling capabilities of DNA microarray and the quantitative reliability and sensitivity of quantitative PCR (qPCR). The results are highly reproducible within the same assay run or between different assay runs. RT2 Profiler Custom PCR Arrays were used to simultaneously examine the mRNA levels of 187 genes, including 7 housekeeping genes in 384-well plates according to the protocol of the manufacturer (SuperArray Bioscience). The genes were chosen based on prior DNA microarray studies as described in the Results section. The qPCR reactions were carried out using an ABI Prism 7900 thermocycler. Six of the seven housekeeping genes on the array were used to normalize the gene expression by the ΔΔCt method. Data were analyzed using a web-based software program provided by the manufacturer with additional analysis using GraphPad Prism 4 for Macintosh.
Data analysis.
All data are presented as mean ± SEM. Statistical analysis was performed using GraphPad Prism 4.0 by one-way ANOVA followed by Dunnett post hoc test. P < 0.05 indicates statistical significance.
RESULTS
Novel hypothalamic hypoglycemia–induced genes.
As shown in Fig. 1, similar levels of hypoglycemia were achieved in all insulin-injected groups.
One purpose of this study was to expand the panel of hypothalamic genes regulated by hypoglycemia (7) to facilitate assessing mechanisms of HAAF. Toward this end, a series of DNA microarray studies were undertaken to discover genes that are regulated by hypoglycemia and fasting. From these microarrays, a set of genes was chosen that were significantly (by uncorrected t test) regulated in the same direction by both hypoglycemia and fasting. The expression of a subset of these genes was assessed in the present study using a custom-designed qPCR array (SABiosciences). This array is now commercially available from SABiosciences and constitutes a powerful resource for the scientific community. After qPCR, expression of these genes was statistically assessed by one-way ANOVA followed by Dunnett post hoc test. Only genes for which the overall ANOVA was significant (P < 0.05) and an effect of either acute or repetitive hypoglycemia was significant by post hoc test, which were also significantly regulated by fasting in a separate study, are presented in this article (Table 1).
TABLE 1.
Gene nomenclature and regulation by acute insulin–induced hypoglycemia
| Gene symbol | Gene name | Reference sequence no. | Fold change (mean ± SE) | P |
|---|---|---|---|---|
| Angptl4 | Angiopoietin-like 4 | NM_020581 | 2.13 ± 0.15 | 0.00003 |
| Cdkn1a | Cyclin-dependent kinase inhibitor 1A (p21) | NM_007669 | 5.67 ± 0.44 | 0.000001 |
| Cpt1a | Carnitine palmitoyltransferase 1a | NM_013495 | 1.10 ± 0.11 | 0.53285 |
| Cxcl14 | Chemokine (C-X-C motif) ligand 14 | NM_019568 | 0.82 ± 0.03 | 0.00133 |
| GLUT1 | facilitated glucose transporter, member 1 | NM_011400 | 1.91 ± 0.32 | 0.01853 |
| Gpd1 | Glycerol 3-phosphate dehydrogenase 1 (soluble) | NM_010271 | 1.67 ± 0.16 | 0.00708 |
| Gpd2 | Glycerol 3-phosphate dehydrogenase 2 (mitocho) | NM_010274 | 0.94 ± 0.01 | 0.03812 |
| Hif3α | Hypoxia inducible factor 3, α subunit | NM_016868 | 2.06 ± 0.27 | 0.00575 |
| Pdk4 | Pyruvate dehydrogenase kinase, isoenzyme 4 | NM_013743 | 1.77 ± 0.23 | 0.01009 |
| Pnpla2 | Patatin-like phospholipase domain containing 2 | NM_025802 | 1.64 ± 0.13 | 0.00084 |
| S3-12 | Perilipin 4 | NM_020568 | 3.04 ± 0.21 | 0.00007 |
| Sox17 | SRY-box containing gene 17 | NM_011441 | 1.46 ± 0.12 | 0.00565 |
| Ucp2 | Uncoupling protein 2 | NM_011671 | 1.98 ± 0.28 | 0.01186 |
Hypoglycemia-induced elevation of plasma corticosterone fails after antecedent hypoglycemia: prevention of failure by naloxone, but not valsartan.
In the present study, mice were killed 4 h after insulin injection to allow examination of hypothalamic molecular responses to gene expression (7). However, this time point is not optimum for the assessment of the failure of hormonal responses, which in mice are usually measured 120 min after the induction of hypoglycemia (19,20), similar to studies in rats (4,21) and humans (22). Thus, although we measured all three counterregulatory hormones, any conclusions based on hormone levels at this late time point must be interpreted with caution. As observed in humans (23), a single acute exposure to hypoglycemia caused a significant elevation of plasma corticosterone, a response completely prevented by antecedent hypoglycemia (Fig. 2A). This counterregulatory failure was not prevented by treatment with valsartan, but was completely prevented by treatment with naloxone. Similarly, a single exposure to hypoglycemia caused a significant elevation of plasma glucagon, and this induction was completely prevented by antecedent hypoglycemia (Fig. 2B). Furthermore, the failure of glucagon to respond to hypoglycemia was not rescued by either drug treatment (Fig. 2B). Finally, plasma epinephrine was also induced by a single episode of hypoglycemia, but this induction was not blocked at this time point by antecedent hypoglycemia (Fig. 2C). However, the induction appears to have been attenuated by valsartan and naloxone (Fig. 2C). Subject to the caveat concerning the late time point, these results do not support that valsartan will prevent counterregulatory failure and may in fact worsen failure. Furthermore, naloxone, while possibly protective for activation of the hypothalmic-adrenal-pituitary axis, may not protect against failure in glucagon and epinephrine secretion after antecedent hypoglycemia.
FIG. 2.

Counterregulatory hormones in mice exposed to antecedent hypoglycemia and naloxone (2 mg/kg) or valsartan (40 mg/kg/day). Trunk blood was collected at the time the animals were killed, 4 h after final insulin or saline injection. Hormone levels were measured by ELISA in the same groups as described in Fig. 1. Data are means ± SE (n = 10 for all groups). *P < 0.05 as compared with the euglycemic group (Dunnett test).
Hif3α, S3-12, and GLUT1 are induced after acute and repetitive hypoglycemia.
As shown in Table 1 and Fig. 3, Hif3α (hypoxia-induced factor 3α), S3-12 (perilipin 4), and GLUT1 (facilitative GLUT isoform 1) were induced by acute hypoglycemia. Induction by hypoglycemia was not influenced by antecedent hypoglycemia, valsartan, or naloxone. Thus, the induction of these genes did not correlate with HAAF or its reversal by naloxone. Furthermore, the induction of these genes appears not to be dependent on hypoglycemia-induced angiotensin since valsartan did not influence the expression of these genes. It should be noted that in this study the induction of angiotensin by hypoglycemia did not reach statistical significance, so these results are not presented here.
FIG. 3.
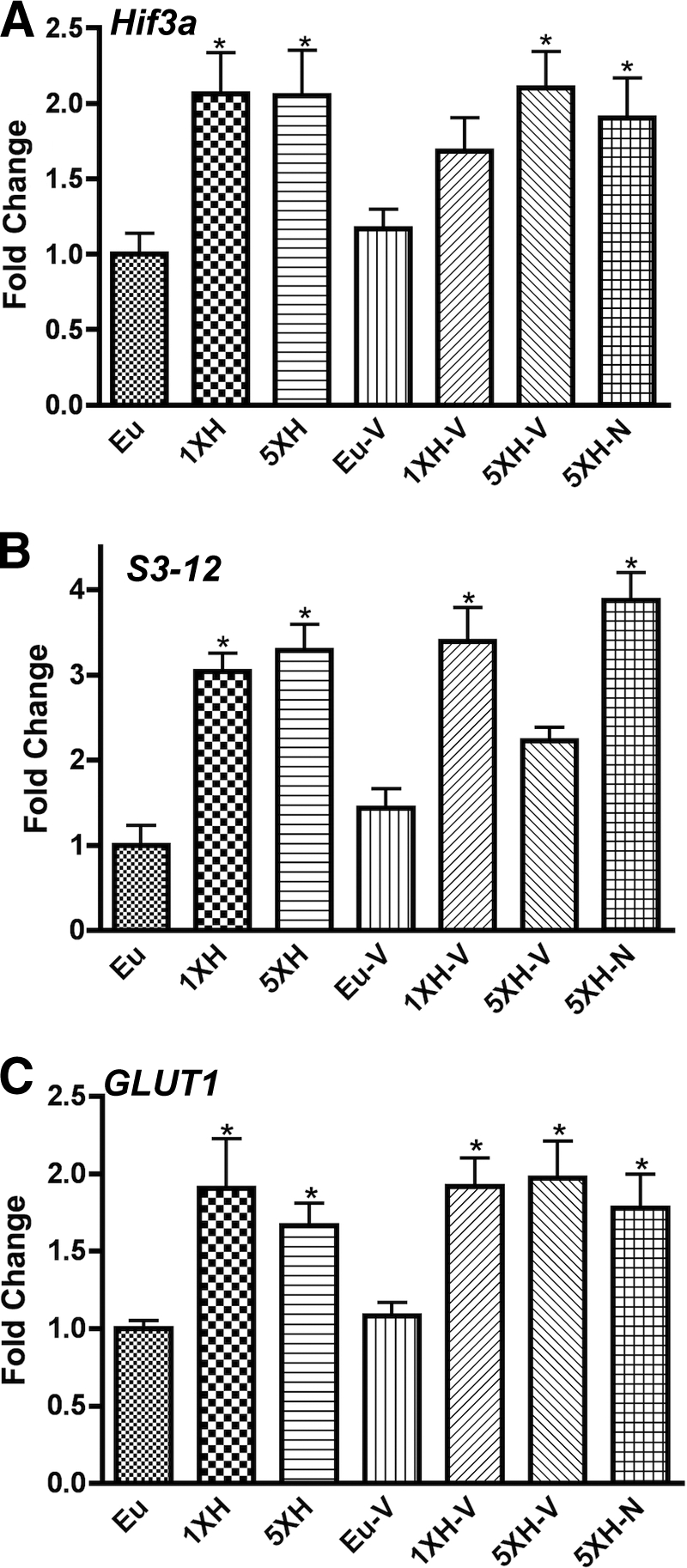
Real-time qPCR data for murine hypothalamic genes that do not correlate with counterregulatory failure or its reversal by naloxone or valsartan. Relative expression levels of (A) Hif3a, (B) S3-12, and (C) GLUT1 were assessed using custom PCR arrays in the same groups described in Fig. 1. Animals were killed 4 h after the final insulin or saline injection. Data for each gene were normalized to a panel of housekeeping transcripts and expressed as fold change compared with the saline-injected (euglycemic) group. Data are means ± SE (n = 6 for all groups). *P < 0.05 as compared with the euglycemic group (Dunnett test).
Induction of Pdk4, Gpd1, Angptl4, and Cdkn1a by acute hypoglycemia fails after antecedent hypoglycemia: prevention of failure by naloxone, not valsartan.
Another set of genes was induced by acute hypoglycemia but not after antecedent hypoglycemia: Pdk4 (pyruvate dehydrogenase kinase 4), Gpd1 (glycerol 3-phosphate dehydrogenase isoform 1), Angptl4 (also known as fasting-induced adipose factor), and Cdkn1a (also known as p21) (Fig. 4). The failure of these genes to respond to hypoglycemia after antecedent hypoglycemia was prevented by treatment with naloxone, but not by treatment with valsartan (Fig. 4).
FIG. 4.
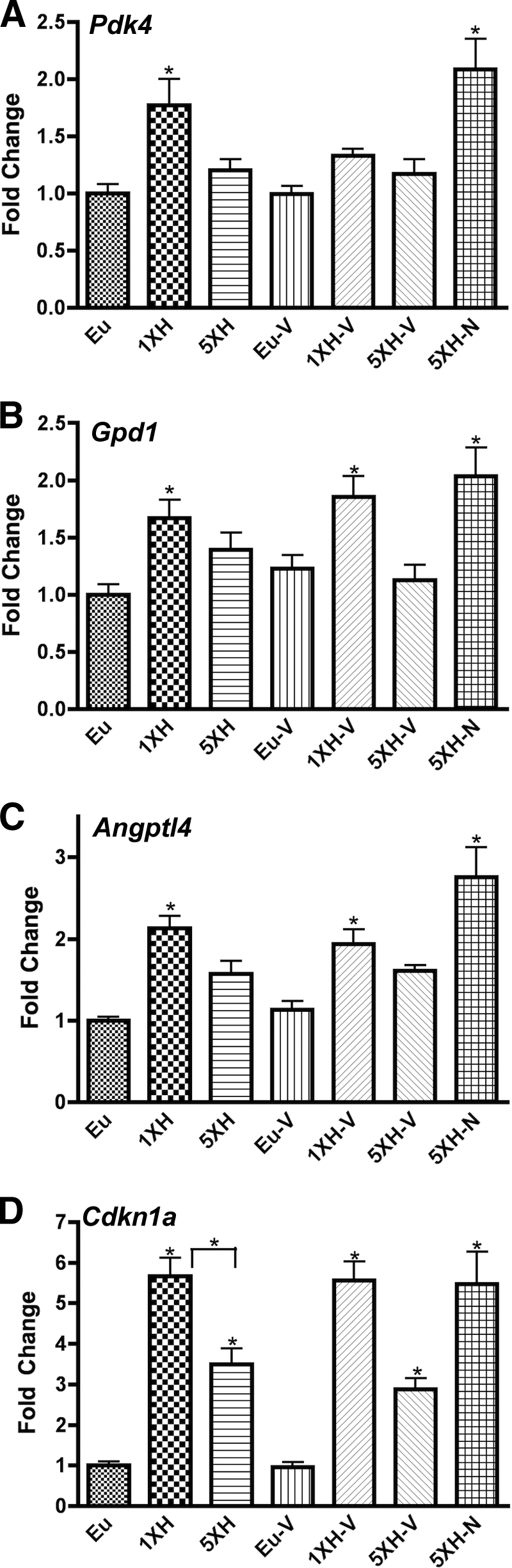
Real-time qPCR data for murine hypothalamic genes that fail to respond after antecedent hypoglycemia, and the failure was prevented by naloxone, but not valsartan. Relative expression levels of (A) Pdk4, (B) Gpd1, (C) Angptl4, and (D) Cdkn1a were assessed using custom PCR arrays from SABiosciences in the same groups described in Fig. 1. Animals were killed 4 h after the final insulin or saline injection. Data for each gene were normalized to a panel of housekeeping transcripts and expressed as fold change compared with the saline-injected (euglycemic) group. Data are means ± SE (n = 6 for all groups). *P < 0.05 as compared with the euglycemic group (Dunnett test).
Naloxone prevents the regulation of Gpd2, Cxcl14, and Sox17 by hypoglycemia.
A different pattern of expression was observed for the genes in Fig. 5: the regulation of these genes by hypoglycemia was not impaired by antecedent hypoglycemia but was prevented by naloxone, though not by valsartan. In contrast to Gpd2 (glycerol 3-phosphate dehydrogenase isoform 2) and Cxcl14 (chemokine [C-X-C] motif ligand14), which were inhibited by hypoglycemia, Sox17 (SRY-box containing gene 17) was induced by hypoglycemia.
FIG. 5.

Real-time qPCR data for murine hypothalamic genes whose regulation by hypoglycemia were not impaired by antecedent hypoglycemia but were prevented by naloxone, though not by valsartan. Relative expression levels of (A) Gpd2 (glycerol 3-phosphate dehydrogenase 2, mitochondrial), (B) Cxcl14, and (C) Sox17 were assessed using custom PCR arrays from SABiosciences in the same groups described in Fig. 1. Animals were killed 4 h after the final insulin or saline injection. Data for each gene were normalized to a panel of housekeeping transcripts and expressed as fold change compared with the saline-injected (euglycemic) group. Data are means ± SE (n = 6 for all groups). *P < 0.05 as compared with the euglycemic group (Dunnett test).
Naloxone prevents the inhibition of Cpt1a by repetitive hypoglycemia.
The only gene in our panel that was significantly regulated by repetitive hypoglycemia but not acute hypoglycemia was Cpt1a. This inhibition was prevented by naloxone (Fig. 6).
FIG. 6.

Hypothalamic expression of Cpt1a was significantly regulated by repetitive hypoglycemia but not acute hypoglycemia, and this inhibition was prevented by naloxone. Relative expression level of Cpt1a (carnitine palmitoyltransferease 1a, liver) was assessed with custom PCR arrays from SABiosciences in the same groups described in Fig. 1. Animals were killed 4 h after the final insulin or saline injection. Data for each gene were normalized to a panel of housekeeping transcripts and expressed as fold change compared with the saline-injected (euglycemic) group. Data are means ± SE (n = 6 for all groups). *P < 0.05 as compared with the euglycemic group (Dunnett test).
Induction of Ucp2 and Pnpla2 by acute hypoglycemia fails after repetitive hypoglycemia, and failure is not reversed by naloxone or valsartan.
Finally, Fig. 7 depicts the genes whose induction by acute hypoglycemia failed after antecedent hypoglycemia and whose failure was not prevented by naloxone. These genes were Ucp2 (Uncoupling Protein 2) and Pnpla2 (also known as adipose triglyceride lipase).
FIG. 7.

Hypothalamic expression of genes whose induction by acute hypoglycemia was prevented by antecedent hypoglycemia but not maintained by exposure to naloxone. Relative expression levels of (A) Ucp2 (uncoupling protein 2) and (B) Pnpla2 (patatin-like phospholipase domain containing 2) were assessed with custom PCR arrays from SABiosciences in the same groups described in Fig. 1. Animals were killed 4 h after the final insulin or saline injection. Data for each gene were normalized to a panel of housekeeping transcripts and expressed as fold change compared with the saline-injected (euglycemic) group. Data are means ± SE (n = 6 for all groups). *P < 0.05 as compared with the euglycemic group (Dunnett test).
DISCUSSION
In these studies, we observed that 4 days of antecedent hypoglycemia in mice completely prevented the elevation of corticosterone produced by acute hypoglycemia (Fig. 2A) and glucagon (Fig. 2B) but not epinephrine (Fig. 2C) when measured 240 min after the injection of insulin. The counterregulatory failure to increase corticosterone was prevented by naloxone, but not valsartan, and the counterregulatory failure of glucagon was not prevented by either treatment.
The pattern of gene expression in these studies suggests a metabolic basis for HAAF and its prevention by naloxone. (We conclude that the effects of insulin-induced hypoglycemia on gene expression are due to hypoglycemia, not insulin, since in every case the effects of insulin-induced hypoglycemia were in the same direction as produced by fasting, a condition characterized by reduced glucose and reduced insulin.) First, it should be noted that acute hypoglycemia–induced Pdk4 (Fig. 4), which inhibits pyruvate dehydrogenase, constitutes a classic mechanism to shift metabolic economy away from glycolysis and toward β-oxidation (24). Such a shift would be expected to enhance sensitivity to hypoglycemia by reducing glucose metabolism. However, the induction of Pdk4 was reversed after antecedent hypoglycemia and was prevented by valsartan, conditions in which glucocorticoid induction was impaired. Most important, naloxone treatment maintained the induction of Pdk4 by hypoglycemia in association with the maintenance of the glucocorticoid response. We have previously reported that estradiol, which impairs counterregulatory responses in humans (25) and rats (26), inhibits hypothalamic Pdk4 in association with impaired hypothalamic responses to hypoglycemia (27). Similarly, Jiang et al. (28) demonstrated with the use of metabolic tracers that recurrent antecedent hypoglycemia caused a robust increase in neuronal, but not glial, pyruvate dehydrogenase activity in association with counterregulatory failure, consistent with a decrease in Pdk4 activity. This set of observations suggests that HAAF is caused by a failure to maintain the shift away from glycolysis toward alternate fuel use in neurons, and that naloxone prevents HAAF by maintaining this shift.
The pattern of expression of other genes supports this metabolic shift hypothesis. For example, like Pdk4, the induction of Angptl4 by hypoglycemia fails after antecedent hypoglycemia, and this failure is prevented by naloxone. Angptl4 stimulates β-oxidation and uncoupling (29), directly supporting that acute hypoglycemia reprograms hypothalamic metabolism away from glycolysis and toward alternate fuel use (β-oxidation), that HAAF is associated with the failure of this reprogramming, and that naloxone prevents HAAF by preventing this failure. Similarly, hypothalamic Ucp2 has recently been shown to mediate an induction of β-oxidation (23). Similarly, repetitive hypoglycemia inhibited Cpt1a (Fig. 6), an effect prevented by naloxone. β-Oxidation mediated by Cpt1 constitutes a key element by which hypothalamic neurons sense nutritional state, plausibly by reducing nutrient flux through pathways that metabolize glucose (23,30). Therefore the reduction in β-oxidation produced by repetitive hypoglycemia would be expected to increase nutrient flux through pathways that metabolize glucose, thus increasing sensitivity to glucose and reducing sensitivity to hypoglycemia. The prevention of this effect by naloxone also supports that HAAF is caused by the failure to produce alternative metabolic pathways to glucose utilization.
Furthermore, the induction of Gpd1 by hypoglycemia (Fig. 4) fails after antecedent hypoglycemia, a failure prevented by naloxone but not valsartan. Gpd1 catalyzes the interconversion of glycerol and dihydrooxyacetone (DHA). DHA is converted to glycerol as part of the glycerol NADH shuttle mechanism. However, this shuttle requires the activity of Gpd2, and in this study we observed that hypoglycemia inhibited Gpd2 (Fig. 5), an effect that was prevented by naloxone but not valsartan. Thus, inhibition of the glycerol shuttle activity (which is active during glycolysis) correlated with the failure of the counterregulatory elevation of corticosterone. This suggests that the key metabolic effect of Gpd1 induction by hypoglycemia is to catalyze conversion of glycerol to DHA, providing an alternative to glucose for fuel; that failure in this conversion is associated with counterregulatory failure; and that naloxone prevents counterregulatory failure by maintaining this alternative metabolic pathway.
The other gene most prominently implicated in HAAF and its reversal by naloxone is Cdnk1a, more commonly known as p21, a major inhibitor of cell division. However, the functional significance of Cdnk1a in counterregulation and its failure is unclear. It is plausible that Cdnk1a expression is a reflection, rather than a cause, of counterregulatory failure since this gene is induced by glucocortiocoids (31). Interestingly, Cxc114 appears to produce insulin resistance (32), so its inhibition by hypoglycemia might enhance the inhibitory effect of insulin on the counterregulatory response (33), and reversal of this inhibition might mediate part of the reversal of impairments by naloxone. Finally, Sox17 is the canonical inhibitor of the Wnt signaling pathway (34), and some evidence suggests that the Wnt pathway promotes glycolysis at the expense of β-oxidation (35). Thus, Sox17 may contribute to the apparent switch away from glycolysis produced by hypoglycemia as indicated above, but since the induction continues even after antecedent hypoglycemia, counterregulatory failure is probably not attributable to the Sox17/Wnt pathway.
It should also be noted that many of the genes associated here with impaired counterregulatory elevation of corticosterone are induced by the metabolic transcription factor Ppar-α, including Pdk4, Cpt1a, Ucp2 (36), and Gpd1 (36,37). Indeed, a major function of Ppar-α is to activate the uptake and oxidation of free fatty acids (36) and glycerol metabolism (37). Furthermore, Ppar-α knockout exhibit hypoglycemia during fasting (38), a phenomenon plausibly similar to HAAF. Of particular interest, whole-body glucose use is reduced by infusing an activator of Ppar-α into the third ventricle (38). We therefore speculate that counterregulatory responses are enhanced by hypothalamic activation of Ppar-α, that this action fails after repetitive hypoglycemia, that this failure is reversed by naloxone, and that Ppar-α activators such as WY13643 might also be useful in reversing counterregulatory failure. It must be emphasized however that this speculation has not been directly tested.
To the extent that naloxone prevented the loss of responsiveness to hypoglycemia by antecedent hypoglycemia (e.g., corticosterone, Pdk4, Angptl4, and Cdkn1a/p21), the precise mechanism mediating these effects remains unclear. It seems very likely that these effects of naloxone are mediated by blockade of μ-opioid receptors since these are the main known mechanisms of action of naloxone (39). One of the most prominent responses to hypoglycemia is the release of the natural μ-opioid agonist β-endorphin into the plasma from the anterior pituitary (40). Conversely, infusion of β-endorphin into the hypothalamus inhibits some hypothalamic responses to hypoglycemia (41), which would have the effect of amplifying glucocorticoid responses to hypoglycemia as observed in this study. However, it remains to be determined if β-endorphin released from the pituitary exerts these effects in the hypothalamus or if other sources of opioids within the central nervous system mediate these effects.
Several major caveats apply to the present studies. Firstly, the counterregulatory hormones were measured 240 min after insulin injection, well after the more typical time point of around 120 min (19,20). Since those studies demonstrated counterregulatory failure of epinephrine after only a single antecedent exposure to hypoglycemia, which is similar to the results in humans (42), further analysis will be required to determine if the protocol used in the present studies is in fact an adequate model for human HAAF. Replicating these studies with glucose clamps and sampling blood earlier will clarify this issue, and the use of different doses of insulin could improve the similarity to human HAAF. Secondly, drug levels were not measured, so failure to produce protective effects could be due to low drug levels in the blood. However, the doses of valsartan were chosen based on previous doses that produced neuroprotection without reducing blood pressure (18). Thirdly, valsartan did in fact appear to impair several responses to hypoglycemia, including glucagon and Pdk4, suggesting that the drug was in fact having effects at the dose used. Similarly naloxone clearly produced several effects on hormonal and molecular responses to hypoglycemia. Whether other doses would have produced a better outcome remains to be determined. Another concern is that the magnitudes of the effects on gene expression were rather small. However the results were reliable because in most cases they were observed in more than one condition, and we have corroborated that fasting similarly and significantly regulates every gene described in this study. A more telling concern is whether the effects observed here could functionally account for counterregulation or its failure given the small magnitude of the effects. With respect to this concern, we anticipate that the most important mode of regulation of these gene products is at the allosteric level (e.g., of Cpt1a by malonyl-CoA) and that the regulation of expression functions mainly as a clue to which gene products are involved in various processes. Nevertheless, as always with studies of gene expression, any conclusions must be considered provisional until corroborated by more direct assessment of function; in this case by analysis of metabolic fluxes.
In conclusion we describe here that naloxone, but not valsartan, prevents counterregulatory failure after antecedent hypoglycemia in mice as in humans (11,12). Preservation of counterregulatory responses was associated with the preservation of responsiveness to hypoglycemia of a subset of hypoglycemia-regulated genes reported here for the first time. The pattern of responses of these genes in relation to counterregulatory failure and its prevention by naloxone suggests that naloxone preserves counterregulatory responses by maintaining a metabolic profile in which alternate fuels are used instead of glucose. Nevertheless, since this study did not include groups in which naloxone was only given once or not on the last day, it remains to be determined if the effect of naloxone was to prevent the failure of hypoglycemia-induced responses or to directly induce corticostereone secretion and related changes in hypothalamic gene expression. In contrast, valsartan did not prevent or reverse loss of responsiveness to hypoglycemia of these genes. Nevertheless, the dose of valsartan used is neuroprotective (18) and did block some responses to hypoglycemia, suggesting that the hypothesis that elevated angiotensin produces HAAF is probably incorrect. Further analysis with mice in which either the AT1 or the AT2 receptor has been ablated may clarify this issue. Taken together, these studies suggest that manipulations causing reprogramming of hypothalamic metabolic processes away from glycolysis and toward alternate fuel use might be useful in preventing or reversing HAAF.
ACKNOWLEDGMENTS
These studies were supported by the Juvenile Diabetes Research Foundation.
No potential conflicts of interest relevant to this article were reported.
M.M.P. designed the studies, carried out the animal work and the qPCR and hormone analysis, and wrote the manuscript. J.W.M. carried out the microarray studies and reviewed the manuscript. C.V.M. conceived the studies and wrote the manuscript.
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
See accompanying commentary, p. 24.
REFERENCES
- 1.Cryer PE: Hypoglycemia-associated autonomic failure in diabetes. Am J Physiol Endocrinol Metab 2001;281:E1115–E1121 [DOI] [PubMed] [Google Scholar]
- 2.Borg MA, Tamborlane WV, Shulman GI, Sherwin RS: Local lactate perfusion of the ventromedial hypothalamus suppresses hypoglycemic counterregulation. Diabetes 2003;52:663–666 [DOI] [PubMed] [Google Scholar]
- 3.Borg MA, Borg WP, Tamborlane WV, Brines ML, Shulman GI, Sherwin RS: Chronic hypoglycemia and diabetes impair counterregulation induced by localized 2-deoxy-glucose perfusion of the ventromedial hypothalamus in rats. Diabetes 1999;48:584–587 [DOI] [PubMed] [Google Scholar]
- 4.Borg MA, Sherwin RS, Borg WP, Tamborlane WV, Shulman GI: Local ventromedial hypothalamus glucose perfusion blocks counterregulation during systemic hypoglycemia in awake rats. J Clin Invest 1997;99:361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borg WP, Sherwin RS, During MJ, Borg MA, Shulman GI: Local ventromedial hypothalamus glucopenia triggers counterregulatory hormone release. Diabetes 1995;44:180–184 [DOI] [PubMed] [Google Scholar]
- 6.Borg WP, During MJ, Sherwin RS, Borg MA, Brines ML, Shulman GI: Ventromedial hypothalamic lesions in rats suppress counterregulatory responses to hypoglycemia. J Clin Invest 1994;93:1677–1682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mastaitis JW, Wurmbach E, Cheng H, Sealfon SC, Mobbs CV: Acute induction of gene expression in brain and liver by insulin-induced hypoglycemia. Diabetes 2005;54:952–958 [DOI] [PubMed] [Google Scholar]
- 8.Pedersen-Bjergaard U, Agerholm-Larsen B, Pramming S, Hougaard P, Thorsteinsson B: Prediction of severe hypoglycaemia by angiotensin-converting enzyme activity and genotype in type 1 diabetes. Diabetologia 2003;46:89–96 [DOI] [PubMed] [Google Scholar]
- 9.Pedersen-Bjergaard U, Agerholm-Larsen B, Pramming S, Hougaard P, Thorsteinsson B: Activity of angiotensin-converting enzyme and risk of severe hypoglycaemia in type 1 diabetes mellitus. Lancet 2001;357:1248–1253 [DOI] [PubMed] [Google Scholar]
- 10.Deininger E, Oltmanns KM, Wellhoener P, Fruehwald-Schultes B, Kern W, Heuer B, Dominiak P, Born J, Fehm HL, Peters A: Losartan attenuates symptomatic and hormonal responses to hypoglycemia in humans. Clin Pharmacol Ther 2001;70:362–369 [PubMed] [Google Scholar]
- 11.Caprio S, Gerety G, Tamborlane WV, Jones T, Diamond M, Jacob R, Sherwin RS: Opiate blockade enhances hypoglycemic counterregulation in normal and insulin-dependent diabetic subjects. Am J Physiol 1991;260:E852–E858 [DOI] [PubMed] [Google Scholar]
- 12.Leu J, Cui MH, Shamoon H, Gabriely I: Hypoglycemia-associated autonomic failure is prevented by opioid receptor blockade. J Clin Endocrinol Metab 2009;94:3372–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Madsen BK, Hølmer P, Ibsen H, Christensen NJ: The influence of captopril on the epinephrine response to insulin-induced hypoglycemia in humans. The interaction between the renin-angiotensin system and the sympathetic nervous system. Am J Hypertens 1992;5:361–365 [DOI] [PubMed] [Google Scholar]
- 14.Worck RH, Frandsen E, Ibsen H, Petersen JS: AT1 and AT2 receptor blockade and epinephrine release during insulin-induced hypoglycemia. Hypertension 1998;31:384–390 [DOI] [PubMed] [Google Scholar]
- 15.Li XC, Widdop RE: AT2 receptor-mediated vasodilatation is unmasked by AT1 receptor blockade in conscious SHR. Br J Pharmacol 2004;142:821–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T: International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev 2000;52:415–472 [PubMed] [Google Scholar]
- 17.Han HJ, Heo JS, Lee YJ: ANG II increases 2-deoxyglucose uptake in mouse embryonic stem cells. Life Sci 2005;77:1916–1933 [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Ho L, Chen L, Zhao Z, Zhao W, Qian X, Humala N, Seror I, Bartholomew S, Rosendorff C, Pasinetti GM: Valsartan lowers brain beta-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J Clin Invest 2007;117:3393–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacobson L, Ansari T, Potts J, McGuinness OP: Glucocorticoid-deficient corticotropin-releasing hormone knockout mice maintain glucose requirements but not autonomic responses during repeated hypoglycemia. Am J Physiol Endocrinol Metab 2006;291:E15–E22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacobson L, Ansari T, McGuinness OP: Counterregulatory deficits occur within 24 h of a single hypoglycemic episode in conscious, unrestrained, chronically cannulated mice. Am J Physiol Endocrinol Metab 2006;290:E678–E684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levin BE, Becker TC, Eiki J, Zhang BB, Dunn-Meynell AA: Ventromedial hypothalamic glucokinase is an important mediator of the counterregulatory response to insulin-induced hypoglycemia. Diabetes 2008;57:1371–1379 [DOI] [PubMed] [Google Scholar]
- 22.Kerr D, Sherwin RS, Pavalkis F, Fayad PB, Sikorski L, Rife F, Tamborlane WV, During MJ: Effect of caffeine on the recognition of and responses to hypoglycemia in humans. Ann Intern Med 1993;119:799–804 [DOI] [PubMed] [Google Scholar]
- 23.Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, Tschöp MH, Shanabrough M, Cline G, Shulman GI, Coppola A, Gao XB, Horvath TL, Diano S: UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature 2008;454:846–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mobbs CV, Mastaitis JW, Zhang M, Isoda F, Cheng H, Yen K: Secrets of the lac operon. Glucose hysteresis as a mechanism in dietary restriction, aging and disease. Interdiscip Top Gerontol 2007;35:39–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sandoval DA, Ertl AC, Richardson MA, Tate DB, Davis SN: Estrogen blunts neuroendocrine and metabolic responses to hypoglycemia. Diabetes 2003;52:1749–1755 [DOI] [PubMed] [Google Scholar]
- 26.Adams JM, Legan SJ, Ott CE, Jackson BA: Modulation of hypoglycemia-induced increases in plasma epinephrine by estrogen in the female rat. J Neurosci Res 2005;79:360–367 [DOI] [PubMed] [Google Scholar]
- 27.Cheng H, Isoda F, Mobbs CV: Estradiol impairs hypothalamic molecular responses to hypoglycemia. Brain Res 2009;1280:77–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang L, Herzog RI, Mason GF, de Graaf RA, Rothman DL, Sherwin RS, Behar KL: Recurrent antecedent hypoglycemia alters neuronal oxidative metabolism in vivo. Diabetes 2009;58:1266–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mandard S, Zandbergen F, van Straten E, Wahli W, Kuipers F, Müller M, Kersten S: The fasting-induced adipose factor/angiopoietin-like protein 4 is physically associated with lipoproteins and governs plasma lipid levels and adiposity. J Biol Chem 2006;281:934–944 [DOI] [PubMed] [Google Scholar]
- 30.Pocai A, Lam TK, Obici S, Gutierrez-Juarez R, Muse ED, Arduini A, Rossetti L: Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J Clin Invest 2006;116:1081–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harms C, Albrecht K, Harms U, Seidel K, Hauck L, Baldinger T, Hübner D, Kronenberg G, An J, Ruscher K, Meisel A, Dirnagl U, von Harsdorf R, Endres M, Hörtnagl H: Phosphatidylinositol 3-Akt-kinase-dependent phosphorylation of p21(Waf1/Cip1) as a novel mechanism of neuroprotection by glucocorticoids. J Neurosci 2007;27:4562–4571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hara T, Nakayama Y: CXCL14 and insulin action. Vitam Horm 2009;80:107–123 [DOI] [PubMed] [Google Scholar]
- 33.Paranjape SA, Chan O, Zhu W, Horblitt AM, McNay EC, Cresswell JA, Bogan JS, McCrimmon RJ, Sherwin RS: Influence of insulin in the ventromedial hypothalamus on pancreatic glucagon secretion in vivo. Diabetes 2010;59:1521–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu DY, Wang ZM, Li-Chen, Wang BL, Shen ZZ, Huang W, Shao ZM: Sox17, the canonical Wnt antagonist, is epigenetically inactivated by promoter methylation in human breast cancer. Breast Cancer Res Treat 2010;119:601–612 [DOI] [PubMed] [Google Scholar]
- 35.Chafey P, Finzi L, Boisgard R, Caüzac M, Clary G, Broussard C, Pégorier JP, Guillonneau F, Mayeux P, Camoin L, Tavitian B, Colnot S, Perret C: Proteomic analysis of beta-catenin activation in mouse liver by DIGE analysis identifies glucose metabolism as a new target of the Wnt pathway. Proteomics 2009;9:3889–3900 [DOI] [PubMed] [Google Scholar]
- 36.Mandard S, Müller M, Kersten S: Peroxisome proliferator-activated receptor alpha target genes. Cell Mol Life Sci 2004;61:393–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patsouris D, Mandard S, Voshol PJ, Escher P, Tan NS, Havekes LM, Koenig W, März W, Tafuri S, Wahli W, Müller M, Kersten S: PPARalpha governs glycerol metabolism. J Clin Invest 2004;114:94–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knauf C, Rieusset J, Foretz M, Cani PD, Uldry M, Hosokawa M, Martinez E, Bringart M, Waget A, Kersten S, Desvergne B, Gremlich S, Wahli W, Seydoux J, Delzenne NM, Thorens B, Burcelin R: Peroxisome proliferator-activated receptor-alpha-null mice have increased white adipose tissue glucose utilization, GLUT4, and fat mass: role in liver and brain. Endocrinology 2006;147:4067–4078 [DOI] [PubMed] [Google Scholar]
- 39.Goodman AJ, Le Bourdonnec B, Dolle RE: Mu opioid receptor antagonists: recent developments. ChemMedChem 2007;2:1552–1570 [DOI] [PubMed] [Google Scholar]
- 40.Nakao K, Nakai Y, Jingami H, Oki S, Fukata J, Imura H: Substantial rise of plasma beta-endorphin levels after insulin-induced hypoglycemia in human subjects. J Clin Endocrinol Metab 1979;49:838–841 [DOI] [PubMed] [Google Scholar]
- 41.Suda T, Sato Y, Sumitomo T, Nakano Y, Tozawa F, Iwai I, Yamada M, Demura H: Beta-endorphin inhibits hypoglycemia-induced gene expression of corticotropin-releasing factor in the rat hypothalamus. Endocrinology 1992;130:1325–1330 [DOI] [PubMed] [Google Scholar]
- 42.Hvidberg A, Fanelli CG, Hershey T, Terkamp C, Craft S, Cryer PE: Impact of recent antecedent hypoglycemia on hypoglycemic cognitive dysfunction in nondiabetic humans. Diabetes 1996;45:1030–1036 [DOI] [PubMed] [Google Scholar]