

Abstract
Serotonin transporter (SERT) is the main target for widely used antidepressant agents. Several of these drugs, including imipramine, citalopram, sertraline, and fluoxetine (Prozac), bound more avidly to SERT in the presence of Cl−. In contrast, Cl− did not enhance cocaine or paroxetine binding. A Cl− binding site recently identified in SERT, and shown to be important for Cl− dependent transport, was also critical for the Cl− dependence of antidepressant affinity. Mutation of the residues contributing to this site eliminated the Cl−-mediated affinity increase for imipramine and fluoxetine. Analysis of ligand docking to a single state of SERT indicated only small differences in the energy of interaction between bound ligands and Cl−. These differences in interaction energy cannot account for the affinity differences observed for Cl− dependence. However, fluoxetine binding led to a conformational change, detected by cysteine accessibility experiments, that was qualitatively different from that induced by cocaine or other ligands. Given the known Cl− requirement for serotonin-induced conformational changes, we propose that Cl− binding facilitates conformational changes required for optimal binding of fluoxetine and other antidepressant drugs.
Introduction
Serotonin transporter (SERT) is responsible for reuptake of the neurotransmitter serotonin [5-hydroxytryptamine (5-HT)] into neurons after its release (Rudnick, 2006). SERT belongs to the neurotransmitter sodium symporter (NSS) (SLC6) family of transporters that also includes transporters for dopamine (DAT), norepinephrine (NET), glycine, and GABA. It is a target for drugs of abuse, including cocaine and 3,4-methylenedioxymethamphet-amine (ecstasy) (Rudnick and Wall, 1992). Furthermore, SERT is associated with several psychiatric disorders, such as major depression, anxiety, and obsessive compulsive disorder (Murphy et al., 2004), and represents the primary target for widely used antidepressants, including fluoxetine (Prozac), sertraline (Zoloft), citalopram (Celexa), and paroxetine (Paxil). Although computational and mutagenesis approaches have identified binding sites for substrates and cocaine (Beuming et al., 2008; Celik et al., 2008; Kaufmann et al., 2009), molecular determinants of antidepressant interaction with target proteins remain to be identified.
Early studies on imipramine binding to SERT suggested that Na+ and Cl−, both required for 5-HT transport, might also determine antidepressant affinity (Talvenheimo et al., 1979; Humphreys et al., 1994). However, binding of cocaine and its analogs (−)-2-β-carbomethoxy-3β-(4-fluorophenyl) tropane (CFT) and (−)-2β-carbomethoxy-3β-(4-iodophenyl) tropane (β-CIT) to SERT was stimulated only by Na+ and not by Cl− (Rudnick and Wall, 1991; Wall et al., 1993). Paroxetine affinity increased with Na+ but not Cl− (Cool et al., 1990), and Na+ increased SERT affinity for fluoxetine and citalopram (Humphreys et al., 1994; Wong et al., 1995), but the effect of Cl− was not reported. Thus, of all these compounds, only imipramine was shown to bind more avidly to SERT in the presence of Cl− (Talvenheimo et al., 1979; Humphreys et al., 1994). Despite these intriguing differences between SERT inhibitors, the mechanism by which ions stimulate their binding has not yet been elucidated.
As stated above, both Na+ and Cl− are absolutely required for 5-HT transport (Lingjaerde, 1969; Sneddon, 1969; Rudnick, 1977). The initial step in 5-HT transport is binding of 5-HT+, Na+, and Cl−. Evidence suggests that all three of these solutes must bind before SERT can undergo the conformational changes leading to translocation (Nelson and Rudnick, 1979; Zhang and Rudnick, 2006). The x-ray crystal structure of the amino acid transporter (LeuT), a bacterial homolog of SERT, includes two bound sodium ions (Na1 and Na2) close to the bound leucine (Yamashita et al., 2005). Evidence for a Cl− site in neurotransmitter transporters has been directly provided by studies with SERT and the GABA transporter GAT-1 (Forrest et al., 2007; Zomot et al., 2007). The proposed Cl− site in SERT is close to the probable Na1 site and is formed by residues in transmembrane (TM) helices 2, 6, and 7. Consistent with its role in coupling, Cl− shares two coordinating residues, Ser336 and Asn368, with the Na1 site (Forrest et al., 2007; Zomot et al., 2007).
Our present work reveals that affinities of several antidepressants (fluoxetine, citalopram, and sertraline) are enhanced by Cl−. We also show that maximal binding of fluoxetine involves formation of a conformational state distinct from that favored by cocaine. We propose that Cl− may stimulate fluoxetine binding by facilitating formation of this novel state.
Materials and Methods
Materials.
[125I]β-CIT was purchased from PerkinElmer Life and Analytical Sciences, and 2-aminoethyl methanethiosulfonate hydrobromide (MTSEA) was from Anatrace. Fluoxetine, imipramine, ibogaine, paroxetine, 5-HT, cocaine, and unlabeled β-CIT were from Sigma, sertraline from Pfizer, and citalopram from Lundbeck.
Expression of rat SERT mutants and preparation of crude membranes.
Site-directed mutagenesis of rat SERT was performed using the Stratagene QuickChange kit. The mutated region was subcloned into rSERT cDNA with a c-Myc epitope tag at the NH2 terminus and a FLAG epitope tag at the COOH terminus. All mutations were confirmed by DNA sequencing.
The VTF7-3 virus/T7 RNA polymerase system was used for heterologous expression of the mutants and has been described in detail previously (Blakely et al., 1991). Briefly, HeLa cells were seeded in 75 cm2 cell culture flasks and allowed to grow to confluency. The confluent cells were infected with recombinant VTF7–3 virus and transfected with plasmid containing rSERT cDNA under control of the T7 promoter. At 24 h after transfection, the cells were rinsed with room temperature binding buffer [10 mm HEPES/150 mm NaCl adjusted to pH 8.0 with N-methyl-d-glucamine (NMDG)], scraped, and collected with 10 ml of binding buffer containing 0.5% protease inhibitor mixture (Sigma) and 100 μm phenylmethylsulfonyl fluoride. Subsequently, the cells were lysed by two cycles of sonication and freeze thawing. The crude membranes were collected by centrifugation at 11,000 rpm (SS34 rotor; Sorvall) for 20 min at 4°C, and the pellet was resuspended thoroughly in 1 ml of the same buffer. Membranes were stored at −80°C until used.
Binding assays.
All binding assays were performed in Multiscreen-FB 96-well filtration plates (Millipore), which were preincubated with 0.1% polyethyleneimine (100 μl/well). Before adding the membrane samples, the plates were rinsed three times with 100 μl of room temperature binding buffer. Four different binding buffers were used in this study, as indicated (in mm): 10 HEPES/150 NaCl, 10 HEPES/150 sodium isethionate (2-hydroxyethanesulfonic acid, monosodium salt) (Cl−-free), 10 HEPES/150 NMDG-Cl (Na+-free), and 10 HEPES/150 NMDG-diatrizoate (NaCl-free).
Crude membranes, prepared as described above, were thawed on ice and diluted in one of the above binding buffers depending on the desired ionic content. One hundred microliter samples of the membrane suspension were added to each well and were washed three times by filtration with 100 μl of the same buffer. Binding was initiated by addition of 100 μl of binding buffer containing the indicated concentration range of each inhibitor and 0.15 nm [125I]β-CIT (RTI-55; PerkinElmer Life and Analytical Sciences) and was allowed to proceed for 1 h at room temperature with gentle shaking. The reaction was stopped by washing all wells with 100 μl of ice-cold binding buffer. The filters were removed from the plate, transferred to Wallac 96-well Isoplates (part number 6005070) soaked with 150 μl of Optifluor and counted with Wallac Microbeta plate counter.
S277C accessibility assays.
To determine the conformational effects of inhibitors, we measured their influence on MTSEA inactivation of [125I]β-CIT binding to membranes of the SERT mutant S277C (Zhang and Rudnick, 2006). This mutant was generated in the X5C [SERT mutant in which the five most reactive cysteines were mutated (C15A/C21A/C109A/C357I/C622A)] (Sato et al., 2004). The cysteine at position 277 becomes accessible when the cytoplasmic permeation pathway opens (Zhang and Rudnick, 2006). The membranes were applied as above to Multiscreen-FB plates and preincubated with cocaine, ibogaine, imipramine, or fluoxetine (as indicated) for 10 min at room temperature with gentle shaking. Subsequently, the medium was replaced with binding buffer (10 mm HEPES, pH 8.0 with 150 mm salt as indicated), containing MTSEA and the inhibitor (at the same concentration as in the preincubation). After 15 min of incubation, the reaction with MTSEA was terminated by washing the membranes five times with binding buffer to make sure that MTSEA and the inhibitors were removed. Residual β-CIT binding was determined as described above.
Data analysis.
Nonlinear regression fits of experimental data were performed with Origin (OriginLab), which uses the Marquardt–Levenberg nonlinear least-squares curve-fitting algorithm. Statistical analysis comparing multiple independent experiments was performed in Origin8 using either a two-sample t test or one-way ANOVA, followed by the Tukey's multiple comparisons test as indicated. Error values were calculated as SEM using values from separate independent experiments in each case. Unless indicated otherwise, error bars in the figures are SDs for replicate measurements.
Computational analysis.
Molecular models of SERT containing two Na+ ions were constructed based on the extracellular-facing substrate-bound conformation of LeuT [Protein Data Base identification number 2a65 (Yamashita et al., 2005)], as described previously (Celik et al., 2008; Forrest et al., 2008), and were energy minimized to a maximum root mean square deviation of 0.18 Å. Docking was performed in the presence and absence of a Cl− ion modeled at the position of E290 from LeuT (Forrest et al., 2008).
Molecular models of imipramine, fluoxetine, paroxetine, and cocaine, with parameters generated using MacroModel 9.5 (Schrödinger), were then docked into the SERT models using the InducedFit protocol (Schrödinger) (Sherman et al., 2006). Evidence of competitive inhibition for, e.g., sertraline, citalopram, paroxetine, and imipramine (Talvenheimo et al., 1979; Thomas et al., 1987; Koe et al., 1990; Apparsundaram et al., 2008), suggests that they either share the same binding site with 5-HT or that their binding sites overlap. This so-called S1 site overlaps with the leucine binding site in LeuT. However, some evidence suggests that antidepressants bind to a second site in the extracellular permeation pathway of LeuT (Singh et al., 2007; Zhou et al., 2007, 2009). The possibility that a second molecule of substrate binds at this S2 site has also been proposed (Shi et al., 2008). Thus, compounds were docked to the region of either S1 or S2 site in the SERT model. The InducedFit protocol consisted of three stages.
First, multiple conformations and orientations of the ligand in the binding site were docked using Glide version 4.5 (Schrödinger) (Friesner et al., 2004; Halgren et al., 2004) and screened using the SP scoring function with a softened van der Waals potential (scaled down by a factor of two). In separate calculations, a Phe-341 to alanine mutation was introduced during docking of all ligands to the S1 site to maximize the search space in this step: Phe-341 is not conserved in the NSS family, and its conformation significantly impacts the shape of the binding pocket in the SERT models used.
Second, the protein structure was refined around these initial docked conformations; side-chains within 5 Å of the drug (including Phe-341) were rebuilt, refined, and energy minimized with the ligand using Prime version 1.6 (Schrödinger). Up to 20 protein–ligand complexes from this step, with energies within 30 kcal/mol of the lowest-energy conformation, were retained. This refinement step was also repeated, while allowing adaptability of nearby uncoiled main-chain regions, using Prime loop refinement immediately before the side-chain refinement step. The flexible main-chain regions in S1 were from TM1 and TM6 (residues 96–99 and 337–343, respectively) and in S2 consisted of the EL4a–EL4b loop (residues 397–402) and the EL5 loop between TM9 and TM10 (residues 484–489).
Third, the ligand was docked back into the newly optimized protein. The final poses were clustered using the average-linkage clustering program NMRCLUST (Kelley et al., 1996).
Electrostatic interaction energies between the Cl− ion and the inhibitor were calculated as the product of the partial charges of the drug and the potential generated by the Cl− ion, in the presence of the dielectric environment of the protein, drug, ions, and a low-dielectric slab mimicking the membrane. Electrostatic potentials were calculated with DelPhi version 4 (Rocchia et al., 2002), as described previously (Forrest et al., 2007), and atomic charges for the drugs were generated with MacroModel.
Results
Chloride modulates antidepressant binding to SERT
Previous observations demonstrated that imipramine binding to SERT was stimulated by Cl− (Humphreys et al., 1994), whereas binding of other inhibitors, such as cocaine and paroxetine, was not (Cool et al., 1990; Rudnick and Wall, 1991; Wall et al., 1993). Sodium, however, stimulated binding of all tested inhibitors (Cool et al., 1990; Humphreys et al., 1994). To further our understanding of the role of ions in binding, we tested the effect of Cl− and Na+ on binding of a set of SERT inhibitors, including the tricyclic antidepressant imipramine, the antidepressants fluoxetine, sertraline, citalopram, and paroxetine, and also cocaine, its analog β-CIT, and ibogaine. Several studies support the proposal that all these compounds bind in the substrate site on neurotransmitter transporters (Beuming et al., 2008; Andersen et al., 2009), although others suggest that a secondary site is involved (Zhou et al., 2007, 2009). In either case, inhibitor binding is believed to completely block transport function.
Binding affinity was determined by competition with [125I]β-CIT binding to membranes from HeLa cells expressing wild-type (WT) SERT. KI values were measured in the presence and absence of Na+ or Cl−, replacing Na+ with the cation NMDG and Cl− with isethionate or diatrizoate. We first calculated KD values for β-CIT in all four possible ionic conditions (Table 1). The KD for β-CIT binding was unaffected by the absence of Cl− but was increased fourfold by removing Na+ (Table 1). Because the Na+ dependence of β-CIT affinity could influence the ability of other ligands to displace this radioligand, we used a concentration of [125I]β-CIT (0.15 nm) well below the KD for binding (0.6–2.6 nm). Moreover, we corrected for the small change in β-CIT affinity using the Cheng–Prusoff equation (Cheng and Prusoff, 1973).
Table 1.
KI values for displacement of β-CIT from wild-type SERT
| NaCl | Na-Ise | NMDG-Cl | NMDG-diatrizoate | |
|---|---|---|---|---|
| β-CIT | 0.6 ± 0.2 | 0.6 ± 0.2 | 2.6 ± 0.4* | 2.5 ± 0.8* |
| Cocaine | 486 ± 44 | 405 ± 27 | 1657 ± 47* | ND |
| Paroxetine | 2.0 ± 0.4 | 2.0 ± 0.8 | 6.4 ± 1.1* | ND |
| Imipramine | 61 ± 4 | 221 ± 13* | 296 ± 36* | 518 ± 69* |
| Fluoxetine | 13 ± 1 | 38 ± 0.8* | 48 ± 3* | 66 ± 8* |
| Sertraline | 4.2 ± 0.7 | 7.0 ± 1.3* | 30 ± 3* | ND |
| Citalopram | 3.1 ± 0.5 | 10.5 ± 3.2* | 16 ± 2* | ND |
| Ibogaine | 3402 ± 387 | 2592 ± 518 | 1112 ± 213* | ND |
Displacement of β-CIT binding from wild-type rSERT was performed as described for Figure 1 under four ionic conditions [NaCl, Na-isethionate (Na-Ise), NMDG-Cl, and NMDG-diatrizoate]. KD values (in bold) (in nanomolar concentrations) were calculated for β-CIT binding and were used to determine KI values (in nanomolar concentrations) of the other inhibitors using the Cheng–Prusoff equation (Cheng and Prusoff, 1973). ND, Not determined. Statistical analysis from three or four independent experiments (2 for Na+ dependences published previously; paroxetine, fluoxetine, and citalopram in NMDG-Cl) was performed in Origin8 using one-way ANOVA, followed by Tukey's multiple comparisons test.
*p < 0.03, statistically significant differences between affinities in NaCl versus the other ionic condition. The difference between NMDG-Cl and NMDG-diatrizoate was statistically significant for imipramine (p = 0.006) but not for fluoxetine (p = 0.06).
The effect of Na+ on binding is shown in Figure 1 and Table 1. Consistent with previous studies, the potency for most of the ligands tested was reduced in the absence of Na+. KI values for cocaine, β-CIT, imipramine, fluoxetine, paroxetine, sertraline, and citalopram increased approximately threefold to sevenfold in NMDG-Cl relative to NaCl. The sole exception was ibogaine, an inhibitor that stabilizes the cytoplasmic-facing conformation of SERT (Jacobs et al., 2007), which was more potent in the absence of Na+ (KI of 3.4 ± 0.4 and 1.1 ± 0.2 μm in NaCl and NMDG-Cl, respectively).
Figure 1.

Displacement of β-CIT binding to wild-type rat SERT by cocaine, ibogaine, imipramine, and fluoxetine in the presence or absence of Na+. Membranes from HeLa cells expressing rSERT were incubated with 0.15 nm [125I]β-CIT and the indicated concentrations of cocaine (0–20 μm), ibogaine (0–80 μm), imipramine (0–1.5 μm), and fluoxetine (0–1 μm) in either 150 mm NaCl (filled circles) or 150 mm NMDG-Cl (open circles). The concentration of β-CIT used (0.15 nm) was below the KD for binding in NMDG-Cl (2.6 nm), so as to minimize the effect of the lower β-CIT affinity in the absence of Na+. Representative results are presented as the percentage of [125I]β-CIT bound in the absence of the inhibitor. KI values calculated from multiple experiments and the corresponding statistical analyses are presented in Table 1.
We then tested the Cl− dependence of the same set of inhibitors. The data in Figure 2 and Table 1 show that fluoxetine, sertraline, and citalopram, like imipramine, bind more tightly in the presence of Cl−. The increase in affinity ranged from 1.7-fold for sertraline to 3.6-fold for imipramine. In agreement with previous studies, affinities for cocaine, β-CIT, and paroxetine were unaffected by Cl− (Cool et al., 1990; Humphreys et al., 1994). We also tested binding of β-CIT, imipramine, and fluoxetine in the absence of NaCl using NMDG-diatrizoate as a replacement. Although β-CIT binding was similar in NMDG-diatrizoate and NMDG-Cl, imipramine bound with reduced affinity in NaCl-free medium, indicating that each ion independently increased affinity for this antidepressant.
Figure 2.

Displacement of β-CIT binding to SERT by cocaine, ibogaine, imipramine, and fluoxetine in the presence or absence of Cl−. The effect of Cl− on displacement of β-CIT was tested in membranes from HeLa cells expressing wild-type rSERT. The membranes were incubated with 0.15 nm [125I]β-CIT and the indicated concentrations of cocaine (0–20 μm), ibogaine (0–80 μm), imipramine (0–1 μm), and fluoxetine (0–1 μm) in either 150 mm NaCl (filled circles) or 150 mm Na-isethionate (open circles). Representative results are presented as percentage of [125I]β-CIT bound in the absence of the inhibitor. KI values calculated from multiple experiments and the corresponding statistical analyses are presented in Table 1.
Mutation of proposed Cl− binding site residues alters the Cl− dependence of antidepressant binding
We recently proposed a binding site that mediates the Cl− dependence of neurotransmitter transport. Mutations at these positions altered the Cl− dependence of 5-HT transport and in some cases removed the requirement for Cl− (Forrest et al., 2007). To determine whether this same Cl− site is responsible for the Cl− dependence of inhibitor binding, we characterized several mutants at positions Ser-372, Asn-368, Ser-336, and Tyr-121 that were predicted to coordinate Cl− (Forrest et al., 2007). We converted these positions to either aspartic acid, which might be expected to substitute for the negatively charged Cl− ion, or to aliphatic residues (alanine for Ser-372 and Ser-336, leucine or alanine for Asn-368, and leucine for Tyr-121 because smaller replacements at 121 were not expressed well), to prevent that position from coordinating chloride ions. Introduction of aspartate or glutamate at position Tyr-121 eliminated β-CIT binding and those mutants were not further examined.
Using the above mutants, we measured the inhibitory potencies of two antidepressants, fluoxetine and imipramine, that bound more tightly in the presence of Cl−, and cocaine, which was not affected by Cl−. The results are shown in Figure 3 and Table 2. Any mutation, to either aspartic acid or an aliphatic residue, at 121, 336, 368, or 372 eliminated the ability of Cl− to increase fluoxetine or imipramine affinity. For both antidepressants, aliphatic substitutions led to marked reductions in binding. For example, fluoxetine affinities in NaCl were reduced to 11, 13, and 15% of wild-type affinity, respectively, in N368L, S372A, and S336A. Significantly, when aspartic acid was introduced at positions S336 or S372, the affinity remained high (90 ± 11 and 56 ± 2% of WT, respectively) or became even higher than WT in the case of N368D (350 ± 50% of WT). Replacement of Tyr-121 with phenylalanine or leucine or Asn-368 with alanine had an intermediate effect, decreasing fluoxetine affinity to 36 ± 3, 43 ± 2, and 40 ± 4% of wild type, respectively. These mutations had generally the same effects on imipramine binding and its Cl− dependence, the largest difference being that imipramine, but not fluoxetine, affinity was markedly reduced in S336D (Table 2). In contrast to mutations at 121, 336, 368, or 372, mutation of Cys-369 to alanine or serine did not affect Cl− stimulation (Table 2). Cys-369 corresponds to a residue proposed to participate in a Cl− site in GAT-1 (Zomot et al., 2007).
Figure 3.
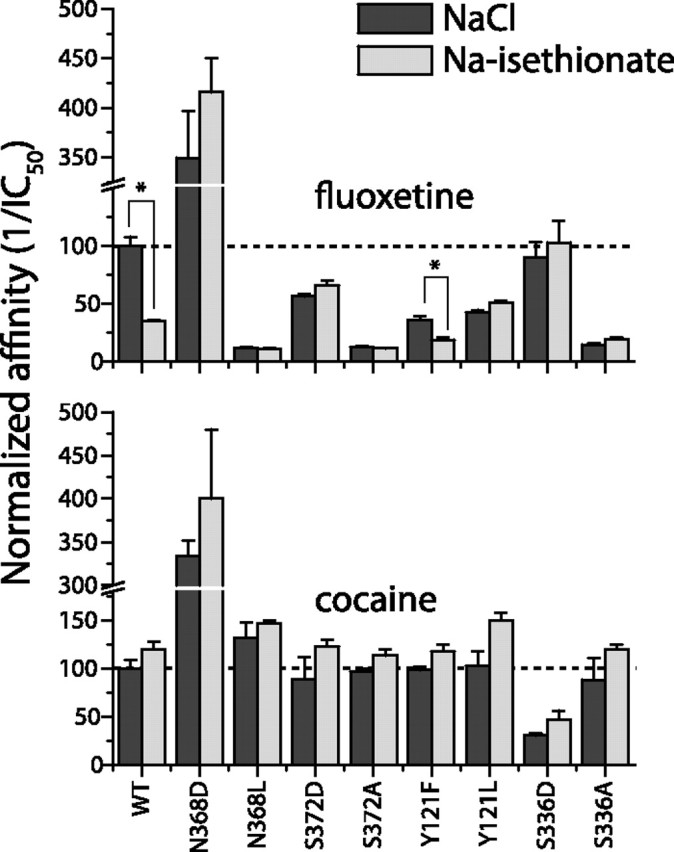
Effect of mutations at the chloride binding site on cocaine and fluoxetine affinity. Binding affinity for fluoxetine (top) and cocaine (bottom) for mutants of Tyr-121, Ser-336, Asn-368, and Ser-372 was determined through displacement of β-CIT binding in the presence and absence of chloride. Crude membranes from cells expressing these mutants were incubated in 150 mm NaCl (darker bars) or 150 mm Na-isethionate (lighter bars) in the presence of 0.15 nm [125I]β-CIT and increasing concentrations of cocaine (0–20 μm) or fluoxetine (0–1 μm). Inhibitor concentrations resulting in half-maximal inhibition of β-CIT binding were calculated and the corresponding affinities (1/IC50) were plotted as percentage of binding affinity for WT SERT in the presence of 150 mm NaCl. Error bars correspond to ±SE from two to four independent experiments. Asterisks indicate a statistically significant increase in affinity in NaCl relative to Na-isethionate.
Table 2.
Effect of Cl− on ligand binding affinity for wild-type and mutant SERT
| Cl− | Cocaine (nm) |
Fluoxetine (nm) |
Imipramine (nm) |
|||
|---|---|---|---|---|---|---|
| + | − | + | − | + | − | |
| WT | 486 ± 44 | 405 ± 27 | 13 ± 1 | 38 ± 1† | 61 ± 4 | 221 ± 13† |
| Y121F | 494 ± 16 | 413 ± 24 | 36 ± 3* | 71 ± 7† | 223 ± 9* | 322 ± 3† |
| Y121L | 474 ± 69 | 324 ± 16 | 32 ± 2* | 26 ± 1 | 160 ± 31* | 147 ± 23 |
| S336A | 551 ± 138 | 405 ± 16 | 91 ± 9* | 70 ± 8 | 744 ± 71* | 564 ± 9 |
| S336D | 1596 ± 113* | 1037 ± 202 | 14 ± 2 | 13 ± 2 | 699 ± 141 | 513 ± 91 |
| N368A | ND | ND | 33 ± 5* | 29 ± 4 | 183 ± 31* | 179 ± 47 |
| N368D | 146 ± 8* | 121 ± 24 | 3.8 ± 0.5* | 3.2 ± 0.2 | 15 ± 3* | 15 ± 2 |
| N368L | 369 ± 44 | 332 ± 8 | 113 ± 5* | 120 ± 3 | 1375 ± 84* | 1158 ± 157 |
| C369A | ND | ND | 20 ± 1* | 41 ± 4† | 144 ± 41* | 329 ± 65† |
| C369S | ND | ND | 25 ± 1 | 75 ± 7† | 40 ± 2 | 213 ± 3†up> |
| S372A | 502 ± 17 | 429 ± 24 | 105 ± 8* | 117 ± 5 | 522 ± 138 | 480 ± 55 |
| S372D | 547 ± 142 | 397 ± 24 | 23 ± 1* | 20 ± 2 | 245 ± 32* | 215 ± 21 |
KI values for displacement of β-CIT binding by cocaine, imipramine, and fluoxetine were determined in the presence and absence of Cl−, as in Table 1, for mutants at proposed chloride binding site residues and Cys-369 as a control (in bold).
*Significant differences (p < 0.05) between wild-type and mutants in NaCl medium.
†Statistically significant differences (p < 0.05) between NaCl and Na-isethionate for a given mutant. Statistical analysis is from three to five independent experiments (2 when the difference between wild-type and mutant was >8-fold: imipramine binding to S336A, D, N368L, and S372A). ND, Not determined. Analysis was performed in Origin8 using the two-sample t test.
Most mutations that had an impact on fluoxetine and imipramine affinity had only a minor influence on cocaine binding (Fig. 3, Table 2), which was not stimulated by Cl−. The only mutations that affected cocaine binding were S336D, for which affinity was reduced to 30 ± 2% of WT (similar to imipramine), and N368D, which increased cocaine affinity 3.5-fold. These results indicate that the ability of chloride to stimulate binding requires the same binding site residues proposed to mediate the Cl− requirement for transport (Forrest et al., 2007).
Flexible-fit docking suggests that the effect of Cl− ions on antidepressant binding is unlikely to be direct
To test the possibility that Cl− stimulates antidepressant binding through a direct interaction, we used molecular modeling and docking approaches. Specifically, we predicted the conformations of the tested compounds bound to a homology model of SERT based on LeuT in an extracellular-facing substrate-occluded conformation (Yamashita et al., 2005). Compounds were docked to either the central S1 binding site that overlaps the leucine in LeuT or the secondary S2 binding site in the extracellular vestibule (Singh et al., 2007; Zhou et al., 2007; Shi et al., 2008). In the S1 site, the large majority of poses generated for all compounds contained a close interaction between the side-chain carboxyl of Asp-98 and the charged amine of the ligand (Fig. 4). This positioning of the amine close to Asp-98 is consistent with experimental evidence (Barker et al., 1999; Celik et al., 2008), as well as with docking results for 5-HT to SERT (Celik et al., 2008; Kaufmann et al., 2009) and for dopamine, CFT, and cocaine to DAT (Beuming et al., 2008).
Figure 4.

Representative configurations of fluoxetine, cocaine, paroxetine, and imipramine after flexible-fit docking. Several representative conformations from highly populated clusters are shown for each drug (brown), along with a single representative protein conformation. Drugs and selected protein side-chains (D98, Y121, Y176, S336, N368, and S372) are shown using sticks. Chloride (magenta) and sodium (dark blue) ions are shown as spheres. The positively charged amine nitrogen atom (blue) of each drug adopts a position close to D98 and Na1. (Note that imipramine contains two nitrogen atoms).
In this arrangement, the amine group was the closest part of the ligand to the Cl− binding site, albeit separated by the Na1 binding site. The mean distance of the position of the Cl− ion to the positively charged amine nitrogen atom of the ligand was ∼10Å for fluoxetine, imipramine, cocaine, and paroxetine (Table 3). These distances suggest that the only possible direct interaction between Cl− and the drug is a relatively weak, favorable electrostatic interaction. Indeed, the electrostatic interaction energy with the Cl− ion, calculated using the Poisson–Boltzmann equation and averaged over all chloride-bound poses, was similarly small for each drug (Table 3). Using a protein dielectric constant of 4, which reflects moderate screening by the protein atoms, the interaction energies are approximately −5 kcal/mol, whereas using a dielectric of 8, which might also be considered reasonable, these energies are approximately −2.5 kcal/mol. The differences in electrostatic interaction energies between the drugs were statistically significant (p < 0.001 with a two-tailed t test over all low-energy poses) only between cocaine and either fluoxetine or paroxetine.
Table 3.
Properties of docked poses in a homology model of WT SERT
| S1 |
S2 |
|||||
|---|---|---|---|---|---|---|
| d | ΔGelec |
d | ΔGelec |
|||
| ϵ = 4 | ϵ = 8 | ϵ = 4 | ϵ = 8 | |||
| Cocaine | 10.0 ± 0.6 | −4.5 ± 0.6 | −2.4 ± 0.3 | 17.3 ± 2.5 | −1.0 ± 0.4 | −0.7 ± 0.2 |
| Paroxetine | 9.3 ± 1.1 | −5.1 ± 0.8 | −2.7 ± 0.4 | 19.6 ± 2.2 | −0.7 ± 0.3 | −0.5 ± 0.2 |
| Imipramine | 12.6 ± 0.8 | −4.6 ± 1.1 | −2.4 ± 0.6 | 20.2 ± 1.7 | −0.9 ± 0.1 | −0.6 ± 0.0 |
| Fluoxetine | 9.1 ± 0.8 | −5.4 ± 0.6 | −2.8 ± 0.3 | 18.2 ± 1.6 | −0.8 ± 0.2 | −0.6 ± 0.1 |
Compounds were docked to either S1 or S2 binding sites. d refers to the mean distance between the charged N atom of the ligand and the position of the Cl− atom (in amgstroms), over all determined poses. The electrostatic component of the interaction energy, ΔGelec (in kilocalories per mole) was determined using a protein dielectric constant (ϵ) of 4 or 8, as described previously (Forrest et al., 2007) and calculated as a mean over all determined poses. SDs about the mean are also given.
In contrast to the results for the primary site, no clear preferences for specific orientations were observed for the ligands docked to the secondary site (data not shown). However, the mean distance of the position of the Cl− ion to the positively charged amine nitrogen atom of the ligand in S2 was consistently in the range 17–20 Å for all compounds tested (Table 3). This is similar to the distance of 16.4 ± 0.1 Å between the amine nitrogen and Glu-290 determined for structures of LeuT with tricyclic antidepressants bound to S2 (Singh et al., 2007; Zhou et al., 2007). Consistent with these large distances, the electrostatic energies of interaction of the ligands with the Cl− ion were small (less than or equal to −1 kcal/mol), with statistically significant differences between cocaine and either fluoxetine or paroxetine, and between imipramine and fluoxetine.
Thus, no clear distinction could be made between those compounds that are affected by Cl−, such as fluoxetine and imipra-mine, and those that are not, such as cocaine and paroxetine, in terms of the docking results or electrostatic interaction energies. Consequently, a direct electrostatic interaction within this extracellular-facing conformation of SERT appears not to be sufficient to explain the markedly different Cl− effects on binding.
Fluoxetine binding stabilizes SERT in a novel conformational state
Cl− was shown previously to allow 5-HT-induced conformational changes in SERT (Zhang and Rudnick, 2006). We considered the possibility that its role in antidepressant binding is to facilitate conformational transitions that allow these compounds to bind with maximal affinity. Previously, we showed that accessibility of residues in the cytoplasmic permeation pathway was altered by ligand binding. To examine whether antidepressant binding alters the conformational state of SERT, we tested the effect of imipramine and fluoxetine on the reactivity of a cysteine at position 277 in the cytoplasmic permeation pathway. This residue is on the face of TM5 that contributes to the pathway, and its reactivity was shown to increase and decrease in response to ligands (Zhang and Rudnick, 2006; Jacobs et al., 2007). Furthermore, it is close to the cytoplasmic end of TM5, a position where it is unlikely to be directly affected by ligand binding at the substrate site.
We measured the ability of MTSEA to inactivate β-CIT binding to membranes prepared from cells expressing SERT S277C (Fig. 5). Under the conditions used, 0.1 mm MTSEA inactivated approximately half of the β-CIT binding in the absence of added ligands. Cocaine decreased this inhibition, resulting in more residual β-CIT binding activity, consistent with the ability of cocaine to stabilize the extracellular-facing conformation of SERT, in which the cytoplasmic pathway is closed. Ibogaine, in contrast, increased the extent of inactivation, leading to less residual activity. Ibogaine has been proposed to stabilize the cytoplasm-facing conformation of SERT, in which the cytoplasmic pathway is open (Jacobs et al., 2007). Fluoxetine and imipramine led to levels of inactivation intermediate between those measured with cocaine or ibogaine. Figure 5 shows that, even at saturating concentrations, the antidepressants affected the reactivity of S277C in a quantitatively different manner than either cocaine or ibogaine. In separate experiments, we found that Cl− affected the concentrations of imipramine and fluoxetine required to influence MTSEA sensitivity, although at saturating concentrations, they had the same effect in the presence or absence of Cl−. The imipramine effect on MTSEA inactivation required approximately eightfold higher concentrations of imipramine in the absence of Cl− (Fig. 5, inset). Because the change in MTSEA sensitivity with fluoxetine was small, it was difficult to accurately measure the effect of Cl− on fluoxetine potency.
Figure 5.
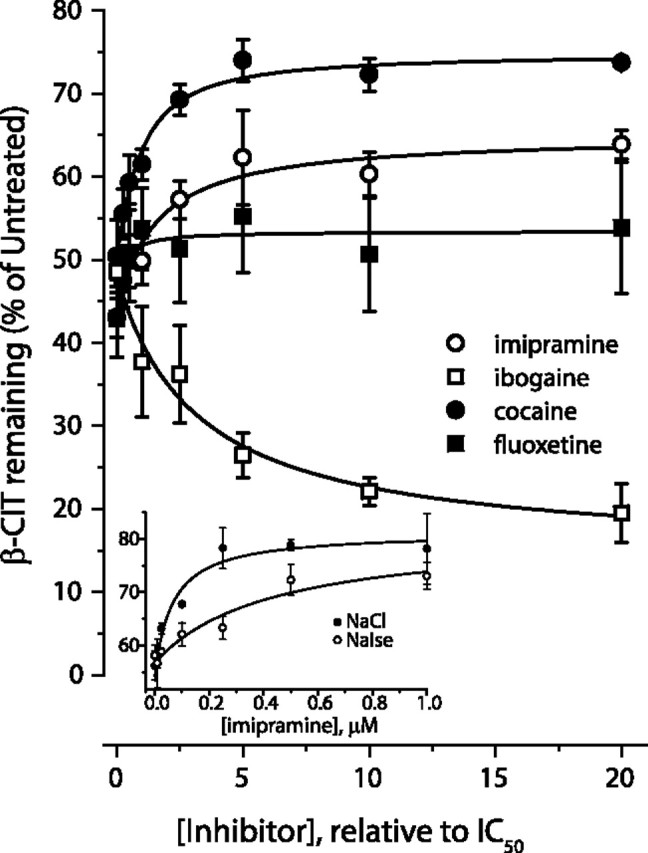
Modulation of MTSEA-induced inactivation of S277C by fluoxetine, imipramine, cocaine, and ibogaine. Membranes from cells expressing S277C were treated for 15 min with 0.1 mm MTSEA in the presence of varying concentrations of cocaine, ibogaine, imipramine, or fluoxetine. After washing membranes free of MTSEA and inhibitors, residual binding activity was determined by incubation with [125I]β-CIT as described in Materials and Methods. The concentration range of each inhibitor is presented relative to its IC50 (x-axis). The typical experiment illustrated was repeated four times with similar results. Inset, Imipramine effect in the presence (filled circles) or absence (open circles) of Cl−. The IC50 for imipramine was 0.4 ± 0.1 μm in the absence of Cl− and 0.05 ± 0.02 μm in its presence. In the absence of imipramine, Cl− had a minor effect, decreasing the IC50 for MTSEA by only ∼30% (comparing NaCl with Na-isethionate).
The same ligands were then tested at saturating concentrations over a range of MTSEA concentrations so as to more precisely determine their effect on S277C reactivity (Fig. 6). The MTSEA concentration leading to half-maximal inactivation provides a measure of reactivity under each condition (Rudnick, 2002). The results show that fluoxetine (300 nm), imipramine (1 μm), and cocaine (10 μm) all decreased the reactivity of S277C relative to MTSEA alone. Each of these inhibitors increased the MTSEA concentration required for half-maximal inactivation, whereas ibogaine (20 μm) decreased that concentration, indicating higher reactivity of S277C. Significantly, the effect of fluoxetine was markedly different from that of cocaine, decreasing the rate constant for S277C modification fourfold, whereas cocaine and imipramine decreased it by 12-fold and 6-fold, respectively (see legend of Fig. 6). We interpret the changes in inactivation rate to indicate that saturating concentrations of fluoxetine and possibly imipramine result in a different conformational equilibrium than cocaine and that those conformational preferences led to different reactivities of S277C with each of the three ligands bound.
Figure 6.

MTSEA dependence of SERT S277C inactivation and effects of fluoxetine, imipramine, cocaine, and ibogaine. Membranes from cells expressing S277C/X5C were incubated for 15 min with the indicated concentrations of MTSEA in the presence of saturating concentrations of cocaine (10 μm), ibogaine (20 μm), fluoxetine (300 nm), or imipramine (1 μm). The MTSEA concentration giving half-maximal inactivation of [125I]β-CIT binding was used to calculate inactivation rate constants. MTSEA concentrations leading to half-maximal inactivation and the corresponding rate constants were as follows: MTSEA alone, 33 ± 4 μm, 25 ± 3 m−1 s−1; cocaine, 390 ± 43 μm, 2 ± 0.2 m−1 s−1; ibogaine, 8 ± 2 μm, 100 ± 15 m−1 s−1; imipramine, 267 ± 70 μm, 3.9 ± 1 m−1 s−1; and fluoxetine, 131 ± 24 μm, 7 ± 2 m−1 s−1. Inactivation rates corresponding to four to six independent experiments were statistically analyzed in Origin8 using a two-sample t test. IC50 values for inactivation in the presence of each inhibitor were significantly different from the values with MTSEA alone. The effect of fluoxetine but not imipramine was significantly different (p < 0.002 for half-maximal inactivation) from that of cocaine. Nevertheless, in four of five experiments, the effect of imipramine was clearly different from that of cocaine, and in only one experiment were the effects of the two drugs indistinguishable.
Discussion
As inhibitors of 5-HT reuptake, antidepressants acting on SERT represent an important therapeutic tool. To understand how these compounds act and to guide the development of new antidepressants, it is essential to understand the structural and mechanistic details underlying the binding process. The work presented here was undertaken to characterize the structural and mechanistic aspects of antidepressant binding to SERT in the light of advances in our understanding of SERT structure and function.
We show here that, in addition to imipramine, fluoxetine, sertraline, and citalopram, which are among the most widely used antidepressants, all bind more avidly to SERT in the presence of Cl−. Although the affinities of cocaine and the cocaine analog β-CIT were not stimulated by Cl−, all but one of the drugs tested bound with higher affinity in the presence of Na+ (Table 1). An interesting exception was ibogaine, which is proposed to stabilize the cytoplasm-facing conformation of SERT (Jacobs et al., 2007) and which bound with increased affinity when Na+ was removed (Table 1). We speculate that this inverse effect may be related to the uniquely different conformation to which ibogaine binds. If so, it would be consistent with evidence that Na+ binding favors the extracellular-facing conformation of transporters in this family (Chen and Reith, 2003; Quick et al., 2006).
We recently identified a Cl− ion binding site in SERT (Forrest et al., 2007) common to Cl−-dependent neurotransmitter transporters, including GAT-1 (Zomot et al., 2007). The work presented here supports the participation of the same site in antidepressant binding. The ability of the proposed Cl− site residues to coordinate a Cl− ion depends on their polar character. Consequently, replacing any of these residues with aliphatic amino acids eliminated the ability of Cl− to stimulate imipramine or fluoxetine binding (Fig. 3, Table 2).
We and others originally proposed that Cl− was needed to stabilize one of the Na+ ions required for transport (Forrest et al., 2007; Zomot et al., 2007). In many NSS transporters that do not require Cl−, such as LeuT and TnaT (a prokaryotic tryptophan transporter), a carboxylic amino acid is found at the position corresponding to Ser-372 in SERT. In S372D and N368D, 5-HT transport became Cl− independent, although these mutants had low transport activity (Forrest et al., 2007). Mutants at these positions retained high β-CIT binding activity, however, allowing more thorough analysis of the role of this Cl− site on antidepressant affinity. For three of the four proposed binding site residues (Y121D and Y121E were inactive), replacement with aspartate led to Cl−-independent fluoxetine binding with affinity close to or better than wild type (Fig. 3, Table 2), consistent with our proposal that the negatively charged carboxylate side chain could replace a bound Cl− ion (Forrest et al., 2007). We considered two ways that Cl− could enhance binding: by direct physical interaction (either steric or electrostatic) or through conformational change.
Analysis of homology models of the outward-facing substrate-bound conformation of SERT indicates that any direct interaction of the Cl− ion would be limited to long-range (>8 Å) electrostatics. We performed flexible-fit docking of the drugs to these SERT homology models to determine whether, in a given state of the protein, such direct electrostatic interactions with Cl− would differ for the various drugs. The results show that any electrostatic energy gained during Cl− binding would be similar for all compounds, which is not consistent with the different effects of Cl− on binding affinity for, e.g., cocaine and fluoxetine.
Conversely, Cl− is known to be required for conformational changes induced by 5-HT, which increased (Sato et al., 2004; Zhang and Rudnick, 2006) or decreased (Mitchell et al., 2004) reactivity of cysteine residues positioned in various parts of the SERT structure. Moreover, Cl− itself increased accessibility of a cysteine at position 277 in the cytoplasmic pathway to a small but significant extent (legend of Fig. 5). Thus, to test whether a conformational change is associated with binding, we measured the effect of ligand binding on the reactivity of a cysteine residue placed in the cytoplasmic permeation pathway. The SERT mutant S277C, like other mutants containing a cysteine in this pathway, reacts with MTSEA at rates that depend on the nature of bound ligands (Zhang and Rudnick, 2006; Forrest et al., 2008). Cocaine is believed to bind to a conformation of SERT, NET, and DAT in which the substrate binding site faces the extracellular medium and the cytoplasmic pathway is closed, leading to low reactivity of S277C (Zhang and Rudnick, 2006; Beuming et al., 2008; Forrest et al., 2008). Ibogaine, in contrast, is thought to stabilize SERT in a conformation with the binding site facing the cytoplasm, leading to an open cytoplasmic pathway and high reactivity of S277C (Jacobs et al., 2007; Forrest et al., 2008). In the membrane preparations used here and in the absence of ligands, S277C reactivity is intermediate, probably because ligand-free SERT is in equilibrium between open and closed conformational states (Figs. 5, 6).
By saturating SERT with a ligand, we shift the equilibrium toward protein conformations optimal for binding that ligand: cytoplasm-facing for ibogaine and extracellular-facing for cocaine. Significantly, fluoxetine (and to a much lesser extent, imipramine) decreased reactivity of S277C to a level distinctly different from that of cocaine, suggesting that fluoxetine binding favors a conformation more cytoplasm facing than cocaine but not as much as with ibogaine (Figs. 5, 6). Because position 277 is near the cytoplasmic end of TM5, over 14 Å from the substrate binding site, it is likely that the effect of antidepressant binding on S277 accessibility is an indirect one mediated by conformational change rather than a direct interaction. Although higher concentrations of both imipramine and fluoxetine were required to decrease S277C reactivity in the absence of Cl−, the extent of the decrease was similar with and without Cl− (Fig. 5, inset).
We recently described a mechanism for neurotransmitter transport based on conformational changes required to expose residues in the cytoplasmic permeation pathway. In this mechanism, the protein alternates between two conformations that differ in the tilt angle of a four-helix bundle composed of TM1, TM2, TM6, and TM7 (Forrest et al., 2008). The Cl− binding site residues are all contained within the bundle, suggesting that Cl−–bundle interactions are important for conformational changes induced by 5-HT. We propose that conformational changes that alter the relationship between the bundle and the rest of the protein are influenced by Cl− binding and are required not only for alternating access in transport but also for forming the optimal binding conformation for fluoxetine and possibly other antidepressants.
These conclusions are consistent with the recently-reported “open-out” structure of LeuT solved in the presence of a competitive inhibitor, tryptophan (Singh et al., 2008), in which the orientation of a large portion of the bundle is altered to adopt a protein conformation more open to the extracellular side than the substrate-bound state. It also may be noteworthy that the arrangement of the residues corresponding to the proposed SERT Cl− binding site differs slightly between the substrate- and inhibitor-bound structures of LeuT. Specifically, the ability to coordinate a Cl− ion may be compromised in the equivalent open-out conformation of SERT, whereas Cl− binding may act to stabilize SERT conformations that are similar to the occluded state of LeuT.
We further surmise from the different level of S277C reactivity with 5-HT, imipramine, and fluoxetine that the Cl−-dependent conformational preferences of the bundle may differ not only in extent but also direction of tilt with different ligands. For example, S277C reactivity was much greater in the presence of 5-HT compared with fluoxetine (Zhang and Rudnick, 2006 vs Fig. 6), consistent with a greater extent of tilt during transport. Additionally, although imipramine and fluoxetine binding are stimulated by Cl− to a similar extent, fluoxetine binding causes a larger increase in S277C reactivity, which may reflect a conformation with a different tilt direction.
Our results provide additional support to the notion that binding of different inhibitors influences the equilibrium between protein conformations (Zhang and Rudnick, 2006; Jacobs et al., 2007; Forrest et al., 2008). The computational results are also consistent with this idea, because by using a single protein structure it is not possible to rationalize the effect of chloride. Interestingly, several lines of evidence suggest that substrates, antidepressants, and psychostimulants have profound and differential effects on monoamine transporter regulation (Ramamoorthy and Blakely, 1999; Saunders et al., 2000; Daws et al., 2002; Little et al., 2002; Cervinski et al., 2005; Chanrion et al., 2007; Lau et al., 2008). It is possible that changes in the conformational equilibrium attributable to antidepressant binding might influence transporter regulation, including posttranslational modification, trafficking, and interaction of the transporter with regulatory proteins.
Footnotes
We thank Drs. Ove Wiborg and Birgit Schiøtt for the atomic coordinates of their model of 5-HT bound to SERT, José Faraldo-Gómez for useful discussions, and Bret Bessac for help with statistical analyses.
References
- Andersen J, Taboureau O, Hansen KB, Olsen L, Egebjerg J, Strømgaard K, Kristensen AS. Location of the antidepressant binding site in the serotonin transporter: importance of Ser-438 in recognition of citalopram and tricyclic antidepressants. J Biol Chem. 2009;284:10276–10284. doi: 10.1074/jbc.M806907200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apparsundaram S, Stockdale DJ, Henningsen RA, Milla ME, Martin RS. Antidepressants targeting the serotonin reuptake transporter act via a competitive mechanism. J Pharmacol Exp Ther. 2008;327:982–990. doi: 10.1124/jpet.108.142315. [DOI] [PubMed] [Google Scholar]
- Barker EL, Moore KR, Rakhshan F, Blakely RD. Transmembrane domain I contributes to the permeation pathway for serotonin and ions in the serotonin transporter. J Neurosci. 1999;19:4705–4717. doi: 10.1523/JNEUROSCI.19-12-04705.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuming T, Kniazeff J, Bergmann ML, Shi L, Gracia L, Raniszewska K, Newman AH, Javitch JA, Weinstein H, Gether U, Loland CJ. The binding sites for cocaine and dopamine in the dopamine transporter overlap. Nat Neurosci. 2008;11:780–789. doi: 10.1038/nn.2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakely RD, Clark JA, Rudnick G, Amara SG. Vaccinia-T7 RNA polymerase expression system: evaluation for the expression cloning of plasma membrane transporters. Anal Biochem. 1991;194:302–308. doi: 10.1016/0003-2697(91)90233-j. [DOI] [PubMed] [Google Scholar]
- Celik L, Sinning S, Severinsen K, Hansen CG, Møller MS, Bols M, Wiborg O, Schiøtt B. Binding of serotonin to the human serotonin transporter. Molecular modeling and experimental validation. J Am Chem Soc. 2008;130:3853–3865. doi: 10.1021/ja076403h. [DOI] [PubMed] [Google Scholar]
- Cervinski MA, Foster JD, Vaughan RA. Psychoactive substrates stimulate dopamine transporter phosphorylation and down-regulation by cocaine-sensitive and protein kinase C-dependent mechanisms. J Biol Chem. 2005;280:40442–40449. doi: 10.1074/jbc.M501969200. [DOI] [PubMed] [Google Scholar]
- Chanrion B, Mannoury la Cour C, Bertaso F, Lerner-Natoli M, Freissmuth M, Millan MJ, Bockaert J, Marin P. Physical interaction between the serotonin transporter and neuronal nitric oxide synthase underlies reciprocal modulation of their activity. Proc Natl Acad Sci U S A. 2007;104:8119–8124. doi: 10.1073/pnas.0610964104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N, Reith ME. Na+ and the substrate permeation pathway in dopamine transporters. Eur J Pharmacol. 2003;479:213–221. doi: 10.1016/j.ejphar.2003.08.070. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Cool DR, Leibach FH, Ganapathy V. High-affinity paroxetine binding to the human placental serotonin transporter. Am J Physiol. 1990;259:C196–C204. doi: 10.1152/ajpcell.1990.259.2.C196. [DOI] [PubMed] [Google Scholar]
- Daws LC, Callaghan PD, Morón JA, Kahlig KM, Shippenberg TS, Javitch JA, Galli A. Cocaine increases dopamine uptake and cell surface expression of dopamine transporters. Biochem Biophys Res Commun. 2002;290:1545–1550. doi: 10.1006/bbrc.2002.6384. [DOI] [PubMed] [Google Scholar]
- Forrest LR, Tavoulari S, Zhang YW, Rudnick G, Honig B. Identification of a chloride ion binding site in Na+/Cl−-dependent transporters. Proc Natl Acad Sci U S A. 2007;104:12761–12766. doi: 10.1073/pnas.0705600104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest LR, Zhang YW, Jacobs MT, Gesmonde J, Xie L, Honig BH, Rudnick G. Mechanism for alternating access in neurotransmitter transporters. Proc Natl Acad Sci U S A. 2008;105:10338–10343. doi: 10.1073/pnas.0804659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Humphreys CJ, Wall SC, Rudnick G. Ligand binding to the serotonin transporter: equilibria, kinetics and ion dependence. Biochemistry. 1994;33:9118–9125. doi: 10.1021/bi00197a014. [DOI] [PubMed] [Google Scholar]
- Jacobs MT, Zhang YW, Campbell SD, Rudnick G. Ibogaine, a noncompetitive inhibitor of serotonin transport, acts by stabilizing the cytoplasm-facing state of the transporter. J Biol Chem. 2007;282:29441–29447. doi: 10.1074/jbc.M704456200. [DOI] [PubMed] [Google Scholar]
- Kaufmann KW, Dawson ES, Henry LK, Field JR, Blakely RD, Meiler J. Structural determinants of species-selective substrate recognition in human and Drosophila serotonin transporters revealed through computational docking studies. Proteins. 2009;74:630–642. doi: 10.1002/prot.22178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Gardner SP, Sutcliffe MJ. An automated approach for clustering an ensemble of NMR-derived protein structures into conformationally related subfamilies. Protein Eng. 1996;9:1063–1065. doi: 10.1093/protein/9.11.1063. [DOI] [PubMed] [Google Scholar]
- Koe BK, Lebel LA, Welch WM. [3H] sertraline binding to rat brain membranes. Psychopharmacology (Berl) 1990;100:470–476. doi: 10.1007/BF02243998. [DOI] [PubMed] [Google Scholar]
- Lau T, Horschitz S, Berger S, Bartsch D, Schloss P. Antidepressant-induced internalization of the serotonin transporter in serotonergic neurons. FASEB J. 2008;22:1702–1714. doi: 10.1096/fj.07-095471. [DOI] [PubMed] [Google Scholar]
- Lingjaerde O. Uptake of serotonin in blood platelets: dependence on sodium and chloride, and inhibition by choline. FEBS Lett. 1969;3:103–106. doi: 10.1016/0014-5793(69)80108-0. [DOI] [PubMed] [Google Scholar]
- Little KY, Elmer LW, Zhong H, Scheys JO, Zhang L. Cocaine induction of dopamine transporter trafficking to the plasma membrane. Mol Pharmacol. 2002;61:436–445. doi: 10.1124/mol.61.2.436. [DOI] [PubMed] [Google Scholar]
- Mitchell SM, Lee E, Garcia ML, Stephan MM. Structure and function of extracellular loop 4 of the serotonin transporter as revealed by cysteine-scanning mutagenesis. J Biol Chem. 2004;279:24089–24099. doi: 10.1074/jbc.M311173200. [DOI] [PubMed] [Google Scholar]
- Murphy DL, Lerner A, Rudnick G, Lesch KP. Serotonin transporter: gene, genetic disorders, and pharmacogenetics. Mol Interv. 2004;4:109–123. doi: 10.1124/mi.4.2.8. [DOI] [PubMed] [Google Scholar]
- Nelson PJ, Rudnick G. Coupling between platelet 5-hydroxytryptamine and potassium transport. J Biol Chem. 1979;254:10084–10089. [PubMed] [Google Scholar]
- Quick M, Yano H, Goldberg NR, Duan L, Beuming T, Shi L, Weinstein H, Javitch JA. State-dependent conformations of the translocation pathway in the tyrosine transporter Tyt1, a novel neurotransmitter: sodium symporter from Fusobacterium nucleatum. J Biol Chem. 2006;281:26444–26454. doi: 10.1074/jbc.M602438200. [DOI] [PubMed] [Google Scholar]
- Ramamoorthy S, Blakely RD. Phosphorylation and sequestration of serotonin transporters differentially modulated by psychostimulants. Science. 1999;285:763–766. doi: 10.1126/science.285.5428.763. [DOI] [PubMed] [Google Scholar]
- Rocchia W, Sridharan S, Nicholls A, Alexov E, Chiabrera A, Honig B. Rapid grid-based construction of the molecular surface and the use of induced surface charge to calculate reaction field energies: applications to the molecular systems and geometric objects. J Comput Chem. 2002;23:128–137. doi: 10.1002/jcc.1161. [DOI] [PubMed] [Google Scholar]
- Rudnick G. Active transport of 5-hydroxytryptamine by plasma membrane vesicles isolated from human blood platelets. J Biol Chem. 1977;252:2170–2174. [PubMed] [Google Scholar]
- Rudnick G. Chemical modification strategies for structure-function studies. In: Quick MW, editor. Transmembrane transporters. Hoboken, NJ: Wiley; 2002. pp. 125–141. [Google Scholar]
- Rudnick G. Serotonin transporters: structure and function. J Membr Biol. 2006;213:101–110. doi: 10.1007/s00232-006-0878-4. [DOI] [PubMed] [Google Scholar]
- Rudnick G, Wall SC. Binding of the cocaine analog 2 beta-[3H] carboxymethoxy-3 beta-(4-fluorophenyl)tropane to the serotonin transporter. Mol Pharmacol. 1991;40:421–426. [PubMed] [Google Scholar]
- Rudnick G, Wall SC. The molecular mechanism of ecstasy [3,4-methylenedioxymethamphetamine (MDMA)]: serotonin transporters are targets for MDMA-induced serotonin release. Proc Natl Acad Sci U S A. 1992;89:1817–1821. doi: 10.1073/pnas.89.5.1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Zhang YW, Androutsellis-Theotokis A, Rudnick G. Analysis of transmembrane domain 2 of rat serotonin transporter by cysteine scanning mutagenesis. J Biol Chem. 2004;279:22926–22933. doi: 10.1074/jbc.M312194200. [DOI] [PubMed] [Google Scholar]
- Saunders C, Ferrer JV, Shi L, Chen J, Merrill G, Lamb ME, Leeb-Lundberg LM, Carvelli L, Javitch JA, Galli A. Amphetamine-induced loss of human dopamine transporter activity: an internalization-dependent and cocaine-sensitive mechanism. Proc Natl Acad Sci U S A. 2000;97:6850–6855. doi: 10.1073/pnas.110035297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49:534–553. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- Shi L, Quick M, Zhao Y, Weinstein H, Javitch JA. The mechanism of a neurotransmitter:sodium symporter–inward release of Na+ and substrate is triggered by substrate in a second binding site. Mol Cell. 2008;30:667–677. doi: 10.1016/j.molcel.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Yamashita A, Gouaux E. Antidepressant binding site in a bacterial homologue of neurotransmitter transporters. Nature. 2007;448:952–956. doi: 10.1038/nature06038. [DOI] [PubMed] [Google Scholar]
- Singh SK, Piscitelli CL, Yamashita A, Gouaux E. A competitive inhibitor traps LeuT in an open-to-out conformation. Science. 2008;322:1655–1661. doi: 10.1126/science.1166777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneddon JM. Sodium-dependent accumulation of 5-hydroxytryptamine by rat blood platelets. Br J Pharmacol. 1969;37:680–688. doi: 10.1111/j.1476-5381.1969.tb08506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talvenheimo J, Nelson PJ, Rudnick G. Mechanism of imipramine inhibition of platelet 5-hydroxytryptamine transport. J Biol Chem. 1979;254:4631–4635. [PubMed] [Google Scholar]
- Thomas DR, Nelson DR, Johnson AM. Biochemical effects of the antidepressant paroxetine, a specific 5-hydroxytryptamine uptake inhibitor. Psychopharmacology (Berl) 1987;93:193–200. doi: 10.1007/BF00179933. [DOI] [PubMed] [Google Scholar]
- Wall SC, Innis RB, Rudnick G. Binding of the cocaine analog 2 β-carbomethoxy-3 β-(4-[125I]iodophenyl)tropane to serotonin and dopamine transporters: different ionic requirements for substrate and 2 β-carbomethoxy-3 β-(4-[125I]iodophenyl)tropane binding. Mol Pharmacol. 1993;43:264–270. [PubMed] [Google Scholar]
- Wong DT, Bymaster FP, Engleman EA. Prozac (fluoxetine, Lilly 110140), the first selective serotonin uptake inhibitor and an antidepressant drug: twenty years since its first publication. Life Sci. 1995;57:411–441. doi: 10.1016/0024-3205(95)00209-o. [DOI] [PubMed] [Google Scholar]
- Yamashita A, Singh SK, Kawate T, Jin Y, Gouaux E. Crystal structure of a bacterial homologue of Na+/Cl−-dependent neurotransmitter transporters. Nature. 2005;437:215–223. doi: 10.1038/nature03978. [DOI] [PubMed] [Google Scholar]
- Zhang YW, Rudnick G. The cytoplasmic substrate permeation pathway of serotonin transporter. J Biol Chem. 2006;281:36213–36220. doi: 10.1074/jbc.M605468200. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Zhen J, Karpowich NK, Goetz RM, Law CJ, Reith ME, Wang DN. LeuT-desipramine structure reveals how antidepressants block neurotransmitter reuptake. Science. 2007;317:1390–1393. doi: 10.1126/science.1147614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Zhen J, Karpowich NK, Law CJ, Reith ME, Wang DN. Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat Struct Mol Biol. 2009;16:652–658. doi: 10.1038/nsmb.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomot E, Bendahan A, Quick M, Zhao Y, Javitch JA, Kanner BI. Mechanism of chloride interaction with neurotransmitter:sodium symporters. Nature. 2007;449:726–730. doi: 10.1038/nature06133. [DOI] [PubMed] [Google Scholar]