

Abstract
Preclinical and clinical data suggest that a modification in GABAB receptor expression and function may contribute to the symptoms of major depression and the response to antidepressants. This includes laboratory animal experiments demonstrating that antidepressants modify brain GABAB receptor expression and function and that GABAB receptor antagonists display antidepressant potential in animal models of this condition. Clinical and post-mortem studies reveal changes in GABAergic transmission associated with depression as well as depression-related changes in GABAB subunit expression that are localized to the cortical depression network. Detailed in this review are the preclinical and clinical data implicating a role for the GABAB receptor system in mediating symptoms of this disorder and its possible involvement in the response to antidepressants. Particular emphasis is placed on clinical and post-mortem studies, including previously unpublished work demonstrating regionally-selective modifications in GABAB receptor subunit expression in brain samples obtained from depressed subjects. Together with the earlier preclinical studies, these new data point to a role for the GABAB system in major depression and support the antidepressant potential of GABAB receptor antagonists.
Keywords: GABAB receptors, depression, antidepressants, human brain, GABAB receptor expression, hippocampus, dentate gyrus
Introduction
It has been over five decades since the discovery of antidepressants (Kuhn, 1958). During that time, neuronal pathways and neurochemical systems that appear critical for mediating the symptoms of this disorder have been identified (Rajkowska et al., 1999; Nestler et al., 2001; Krystal et al., 2002; Mayberg, 2003; Seminowicz et al., 2004). These discoveries were due, in part, to the insights gained on the mechanisms of action of these drugs. Even with this progress, however, little has changed with regard to the types of agents employed to treat this condition, with this class still dominated by drugs that directly interact with monoamine systems (Kelsey and Nemeroff, 1998). This is not due to a lack of effort, as there remains a need for novel treatments (Enna and Williams, 2009). While newer antidepressants are generally safer than older agents, little progress has been made in decreasing the percentage of non-responders and in speeding the onset of action. Inasmuch as the first antidepressants were discovered empirically in the clinic, and there is no consistently identifiable neuropathology associated with this condition, the difficulties associated with discovering more efficacious antidepressants suggests that fundamental information is still lacking concerning the underlying neurobiological abnormalities responsible for this disorder. Indeed, it is possible that major depression is not a single entity unto itself, but rather a constellation of symptoms that only manifest in association with other psychiatric conditions. This could explain why no single mechanistic approach, such as enhancement of monoaminergic transmission, would be effective in all, or even the majority, of depressed patients because of the variable nature of the underlying cause.
Numerous attempts have been made to identify and develop antidepressants that target neurotransmitter systems other than those directly associated with the monoamines. Included have been antagonists for neurokinin-1 receptors (Herpfer and Lieb, 2005; Alvaro and Di Fabio, 2007), melanin-concentrating hormone-1 receptors (Shimazaki et al., 2006), corticotrophin-releasing factor-1 receptors (Valdez, 2009) and metabotropic glutamate receptors (Lesage and Steckler, 2010). While there are substantial preclinical data supporting the antidepressant potential of these agents, clinical studies have not as yet demonstrated their superiority over conventional therapies. This lack of success is likely due to several factors. One is that the animal models typically employed for screening antidepressant candidates were developed on the basis of their response to the clinically effective monoaminergic drugs. Although many of these models appear to have face validity, construct validity remains questionable given the lack of understanding of the disease process, as does the predictive validity for candidates that do not directly activate monoaminergic transmission. Moreover, as the response to many clinically effective psychotherapeutics involves interactions with multiple sites, it is possible that the most effective antidepressant may be one that targets several transmitter systems. As designing this type of drug is difficult given the number of possible target combinations, such an agent is more likely to be discovered with a pharmacometric approach (Enna and Williams, 2009).
The chances of successfully developing novel antidepressants are likely to be enhanced if there is direct evidence from preclinical, clinical or post-mortem studies directly linking the intended target with major depression in humans. The GABAB receptor is a candidate that fulfills this criterion (Table 1). Thus, since its discovery in the early 1980s (Bowery, 2010), animal studies have indicated that the GABAB receptor system is modified by chronic administration of antidepressants (Enna and Bowery, 2004). These findings, in turn, led to clinical studies aimed at identifying the consequences of stimulating this receptor system in depressed patients. Moreover, post-mortem brain studies demonstrate alterations in GABAergic neurons and possible changes in brain GABA synthesis in depressed subjects (Cryan and Slattery, 2010). Most recently, post-mortem analyses has indicated changes in GABAB receptor subunit gene expression in discrete areas of the brain within the proposed cortical depression network (Klempan et al., 2009; Sequeira et al., 2009).
Table 1.
Chronological listing of selected reports on the relationship between GABAB receptors and major depression
| Authors | Date | Type of report | Journal | Findings |
|---|---|---|---|---|
| Pilc and Lloyd | 1984 | Preclinical | Life Sci | Chronic antidepressant administration increases GABAB receptors binding in the rat brain |
| Lloyd et al. | 1985 | Preclinical | J Pharmacol Exp Ther | GABAB receptor number in brain is increased by electroshock or antidepressant administration in the rat |
| Suzdak and Gianutsos | 1986 | Preclinical | Eur J Pharmacol | Repeated antidepressant or GABAB agonist administration modifies GABAB receptor binding and function in rat brain |
| Gray and Green | 1987 | Preclinical | Br J Pharmacol | Antidepressants or electroconvulsive shock increases GABAB receptor function in mouse brain |
| Cross et al. | 1988 | Clinical | Psychiatry Res | GABAB receptor binding profiles similar between suicide and control |
| Martin et al. | 1989 | Preclinical | Neuropsychobiology | Antidepressant drugs reverse decrements in GABAB receptor expression in a rat model of major depression |
| Marchesi et al. | 1991 | Clinical | Psychoneuroendocrinology | GABAB regulation of growth hormone in major depression |
| Post et al. | 1991 | Clinical | Int Clin Psychopharmacol | Baclofen exacerbates symptoms of major depression |
| Arranz et al. | 1992 | Clinical | Neuropsychobiology | GABAB receptor binding profiles in suicide and control subjects |
| O'Flynn and Dinan | 1993 | Clinical | Am J Psychiatry | GABAB regulation of growth hormone release in major depression |
| Pratt and Bowery | 1993 | Preclinical | Br J Pharmacol | Repeated administration of GABAB antagonist or desipramine up-regulates GABAB receptor binding in rat brain |
| Petty | 1995 | Review | J Affect Disord | A GABAergic hypothesis of depression |
| Nakagawa et al. | 1996 | Preclinical | Brain Res | Baclofen displays antidepressant activity in a rat model of depression |
| Davis et al. | 1997 | Clinical | Psychoneuroendocrinology | Lack of growth hormone response to baclofen in patients with major depression |
| Nakagawa et al. | 1999 | Preclinical | Eur J Pharmacol | GABAB antagonist reduces helplessness in a rat model of major depression |
| Sanacora et al. | 2000 | Clinical | Crit Rev Neurobiol | Neuroimaging and GABAergic function in major depression |
| Krystal et al. | 2002 | Clinical | Mol Psychiatry | Magnetic resonance spectroscopy indicates reduced cortical GABA levels in depression |
| Sands et al. | 2003b | Preclinical | Life Sci | GABAB receptor subunit gene expression in rat brain is differentially modified in models of major depression and schizophrenia |
| Froestl et al. | 2004 | Preclinical | Biochem Pharmacol | GABAB antagonist SGS742 displays antidepressant activity in animal models of depression |
| Mombereau et al. | 2004 | Preclinical | Neuropsychopharmacology | GABAB1 subunit deletion mutant mice displays antidepressant phenotype in the forced-swim test |
| Sands et al. | 2004 | Preclinical | Biochem Pharmacol | Repeated administration of antidepressants modifies GABAB receptor function in rat hippocampus |
| Enna and Bowery | 2004 | Review | Biochem Pharmacol | Review of physiological and pharmacological manipulations that alter GABAB receptor expression and function |
| Fatemi et al. | 2005 | Clinical | Schizophr Res | GAD expression is altered in mood disorders and schizophrenia |
| Mombereau et al. | 2005 | Preclinical | Neuroreport | GABAB2 receptor subunit deletion mutant mice displays antidepressant phenotype in forced-swim test |
| Slattery et al. | 2005 | Preclinical | J Pharmacol Exp Ther | GABAB receptor antagonists display antidepressant activity in rodent models of major depression |
| McCarson et al. | 2005 | Preclinical | Biochem Pharmacol | Effect of antidepressants on GABAB receptor expression in rat spinal cord is state-dependent |
| Frieling and Bleich | 2006 | Review | Eur Arch Psychiatry Clin Neurosci | Tranylcypromine and GABAB receptor function |
| McCarson et al. | 2006 | Preclinical | Brain Res | Antidepressant administration or repeated stress alters GABAB receptor expression and function in rat spinal cord |
| Nowak et al. | 2006 | Preclinical | Br J Pharmacol | GABAB receptor antagonists display antidepressant activity in rodent models of major depression |
| Vigot et al. | 2006 | Preclinical | Neuron | GABAB receptor subunits regulate receptor location and function in mouse hippocampal neurons |
| Bielau et al. | 2007 | Clinical | Ann NY Acad Sci | GAD staining reveals altered GABAergic neuron terminal density in post-mortem brain samples from depressed patients |
| Rajkowska et al. | 2007 | Clinical | Neuropsychopharmacology | Reduced GABAergic neuron number in prefrontal cortex in major depression |
| Cornelisse et al. | 2007 | Preclinical | J Neurophysiol | Selective serotonin reuptake inhibitor treatment reduces GABAB receptor function in rat brain |
| Frankowska et al. | 2007 | Preclinical | Pharmacol Rep | Baclofen displays antidepressant activity in animal models of major depression |
| Klempan et al. | 2009 | Clinical | Mol Psychiatry | GABAA and GABAB receptor expressions are altered in the prefrontal cortex of suicides |
| Maciag et al. | 2009 | Clinical | Biol Psychiatry | Reductions in cortical GABAergic neurons in major depression |
| Sequeira et al. | 2009 | Clinical | PLoS One | Gene expression linkage analysis implicates GABAB mechanisms in major depression |
| Levinson et al. | 2010 | Clinical | Biol Psychiatry | GABAB-mediated cortical silence is prolonged in major depression |
| Cryan and Slattery | 2010 | Review | Advances in Pharmacology | Overview of research implicating a role for GABAB receptors in major depression |
Outlined in this review are the preclinical and clinical data implicating a role for the GABAB receptor system in mediating symptoms of major depression, and its possible involvement in the response to antidepressants. Particular emphasis is placed on clinical and post-mortem studies, with a detailed description of previously unpublished work demonstrating regionally selective modifications in GABAB receptor subunit expression in brain samples obtained from depressed subjects. Together with the earlier clinical and preclinical work (Table 1), these findings point to a role for the GABAB system in major depression and support the antidepressant potential for GABAB receptor antagonists.
GABAB receptors
Two pharmacologically and molecularly distinct GABA receptors have been identified, GABAAand GABAB (Enna, 2007). Whereas the GABAAsite is a pentameric, ligand-gated ion channel allosterically modulated by benzodiazepines and other anxiolytic and hypnotic agents, the GABAB receptor, a G protein-coupled heterodimer, is the site of action for baclofen, a muscle relaxant (Bowery, 2010). A class III metabotropic site, the GABAB receptor is composed of two 7-transmembrane spanning proteins (Kubo and Tateyama, 2005; Binet et al., 2006). While there are multiple subunit isoforms in various animal species, GABAB1a, GABAB1b and GABAB2 predominate, with coupling of either of the GABAB1 subtypes with GABAB2 essential for insertion into the plasma membrane and receptor function (Kaupmann et al., 1998; Chronwall et al., 2001; Enna and Bowery, 2010). Although GABAB receptors are widely distributed throughout the neuroaxis, subunit expression differs among brain regions (Bischoff et al., 1999; Towers et al., 2001; Vigot et al., 2006; Farb et al., 2007), suggesting the possible existence of regionally and molecularly distinct GABAB receptor subtypes.
The activation of GABAB receptors causes neuronal hyperpolarization by decreasing Ca2+ and increasing K+ membrane efflux, the latter through direct coupling to Kir3 K+ channels (Enna, 2001; Ladera et al., 2008; Fernandez-Alacid et al., 2009; Pinard et al., 2010). Located both pre- and post-synaptically, GABAB receptors influence cAMP production through coupling to Gi and Go. The stimulation of this site can either inhibit or enhance formation of this second messenger depending upon whether there is a simultaneous activation of a Gs-coupled site in the same neuronal compartment (Karbon and Enna, 1985). Thus, the response to GABAB receptor stimulation or inhibition may vary as a function of the pre-existing state of the affected cell. Given their widespread distribution, and myriad of effects on second messenger production and ion channel activity, it is not surprising that laboratory animal and human studies indicate that alterations in the GABAB receptor system contribute to the symptoms of a host of clinical conditions, including seizures, cognitive deficits, depression, anxiety, spasticity, drug abuse, schizophrenia, pain and gastro-oesophageal reflux disease (Enna, 2001; Enna and Bowery, 2004; Froestl, 2010).
Because of these findings, efforts have been expended to develop orthosteric GABAB receptor agonists and antagonists, and allosteric modulators (Froestl, 2010). Included are agonists such as arbaclofen placarbil (Gerson et al., 2010; Lal et al., 2009), a baclofen prodrug, and lesogabaran (Bredenoord, 2009), and orthosteric antagonists such as CGP36742, CGP54626 and SCH50911 (Froestl, 2010). Positive allosteric modulators, including CGP7930 and GS39783, have been designed to more selectively activate subsets of the GABAB receptor system and thereby minimize the side effects encountered with orthosteric agonists (Cryan et al., 2004; Mannoury la Cour et al., 2008).
While evidence suggests the possibility of pharmacologically distinct GABAB receptors (Bonanno and Raiteri, 1993a,b; Cunningham and Enna, 1996), their existence has been a matter of dispute (Waldmeier et al., 1994). Indeed, the discovery that heterodimerization is required for receptor function, and the fact that the number of GABAB receptor subunits is limited, argue against variability among orthosteric binding sites, which are exclusively located on the GABAB1subunit. The identification of pharmacologically distinguishable sites is important therapeutically as generalized activation or inhibition of GABAB receptors would be anticipated to be accompanied by numerous side effects, as is the case with baclofen, an orthosteric GABAB receptor agonist. It is encouraging therefore that in recent years the evidence supporting pharmacologically distinct GABABreceptors has grown (Pinard et al., 2010). Data are accumulating to suggest that the differential relative affinities and responses reported for GABAB receptor agonists and antagonists could be due to differences in the expression or function of regulators of G-protein signaling (RGS) proteins, which can influence GABAB receptor and K+ channel responsiveness (Mutneja et al., 2005). Moreover, four sequence-related cytosolic proteins have been discovered that bind as tetramers to the C-terminal domain of the GABAB2 subunit, influencing the pharmacology and kinetics of the receptor response (Pinard et al., 2010; Schwenk et al., 2010).
Thus, the functional responsiveness of GABAB receptors is dependent upon the production of GABAB1 and GABAB2 subunits, the coupling of the latter to G proteins, the phosphorylation state of the receptor, and the scaffolding provided by RGS and cytosolic proteins. Taken together, these observations, along with earlier studies, provide evidence for possible pharmacological heterogeneity among GABAB receptors. Such findings are crucial for customizing compounds to selectively interact with subsets of GABAB receptors in developing new therapies for the treatment of CNS disorders, including major depression.
GABAB receptors and depression
It has been speculated for some time that GABAB receptors are modified in depression and in response to antidepressant therapies (Enna and Bowery, 2004) (Table 1). While there have been conflicting findings (Cryan and Slattery, 2010), the preclinical studies generally indicate that chronic, but not acute, administration of antidepressants or electroconvulsive shock increases the number and function of GABAB receptors in rodent brain (Pilc and Lloyd, 1984; Lloyd et al., 1985; Suzdak and Gianutsos, 1986; Gray and Green, 1987; Martin et al., 1989; Pratt and Bowery, 1993; Frieling and Bleich, 2006; Cornelisse et al., 2007), and that brain GABAB receptor number is decreased in rat models of depression (Martin et al., 1989). Notably, antidepressant-induced increases in GABAB receptor binding occur in only certain rat brain regions (Pratt and Bowery, 1993).
Although these manipulations generally increase GABABreceptor sensitivity, changes in the magnitude and direction of GABAB subunit expression vary with the central nervous system area examined. Thus, it appears that regionally selective changes in the production, assembly and processing of brain GABAB subunits may be important homeostatic mechanisms for controlling CNS activity. For example, antidepressant-induced increases in GABAB receptor binding occur in only certain areas of the rat brain frontal cortex, including laminas I and VI. In contrast, chronic administration of these agents causes no changes in GABAB binding in laminas II, III and V (Pratt and Bowery, 1993). This regional specificity may explain why some have been unable to detect such changes and suggests this receptor modification is not due to a direct interaction of the drug with GABAB receptors, but rather is secondary to an effect on some other, most likely monoaminergic, system (Slattery et al., 2005).
With the cloning of the GABAB receptor subunit genes (Kaupmann et al., 1997; 1998; Jones et al., 1998; White et al., 1998; Chronwall et al., 2001), it became possible to examine whether the changes noted in GABAB receptor binding and function are due to modifications in the transcription or translation of these proteins (McCarson and Enna, 1999; Sands et al., 2003a,b; 2004; McCarson et al., 2005; 2006;). Studies indicate that chronic administration of antidepressants, stress or pain differentially modifies GABAB receptor subunit gene expression and receptor responsiveness in rat spinal cord and hippocampus, and alters GABAB receptor responsiveness in these subjects. The chronic administration of classical antidepressants generally increases GABAB receptor function and GABAB1a gene expression in rat hippocampus and dorsal horn of the spinal cord, while having a variable effect on the expression of the GABAB2 subunit gene. These data suggest that antidepressants cause an up-regulation in GABAB receptor expression and function by decreasing GABAergic tone, supporting the notion that depression is characterized by an overabundance of brain GABAergic activity (Sands et al., 2004).
The antidepressant-induced increase in GABAB receptor function is evidenced by an enhancement in baclofen-stimulated cAMP production in brain and spinal cord tissue obtained from animals that are chronically administered any one of a number of such agents (Sands et al., 2003a; 2004; McCarson et al., 2006) Likewise, repeated administration of amitryptyline or electroconvulsive shock enhances baclofen-induced inhibition of K+-stimulated serotonin release from mouse frontal cortex (Gray and Green, 1987). Both of these findings are consistent with other data indicating these drugs increase GABAB receptor number in this brain area.
An enhancement of receptor number and responsiveness could suggest that antidepressants either correct a depression-related underactive system or decrease GABAergic tone, leading to a supersentive receptor state. While there has been a report suggesting that baclofen displays antidepressant activity in some animal models of depression (Frankowska et al., 2007), the overwhelming weight of evidence suggests that a decrease in GABAB receptor activity is more typically associated with an antidepressant response. Thus, GABAB receptor antagonists display antidepressant properties in most, but not all (Mombereau et al., 2004), animal models of this condition (Nakagawa et al., 1999; Froestl et al., 2004; Mombereau et al., 2005; Nowak et al., 2006), and GABABreceptor stimulation exacerbates learned helplessness in rats (Nakagawa et al., 1996), a behaviour interpreted as a model for human depression. Also, mice lacking functional GABAB receptors behave as though they were receiving an antidepressant, suggesting regionally selective enhancements in brain GABAergic function in depression (Mombereau et al., 2004). Interpretation of these findings is limited, however, by the possibility that this behavioural response is due to secondary adaptive changes in these genetically modified animals rather than being a faithful representation of a phenotype that results solely from a selective decline in GABAB receptor activity.
It has been reported that GABAB receptor positive allosteric modulators display weak anxiolytic activity in some (Cryan et al., 2004; Jacobson and Cryan, 2008), but not all (Jacobson and Cryan, 2008; Paterson and Hanania, 2010), animal models of anxiety. Moreover, anxiety-like behaviour has been noted in GABAB receptor-deficient mice (Mombereau et al., 2005).These findings suggest that GABAB receptor blockade might exacerbate or precipitate an anxiety disorder in susceptible subjects, such as individuals with major depression. However, the clinical importance of this finding remains questionable given the inconsistency of the anxiolytic response to GABAB positive allosteric modulators in animal models, and the lack of any reported anxiogenic effect of an orthosteric GABAB receptor antagonist following administration to humans (Froestl et al., 2004).
Other preclinical data supporting the hypothesis that GABAB receptor blockade may alleviate depression include the finding that GABAB receptor antagonists increase gene expression and protein levels of nerve growth factor and brain-derived neurotophic factor (BDNF) in various regions of the rat brain (Heese et al., 2000; Enna et al., 2006). The relevance of this discovery to depression is based on reports that various classes of antidepressants, as well as electroconvulsive shock, increase the expression of BDNF in the rat hippocampus, and that BDNF displays antidepressant-like activity when placed directly into this brain region (Duman and Monteggia, 2006). In addition, hippocampal levels of BDNF are decreased in a mouse model of depression (Tsankova et al., 2006). It has been proposed that this effect of antidepressants on BDNF expression induces hippocampal neurogenesis, which is thought to be an important factor in alleviating depression (Miller et al., 2008). However, as noted by Tanti and Belzung (2010), BDNF polymorphisms are associated with a number of conditions, the effect of antidepressants on hippocampal neurogenesis is species-dependent and this response is not observed with all agents that display antidepressant activity in other models. Thus, the predictive value of enhanced BDNF production in assessing antidepressant potential remains uncertain.
Nonetheless, the results from nearly 30 years of research suggest that antidepressants cause an up-regulation of GABAB receptor number and function secondary to a decrease in GABAergic activity that may result from prolonged activation of monoaminergic systems (Sands et al., 2004). Serotonergic transmission appears to be particularly important in this regard in that GABAB receptor antagonists no longer display antidepressant properties following administration of para-chlorophenalanine, an inhibitor of tryptophan hydroxylase (Slattery et al., 2005). It has also been reported that antidepressants decrease the function of presynaptic serotonin-3 receptors on GABA neurons, resulting in a decrease in GABA release (Nakagawa and Ishima, 2003). Such findings have led to speculation that depression is characterized by an enhanced GABAB tone, perhaps as a result of a decrease in serotonergic activity, and that the response to antidepressants is dependent upon a reduction in GABAB receptor stimulation which, in turn, leads to a supersensitive GABAB receptor system.
Human studies on the GABAB system and neuropsychiatric disorders
A number of clinical and post-mortem studies support a causal relationship between the GABAergic system and depression (Cryan and Slattery, 2010) (Table 1). Thus, GABA levels in the cerebral cortex, plasma and CSF are lower than normal in depressed patients, as is the number of GABA neurons in layer II of the orbitofrontal cortex (Petty, 1995; Rajkowska et al., 1999; Sanacora et al., 2000; Krystal et al., 2002). A GABAB receptor involvement is suggested by the findings of some (Marchesi et al., 1991; O'Flynn and Dinan, 1993; Lucey et al., 1994), but not others (Davis et al., 1997), that the growth hormone response to baclofen is blunted in depressed individuals as compared with controls, suggesting altered GABAB receptor responsiveness in these patients. Also, an efficacy study with baclofen indicates that this GABAB receptor agonist worsens symptoms of depression (Post et al., 1991). Although the sample size is too small for drawing firm conclusions from this study, these data are interesting in light of the subsequent preclinical work suggesting that depression may be associated with an overstimulation of the GABAB system.
Post-mortem studies reveal that those diagnosed with an affective disorder display a decreased expression of cerebellar glutamic acid decarboxylases (GAD) (Fatemi et al., 2005), the enzymes responsible for the synthesis of GABA, and differences in GAD immunohistochemistry in various regions of the cerebral cortex and the hippocampus as compared with controls (Bielau et al., 2007). There have also been reports of differences between depressed and control subjects in the size and density of cerebral cortical GABA neurons (Rajkowska et al., 2007; Maciag et al., 2009). While such studies are important for establishing a GABAergic dysfunction in depression, they do not directly address whether, and to what extent, these changes influence, or are related to, the GABAB receptor system.
This issue was addressed directly by a study showing that the cortical silent period, a measure of cortical inhibition thought to be a reflection of GABAB receptor function, is prolonged in depressed individuals (Levinson et al., 2010). As baclofen administration lengthens the cortical silent period in normal subjects, the finding with depressed patients supports the preclinical work suggesting that this disorder is characterized by an enhancement in GABAB receptor activity. In contrast, however, earlier binding studies on post-mortem tissue (Cross et al., 1988; Arranz et al., 1992) found no differences in GABAB receptor number or affinity between controls and suicide subjects in frontal and temporal cortices and hippocampal samples. However, the interpretation of these results is compromised by the fact that these studies were conducted using relatively gross brain regions, which may dilute any changes that occur in highly discreet brain areas, and that ligand binding alone reveals nothing about the functional state of the receptor.
Efforts have been made to determine whether modifications in the expression of GABAB receptor subunits are associated with neuropsychiatric illness. Inasmuch as both GABAB1 and GABAB2 must be present to form a functional receptor, a change in the production of either could signal an alteration in the responsiveness of this system. As detailed previously, laboratory animal studies indicate that antidepressant administration, as well as chronic pain and stress, alters GABAB subunit expression and receptor function in rat brain and spinal cord (Sands et al., 2003a; 2004; McCarson et al., 2006), demonstrating the utility of analysing subunit expression as an indicator of receptor modifications. As both GABAB receptor subunits are found throughout the human brain (Billinton et al., 2000; Berthele et al., 2001; Waldvogel et al., 2004), it is likely that alterations in their expression could result in CNS disturbances, the nature of which would depend on the specific brain region involved. Indeed, reports indicate regionally selective changes in GABAB subunit expression in association with schizophrenia (Mizukami et al., 2002), temporal lobe epilepsy (Furtinger et al., 2003; Princivalle et al., 2003) and autism (Fatemi et al., 2009). Microarray studies of post-mortem brain tissue obtained from depressed and non-depressed suicide victims indicate modifications in the expression of genes responsible for the production of both GABAA and GABAB receptors in various regions of the prefrontal cortex and in selected subcortical areas (Klempan et al., 2009; Sequeira et al., 2009). The GABAB2 subunit expression appeared to be particularly affected, being elevated in the depressed suicide group relative to non-depressed individuals (Sequeira et al., 2009). A full appreciation of the significance of these findings awaits replication of this work and a determination as to whether these alterations in gene expression are indicative of a change in subunit protein. Besides hinting at an association between changes in the production of GABAB receptor subunits and depression, these studies confirm the importance of examining this issue in well-defined regions, as the gene alterations are not global, but rather circumscribed to rather discreet brain areas.
Human GABAB receptor subunit expression in depression
To confirm that depression is associated with selective, regionally defined changes in human brain GABAB receptor subunits, a preliminary study was undertaken with post-mortem brain samples obtained from depressed individuals and control subjects. The primary hypothesis was that regionally selective alterations in brain GABAB receptor subunit gene expression are a characteristic of this disorder. The brain areas examined were the hippocampus, subgenual cingulate and orbitofrontal cortex, regions implicated in the pathophysiology of depression (Sheline, 1996; Sheline et al., 1999; Mayberg, 2003; Seminowicz et al., 2004; Pittenger and Duman, 2008; Hajszan et al., 2009; Koolschijn et al., 2009; Yucel et al., 2009; Price and Drevets, 2010). Particular emphasis was placed on subsections of the hippocampus as this is a region where the balance between excitatory and inhibitory inputs appears to be particularly critical. Examining the molecular contribution of GABAB alterations within the extended cortical depression system complements a focus on the reward regions in depression. The entire extended network includes the subgenual cingulate, the dorsolateral prefrontal cortex, orbitofrontal cortex, pregenual anterior cingulate and frontal pole, as well as the amygdala, hippocampus and insula. The regions of greatest importance to major depression are prefrontal cortex, anterior cingulate, amygdala, anterior thalamus and regions within the hippocampus. Inasmuch as these brain areas are richly innervated by monoaminergic and GABAergic neurons, they are most likely to display changes in GABAB receptor subunit expression if, as indicated by the preclinical studies, there is a functional interplay between these transmitter systems in depression and the response to antidepressants.
Because major depression is a neuroanatomically complex condition, it is critical to examine in discrete brain regions the possible molecular and neurochemical changes associated with this condition to reduce the possibility of overlooking a meaningful alteration because of a diluting effect of adjacent tissue. Changes in the volume, function and interaction among cortical-limbic brain areas are particularly evident in depression (Sheline, 1996; Sheline et al., 1999; Mayberg, 2003; Seminowicz et al., 2004; Pittenger and Duman, 2008). Within the hippocampus, alterations in the morphology of the dentate gyrus (DG), CA1 and CA3 regions occur in association with stress, depression and antidepressant therapy (Malberg et al., 2000; Pittenger and Duman, 2008; Hajszan et al., 2009). Likewise, depression-related volume changes occur in the subgenual cingulate cortex (Yucel et al., 2009) and the orbital frontal cortex (Koolschijn et al., 2009). Thus, these brain regions, along with cerebellar cortex, an area not believe to contribute to the symptoms of this disorder, were selected for studying possible changes in GABAB subunit expression in depression.
Human brain tissue from depression and control cases was obtained from the Dallas Brain Collection (Stan et al., 2006). The tissue was collected only after acquiring consent from the next of kin along with permission to review medical records and to conduct a telephone interview with a primary caregiver. All clinical information on each case was evaluated by at least three research psychiatrists and diagnoses were made using DSM-IV criteria. Blood screens for drugs of abuse, alcohol and prescription medications were conducted on each subject. Cases were excluded when there was a known history of neurological disorders or of an axis I psychiatric condition other than major depression.
The human post-mortem material was obtained from 12 cases diagnosed with major depressive disorder and 12 control subjects. The two groups were matched as closely as possible for age, brain pH, post-mortem interval (PMI), and RNA integrity number (RIN), an indicator of human post-mortem tissue RNA quality (Stan et al., 2006) (Table 2). While half the members of the depression group were suicides, only two subjects in this total cohort had detectible blood levels of medication at the time of death (Table 2). The groups were analysed in a paired design. The hippocampus, anterior cingulate and orbitofrontal cortices were selected for study because of the in vivo imaging data suggesting their involvement in the clinical manifestations of depression and the response to antidepressant treatments. The tissue samples were dissected from the anterior cingulate (BA24) and orbitofrontal cortex (BA11), as well as hippocampal subfields and cerebellum. Other than for the hippocampus, the samples were frozen immediately in a mixture of dry ice and isopentane (1:1, v : v), pulverized on dry ice and stored at −80°C until analysed.
Table 2.
Characteristics of depressed and control group subjects
| Controls | Depression | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case | Age (years) | PMI (hours) | RIN | Gender | Race | Cause of death | Case | Age (years) | PMI (hours) | RIN | Gender | Race | Meds | Cause of death |
| C1 | 48 | 20 | 6.8 | M | C | HT CVD | D1 | 46 | 11 | 8.3 | F | C | yes | HT CVD |
| C2 | 60 | 11 | 9.3 | M | AA | HT CVD | D2 | 54 | 6 | 8.9 | M | C | no | AS CVD |
| C3 | 20 | 21 | 8.2 | M | C | Blunt force injury | D3 | 35 | 9 | 7.4 | M | C | no | suicide |
| C4 | 31 | 16 | 8.1 | M | C | HT CVD | D4 | 33 | 18 | 8.5 | M | C | no | suicide |
| C5 | 60 | 20 | 8.5 | M | C | Myocardial infarction | D5 | 61 | 20 | 8.6 | M | C | no | suicide |
| C6 | 43 | 15 | 6.1 | M | C | HT CVD | D6 | 50 | 23 | 8.8 | M | C | no | suicide |
| C7 | 65 | 11 | 8 | F | C | Aortic dissection | D7 | 57 | 16 | 6.6 | F | C | no | suicide |
| C8 | 83 | 13 | 8.5 | F | C | Sharp force injury | D8 | 59 | 10 | 9 | F | C | no | HT CVD |
| C9 | 34 | 23 | 6.2 | M | C | HT CVD | D9 | 40 | 18 | 6.9 | M | C | no | suicide |
| C10 | 48 | 15 | 9.5 | M | C | HT CVD | D10 | 41 | 13 | 8 | F | AA | yes | Diabetic ketoacidosis |
| C11 | 63 | 14 | 7.7 | M | C | AS CVD | D11 | 65 | 14 | 7.6 | M | C | no | HT CVD |
| C12 | 19 | 20 | 9 | M | C | Gunshot wound to chest | D12 | 26 | 19 | 8.1 | F | C | no | Accidental drowning |
| av | 47.83 | 16.58 | 7.99 | 2F/10M | 11C/1AA | av | 47.25 | 14.75 | 8.06 | 5F/7M | 11C/1AA | |||
| SD | 19.45 | 4.08 | 1.12 | SD | 12.39 | 5.10 | 0.79 | |||||||
PMI, post-mortem interval; RIN, RNA integrity number; meds, antidepressant medications at time of death; HT CVD, hypertensive cardiovascular disease; AS CVD, atherosclerotic cardiovascular disease; F, female; M, male; C, Caucasian; AA, African American; av, average; SD, standard deviation.
The entire hippocampus was removed from the fresh brain, embedded longitudinally into a mold with Histomer polymer (Histotech, Frederiksberg, Denmark). Tissue blocks were then taken at 5 mm intervals and frozen immediately in a mixture of dry ice and isopentane (1:1, v : v). Blocks from the mid-level of the hippocampus were used for the study. Four samples, each 300 µm thick, were cryostat sectioned at −20°C, then stored at −80°C. Nissl staining of 14 µm sections adjacent to the samples was used to determine orientation.
In each of the 300 µm sections, the parahippocampal gyrus was first dissected away from the hippocampus proper, then a series of cuts was made to isolate the CA3, CA1, subiculum and DG (Figure 1).
Figure 1.

Representation of the coordinates used to dissect the dentate gyrus (DG), CA1 and CA3 regions of the hippocampus.
The GABAB receptor subunit expression assays were performed blind to diagnosis (depressed or control) using paired samples from the two groups of subjects. Sample pairing was performed by someone not involved with the biochemical assay to ensure that tissues tested on any given day included an equal number of samples from the same brain regions of depressed and control individuals.
Modified versions of human GABAB1a and GABAB2 expression vectors provided by Dr Klemens Kaupmann (Novartis, Basel, Switzerland) were used for probe synthesis. The probe sequences were bases 276-613 of human GABAB1a cDNA (Accession Number AJ225028) and bases 2746-3188 of human GABAB2cDNA (Accession Number BC035071.2). Human β-actin mRNA expression was quantified with probes generated using a pGEM-T vector containing bases 374-1093 (Accession Number NM_001101) of the human β-actin sequence (Nandan and Reiner, 1997).
Total RNA was isolated from tissue samples using a rapid-guanidinium method (Chomczynski and Sacchi, 1987) and assayed separately for GABAB1a, GABAB2 and β-actin mRNAs using solution hybridization–nuclease protection assays (McCarson and Krause, 1994).
The primary analyses were designed to test whether regionally selective alterations occur in GABAB receptor subunit expression in the hippocampus, anterior cingulate and orbitofrontal cortex in cases of depression compared with controls. Secondary analyses were conducted to explore the influence of age and gender on receptor subunit expression. An independent samples t-test was employed for comparing these results given the relatively large sample size, the continuity and normal distribution of the gene expression values, the convergence of measures of central tendency, and the similar variance in each data set. Spearman rank correlations were used to assess possible correlations between mRNA levels with RIN and PMI. Unpaired t-tests were conducted to verify that the two diagnostic groups were matched on demographic variables, age, RIN and PMI. In all analyses, differences were considered statistically significant with P≤ 0.05.
Both GABAB1aand GABAB2 receptor subunit gene expression were detectable in all brain regions examined (Table 3). Of the hippocampal subfields studied, only the DG displayed a significant difference between depressed and control groups. The expression of both subunit genes in the DG differed significantly between the depressed subjects and controls, with a 30% decrease in GABAB1a(t= 2.18, df19, P= 0.04) and a 50% increase in GABAB2gene expression (t = 2.21, df20, P= 0.04) in this brain region (Table 3). The latter result confirms an earlier finding from a microarray study indicating that GABAB2 subunit expression is elevated in brain tissue from depressed suicide subjects as compared with non-depressed individuals (Sequeira et al., 2009). Differential modifications in the expression of GABAB1 and GABAB2 subunit gene expression have been found in rat brain (Sands et al., 2003b) and spinal cord (McCarson et al., 2006) tissue. It is unknown whether these changes are occurring in the same or different cellular elements. Regardless, an increase or decrease in gene expression of either one or both subunits in a single cell is likely to cause, and reflect, an alteration in GABAB receptor function. The results also indicated a significant negative correlation between GABAB1asubunit gene expression and age (R =−0.43, P= 0.04) in the CA3 region of the hippocampus. Covarying for age did not alter the absence of a group difference in GABAB1a expression in CA3. Comparisons between groups revealed a significant decrease in GABAB1a subunit expression in the CA3 subfield in depressed male subjects compared with control males (t = 2.55, df11, P= 0.03; Figure 2).
Table 3.
GABAB1a and GABAB2 subunit gene expressions in various brain regions of depressed and control subjects
| GABAB1a | GABAB2 | |||
|---|---|---|---|---|
| Brain region | Depressed | Control | Depressed | Control |
| CA1 | 17 ± 4 | 23 ± 3 | Not measured | |
| CA3 | 86 ± 18 | 83 ± 8 | Not measured | |
| Dentate gyrus | 25 ± 2* | 37 ± 5 | 17 ± 2* | 11 ± 1 |
| Subgenual cingulate cortex | 14 ± 2 | 16 ± 2 | 13 ± 2 | 17 ± 3 |
| Orbital frontal cortex | 9 ± 2 | 7 ± 1 | 64 ± 13 | 36 ± 6 |
| Cerebellum | 4 ± 1 | 4 ± 1 | 19 ± 6 | 23 ± 5 |
Values are the mean pg subunit specific mRNA/ng β-actin ± SEM.
P < 0.05 compared with corresponding control, two-tailed Student's t-test.
Figure 2.
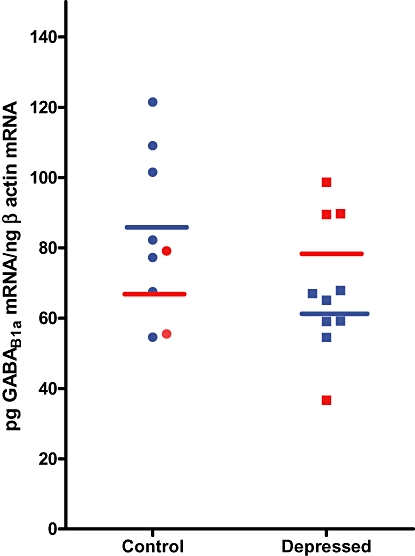
GABAB1a subunit gene expression in the CA3 region of the hippocampus of depressed (n= 6) and control (n= 7) male (blue) and depressed (n= 4) and control (n= 2) female (red) subjects. Horizontal lines indicate the means for each group. The level of significance for the difference between means for the male subjects is P= 0.03, as determined by an independent samples t-test.
No significant differences were noted between depressed and control groups in GABAB1aor GABAB2subunit gene expression in the orbital frontal and anterior cingulate cortices or cerebellum (Table 3). The GABAB2 receptor subunit expression in the orbital frontal cortex was, however, nearly twice as high in male depressed subjects as in male controls (t = 2.3, P= 0.04; Figure 3), and there was a strong trend towards a significant increase in the expression of the GABAB2 subunit (P < 0.06) in this brain region of depressed subjects compared with controls (Cohen's d effect size = 0.83) (Table 3). While there were no significant correlations between age, RIN or PMI and GABAB2 receptor subunit gene expression for these three brain regions, and for GABAB1aexpression in the orbital frontal cortex, a correlation was noted between GABAB1aexpression and PMI (R = 0.41, P= 0.05) and RIN (R = 0.46, P= 0.02) for the anterior cingulate cortex and cerebellum respectively.
Figure 3.

GABAB2 subunit gene expression in the orbital frontal cortex of depressed (n= 7) and control (n= 9) male (blue) and depressed (n= 5) and control (n= 2) female (red) subjects. Horizontal lines indicate the means for each group. The level of significance for the difference between means for the male subjects is P= 0.04, as determined by an independent samples t-test.
Because major depression has been associated with changes in the volume of some brain regions (Rajkowska, 2000; Rajkowska et al., 2007; Maciag et al., 2009), it is possible that measurement of beta-actin gene expression may not be an appropriate reference for control as the quantity of this marker could differ between the two groups as a result of cell loss. However, analysis of the beta-actin gene revealed no significant differences in expression in the brain areas examined in the depressed and control groups (data not shown). This finding indicates that despite any loss of volume or decrease in cell number, the expression of this marker relative to total RNA levels remains unchanged in depression. Accordingly, beta-actin appears to be an appropriate control gene for normalizing the levels of GABAB receptor subunit gene expression under these circumstances.
These results suggest a decrease in GABAB1a and an increase in GABAB2 subunit expression in the DG of depressed individuals as compared with controls. Because dysfunction in this brain region has been previously linked with depression, this change in GABAB receptor subunit expression could be associated with the illness itself. Because of the size of the tissue samples, it was not possible to analyse GABAB2subunit expression in the CA1 and CA3 regions of the hippocampus, leaving open the possibility of a depression-related modification in the expression of this GABAB receptor subunit in these areas. Nonetheless, the fact that no significant changes in GABAB1a subunit gene expression were noted in the CA1 or CA3 regions of the hippocampus, or in GABAB1a and GABAB2expression in the subgenual cingulate cortex, two areas also thought to be involved in the depression brain circuit, nor in the cerebellum, an area outside this circuit (Pittenger and Duman, 2008), suggests that the changes observed in the DG are selective and not a reflection of a generalized abnormality in GABABsubunit gene expression as a result of the disorder, drug treatment or death.
The finding of an apparent decrease in GABAB1a subunit expression in the CA3 region of male subjects, and a doubling of the GABA2subunit expression in the orbital frontal cortex of these individuals as compared with male controls, suggests that the GABAB system may be modified in these depression circuit brain areas as well (Figures 2 and 3). Inasmuch as the GABAB1 subunit gene expression data for the CA3 and orbital frontal cortex nearly attained statistical significance when comparing all samples (Table 3), it seems probable these areas are affected, with the lack of statistical significance for the present findings possibly being due to the influence of variation because of the sample size. Given the small number of female subjects, it is impossible to determine whether these changes are related to gender (Figures 2 and 3). Nevertheless, the modifications noted in the DG alone demonstrate that the GABABreceptor system is altered in a critical brain region associated with major depressive illness.
It is also noteworthy that a negative correlation was found between GABAB1a subunit gene expression in the CA3 region and age, while no significant correlations were detected between GABAB receptor subunit gene expression and drug history or post-mortem delay. It could be speculated that this age-related decline in GABAB1a expression in the CA3 may contribute to the increase in susceptibility to depression in the elderly.
From these data, it is impossible to know whether the changes in GABAB receptor subunit expression lead to a change in the production of subunit protein or receptor function. However, numerous studies have indicated that alterations in GABAB subunit expression are usually accompanied by a change in receptor sensitivity (Sands et al., 2003a; 2004; McCarson et al., 2005; 2006; Merlo et al., 2007). As the production of GABAB2 subunits appears to be the rate-limiting step in the formation of functional GABAB receptors (Thuault et al., 2004), these results, like previous work in laboratory animals, suggest that the system is up-regulated in depressed subjects in the DG, and possibly the CA3 region of the hippocampus and in the orbital frontal cortex. As a change in GABAB receptor activity alters the expression of brain-derived and glial cell line-derived neurotrophic factors (Heese et al., 2000; Enna et al., 2006; Fiorentino et al., 2009), it is possible that alterations in GABAB receptor expression and function influences hippocampal neurogenesis, which may be a component of the clinical response to antidepressants (Pittenger and Duman, 2008).
As these data were obtained from a predominantly antidepressant-free cohort of cases (Table 2), the results suggest that the changes in GABAB receptor subunit expression may be part of the molecular phenotype of this psychiatric condition. It is, however, possible that they are long-lasting responses to prior antidepressant treatment. This is a critical issue as it has been reported that the GABAB receptor response to antidepressants is state-dependent (McCarson et al., 2005), making it impossible to predict the effect of these drugs on human brain GABAB receptors without knowing whether this system is modified by the condition itself. As discussed previously, an increase in GABAB function could be a response to a persistent antidepressant-induced reduction in GABAergic tone, or it might be an evidence of a disease-related supersensitive GABAB receptor system. The latter possibility is consistent with the discovery that GABAB receptor antagonists display an antidepressant profile in animal models of this condition (Nakagawa et al., 1999; Froestl et al., 2004; Slattery et al., 2005; Nowak et al., 2006).
The findings that baclofen, a GABAB receptor agonist, worsens the symptoms of depression (Post et al., 1991) and, like depression, prolongs the cortical silent period in humans (Levinson et al., 2010), and that mice lacking functional GABAB receptors behave as though they have been administered an antidepressant, all support the notion that an overactive GABA system contributes to the symptoms of this disorder. They also argue strongly against the idea that the antidepressant response to GABAB receptor antagonists might be due to an enhancement in GABA release secondary to the blockade of GABAB autoreceptors.
These data confirm and extend previous studies (Klempan et al., 2009; Sequeira et al., 2009) indicating a direct relationship between a modification in GABABreceptor subunit gene expression and major depression, with receptor subunit changes being most evident in the DG. Given the proposed relationship between the DG and affective illness (Malberg et al., 2000), these findings suggest a direct link between modifications in human brain GABAB receptor subunit gene expression and depression, and provide insights into the molecular mechanisms that may be responsible, at least in part, for some of the neurochemical and behavioural changes associated with this condition. These discoveries support the notion that GABAergic medications, in particular GABAB receptor antagonists, may represent a novel approach for the treatment of this disorder.
While it has been some time since the development of orally active GABAB receptor antagonists (Froestl et al., 1995), only one of these phosphinic acid GABA analogues has been examined clinically (Froestl et al., 2004). In this study, SGS742 progressed through Phase II clinical trials as a potential treatment for cognitive deficits. Although no serious side effects were noted at the doses tested, and some benefits were reported for patients diagnosed with mild cognitive impairment, clinical trials were halted because the efficacy was insufficient to warrant commercial development. Given the difficulties associated with demonstrating clinical antidepressant activity, and the low affinity of SG742 for the GABAB receptor site, no effort has yet been made to test the hypothesis that GABAB receptor antagonists are antidepressants. Proof of principle must await the development of more potent, orally active and pharmacokinetically appropriate members of this class.
To fully exploit these preliminary findings on receptor subunit expression in post mortem brain-tissue, future work should focus on determining the functional correlate of these changes and on whether these alterations are drug induced or part of the pathophysiological process. Ultimately, the relationship between GABAB receptors and depression can only be conclusively tested by a thorough clinical assessment of the antidepressant properties of GABAB receptor antagonists.
Acknowledgments
We thank Dr Klemens Kaupmann for the donation of GABAB receptor subunit expression vectors, Ms Kandace Fleming (K-IDDRC HD 002528) for statistical analyses, and Ms Lynn LeCount for editorial support.
Glossary
Abbreviations
- BDNF
brain-derived neurotophic factor
- GABA
γ-aminobutyric acid
- GABAB1a
GABAB1b, GABAB2, γ-aminobutyric acidB receptor subunits
- PMI
postmortem interval
- RGS
regulators of G-protein signaling
- RIN
RNA integrity number
Conflict of interest
The authors declare no competing financial interests in relation to the work described in this report.
References
- Alvaro G, Di Fabio R. Neurokinin 1 receptor antagonists – current prospects. Curr Opin Drug Discov Devel. 2007;10:613–621. [PubMed] [Google Scholar]
- Arranz B, Cowburn R, Eriksson A, Vestling M, Marcusson J. Gamma-aminobutyric acid-B (GABAB) binding sites in postmortem suicide brains. Neuropsychobiology. 1992;26:33–36. doi: 10.1159/000118893. [DOI] [PubMed] [Google Scholar]
- Berthele A, Platzer S, Weis S, Conrad B, Tolle TR. Expression of GAB(B1) and GABA(B2) mRNA in the human brain. Neuroreport. 2001;12:3269–3275. doi: 10.1097/00001756-200110290-00025. [DOI] [PubMed] [Google Scholar]
- Bielau H, Steiner J, Mawrin C, Trubner K, Brisch R, Meyer-Lotz G, et al. Dysregulation of GABAergic neurotransmission in mood disorders: a postmortem study. Ann NY Acad Sci. 2007;1096:157–169. doi: 10.1196/annals.1397.081. [DOI] [PubMed] [Google Scholar]
- Billinton A, Ige AO, Wise A, White JH, Disney GH, Marshall FH, et al. GABAB receptor heterodimer-component localization in human brain. Mol Brain Res. 2000;77:111–124. doi: 10.1016/s0169-328x(00)00047-4. [DOI] [PubMed] [Google Scholar]
- Binet V, Goudet C, Brajon C, Le Corre L, Archer F, Pin J-P, et al. Molecular mechanisms of action of GABAB receptor activation: new insights from the mechanism of action of CGP7930, a positive allosteric modulator. Biochem Pharmacol. 2006;1068:109–117. doi: 10.1042/BST0320871. [DOI] [PubMed] [Google Scholar]
- Bischoff S, Leonhard S, Raymann N, Schuler V, Shigemoto R, Kaupmann K, et al. Spatial distribution of GABA(B)R1 receptor mRNA and binding sites in the rat brain. J Comp Neurol. 1999;412:1–16. [PubMed] [Google Scholar]
- Bonanno G, Raiteri M. Multiple GABAB receptors. Trends Pharmacol Sci. 1993a;14:259–261. doi: 10.1016/0165-6147(93)90124-3. [DOI] [PubMed] [Google Scholar]
- Bonanno G, Raiteri M. gamma-Aminobutyric acid (GABA) autoreceptors in rat cerebral cortex and spinal cord represent pharmacologically distinct subtypes of the GABAB receptor. J Pharmacol Exp Ther. 1993b;265:765–770. [PubMed] [Google Scholar]
- Bowery NG. Historical perspective and emergence of the GABAB receptor. In: Blackburn TP, editor. GABAB Receptor Pharmacology: A Tribute to Norman Bowery. Advances in Pharmacology. Vol. 58. New York: Academic Press; 2010. pp. 1–18. [DOI] [PubMed] [Google Scholar]
- Bredenoord AJ. Lesogaberan, a GABAB agonist for the treatment of gastroesophageal reflux disease. Drugs. 2009;12:576–584. [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step methods of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Chronwall BM, Davis TD, Severidt MW, Wolfe SE, McCarson KE, Beatty DM, et al. Constitutive expression of functional GABAB receptors in mIL-tsA58 cells requires both GABA(B(1)) and GABA(B(2)) genes. J Neurochem. 2001;77:1237–1247. doi: 10.1046/j.1471-4159.2001.00323.x. [DOI] [PubMed] [Google Scholar]
- Cornelisse LN, Van der Harst JE, Lodder JC, Baarendse PJ, Timmerman AJ, Mansvelder HD, et al. Reduced 5-HT1A and GABAB receptor function in dorsal raphe neurons upon chronic fluoxetine treatment of socially stressed rats. J Neurophysiol. 2007;98:196–204. doi: 10.1152/jn.00109.2007. [DOI] [PubMed] [Google Scholar]
- Cross JA, Cheetham SC, Crompton MR, Katona CL, Horton RW. Brain GABAB binding sites in depressed suicide victims. Psychiatry Res. 1988;26:119–129. doi: 10.1016/0165-1781(88)90066-2. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Slattery DA. GABAB receptors and depression: current status. In: Blackburn TP, editor. GABAB Receptor Pharmacology: A Tribute to Norman Bowery. Advances in Pharmacology. Vol. 58. New York: Academic Press; 2010. pp. 427–451. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Kelly PH, Chaperon F, Gentsch C, Mombereau C, Lingenhoehl K, et al. Behavioral characterization of the novel GABAB receptor-positive modulator GS39783 (N,N'-dicyclopentyl-2-methylsulfanyl-5-nitro-pyrimidine-4,6-diamine): anxiolytic-like activity without side effects associated with baclofen or benzodiazepines. J Pharmacol Exp Ther. 2004;310:952–963. doi: 10.1124/jpet.104.066753. [DOI] [PubMed] [Google Scholar]
- Cunningham MD, Enna SJ. Evidence for pharmacologically distinct GABAB receptors associated with cAMP production in rat brain. Brain Res. 1996;720:220–224. doi: 10.1016/0006-8993(96)00120-5. [DOI] [PubMed] [Google Scholar]
- Davis LL, Trivedi M, Choate A, Kramer GL, Petty F. Growth hormone response to the GABA agonist baclofen in major depressive disorder. Psychoneuroendocrinology. 1997;22:129–140. doi: 10.1016/s0306-4530(96)00048-0. [DOI] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Enna SJ. GABA-B mystery. The search for pharmacologically distinct GABA-B receptors. Mol Interv. 2001;1:208–218. [PubMed] [Google Scholar]
- Enna SJ. The GABA receptors. In: Enna SJ, Mohler H, editors. The GABA Receptors. 3rd. Totowa, NJ: Humana Press; 2007. pp. 1–21. [Google Scholar]
- Enna SJ, Bowery NG. GABAB receptor alterations as indicators of physiological and pharmacological function. Biochem Pharmacol. 2004;68:1541–1548. doi: 10.1016/j.bcp.2004.06.037. [DOI] [PubMed] [Google Scholar]
- Enna SJ, Bowery NG. GABAB receptor. In: Lennarz W, Lane MD, editors. Encyclopedia of Biological Chemistry. Vol. 3. New York: Elsevier; 2010. in press. [Google Scholar]
- Enna SJ, Williams M. Challenges in the search for drugs to treat central nervous system disorders. J Pharmacol Exp Ther. 2009;329:1–8. doi: 10.1124/jpet.108.143420. [DOI] [PubMed] [Google Scholar]
- Enna SJ, Reisman SA, Stanford JA. CGP56999A, a GABAB receptor antagonist, enhances expression of brain-derived neurotrophic factor and attenuates dopamine depletion in the rat corpus striatum following a 6-hydroxydopamine lesion of the nigrostriatal pathway. Neurosci Lett. 2006;406:102–106. doi: 10.1016/j.neulet.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Farb DH, Steiger JL, Martin SC, Gravielle MC, Gibbs TT, Russek SJ. Mechanisms of GABAA and GABAB receptor gene regulation and cell surface expression. In: Enna SJ, Mohler H, editors. The GABA Receptors. 3rd. Totowa, NJ: Humana Press; 2007. pp. 169–238. [Google Scholar]
- Fatemi SH, Stary JM, Earle JA, Araghi-Nikman M, Eagan E. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr Res. 2005;72:109–122. doi: 10.1016/j.schres.2004.02.017. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Folsom TD, Reutiman TJ, Thuras PD. Expression of GABAB receptors is altered in brains of subjects with autism. Cerebellum. 2009;8:64–69. doi: 10.1007/s12311-008-0075-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Alacid L, Aguado C, Ciruela F, Martin R, Colon J, Cabanero MJ. Subcellular compartment-specific molecular diversity of pre- and post-synaptic GABA-activated GIRK channels in Purkinje cells. J Neurochem. 2009;110:1363–1376. doi: 10.1111/j.1471-4159.2009.06229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorentino H, Kuczewski N, Diabira D, Ferrand N, Pangalos MN, Porcher C, et al. GABAB receptor activation triggers BDNF release and promotes the maturation of GABAergic synapses. J Neurosci. 2009;29:11650–11661. doi: 10.1523/JNEUROSCI.3587-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankowska M, Filip M, Przegalinski E. Effects of GABAB receptor ligands in animal tests of depression and anxiety. Pharmacol Rep. 2007;59:645–655. [PubMed] [Google Scholar]
- Frieling H, Bleich S. Tranylcypromine: new perspectives on an old drug. Eur Arch Psychiatry Clin Neurosci. 2006;256:268–273. doi: 10.1007/s00406-006-0660-8. [DOI] [PubMed] [Google Scholar]
- Froestl W. Chemistry & pharmacology of GABAB receptor ligands. In: Blackburn TP, editor. GABAB Receptor Pharmacology: A Tribute to Norman Bowery. Advances in Pharmacology. Vol. 58. New York: Academic Press; 2010. pp. 19–62. [DOI] [PubMed] [Google Scholar]
- Froestl W, Mickel SJ, von Sprecher G, Diel PJ, Hall RG, Maier L, et al. Phosphinic acid analogues of GABA. 2. Selective, orally active GABAB antagonists. J Med Chem. 1995;38:3313–3331. doi: 10.1021/jm00017a016. [DOI] [PubMed] [Google Scholar]
- Froestl W, Gallagher M, Jenkins H, Madrid A, Melcher T, Teichman S, et al. SGS742: the first GABA(B) receptor antagonist in clinical trials. Biochem Pharmacol. 2004;68:1469–1487. doi: 10.1016/j.bcp.2004.07.030. [DOI] [PubMed] [Google Scholar]
- Furtinger SH, Pirker S, Czech T, Baumgartner C, Sperk G. Increased expression of γ-aminobutyric acid type B receptors in the hippocampus of patients with temporal lobe epilepsy. Neurosci Lett. 2003;352:141–145. doi: 10.1016/j.neulet.2003.08.046. [DOI] [PubMed] [Google Scholar]
- Gerson LB, Huff FJ, Hila A, Hirota WK, Reilley S, Agrawal A, et al. Arbaclofen placarbil decreases postprandial reflux in patients with gastroesophageal reflux disease. Am J Gastroenterol. 2010;105:1266–1275. doi: 10.1038/ajg.2009.718. [DOI] [PubMed] [Google Scholar]
- Gray JA, Green AR. Increased GABAB receptor function in mouse frontal cortex after repeated administration of antidepressant drugs or electroconvulsive shock. Br J Pharmacol. 1987;92:357–362. doi: 10.1111/j.1476-5381.1987.tb11331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajszan T, Dow A, Warner-Schmidt JL, Szigeti-Buck K, Sallam NL, Parducz A, et al. Remodeling of hippocampal spine synapses in the rat learned helplessness model of depression. Biol Psychiatry. 2009;65:392–400. doi: 10.1016/j.biopsych.2008.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heese K, Otten U, Mathivet P, Raiteri M, Marescaux C, Bernasconi R. GABAB receptor antagonists elevate both mRNA and protein levels of the neurotrophins nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) but not neurotrophin-3 (NT-3) in brain and spinal cord of rats. Neuropharmacology. 2000;39:449–462. doi: 10.1016/s0028-3908(99)00166-5. [DOI] [PubMed] [Google Scholar]
- Herpfer I, Lieb K. Substance P receptor antagonists in psychiatry: rationale for development and therapeutic potential. CNS Drugs. 2005;19:275–293. doi: 10.2165/00023210-200519040-00001. [DOI] [PubMed] [Google Scholar]
- Jacobson LN, Cryan JF. Evaluation of the anxiolytic-like profile of the GABAB receptor positive modulator CGP7930 in rodents. Neuropharmacology. 2008;54:854–862. doi: 10.1016/j.neuropharm.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, et al. GABA(B) receptors function as a heteromeric assembly of the subunits GABA(B)R1 and GABA(B)R2. Nature. 1998;396:674–679. doi: 10.1038/25348. [DOI] [PubMed] [Google Scholar]
- Karbon EW, Enna SJ. Characterization of the relationship between γ-aminobutyric acid B (GABAB) agonists and transmitter-coupled cyclic nucleotide generating systems in rat brain. Mol Pharmacol. 1985;27:53–59. [PubMed] [Google Scholar]
- Kaupmann K, Huggel K, Heid J, Flor PJ, Bischoff S, Mickel SJ, et al. Expression cloning of GABA(B) receptors uncovers similarity to metabotropic glutamate receptors. Nature. 1997;386:239–246. doi: 10.1038/386239a0. [DOI] [PubMed] [Google Scholar]
- Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, et al. GABAB receptor subtypes assemble into functional heteromeric complexes. Nature. 1998;396:683–687. doi: 10.1038/25360. [DOI] [PubMed] [Google Scholar]
- Kelsey JE, Nemeroff CB. Affective disorders. In: Enna SJ, Coyle JT, editors. Pharmacological Management of Neurological and Psychiatric Disorders. New York: McGraw-Hill; 1998. pp. 95–136. [Google Scholar]
- Klempan TA, Sequeira A, Canetti L, Lalovic A, Ernst C, ffrench-Mullen J, et al. Altered expression of genes involved in ATP biosynthesis and GABAergic neurotransmission in the ventral prefrontal cortex of suicides with and without major depression. Mol Psychiatry. 2009;14:175–189. doi: 10.1038/sj.mp.4002110. [DOI] [PubMed] [Google Scholar]
- Koolschijn PC, van Haren NE, Lensvelt-Mulders GJ, Hulshoff Pol HE, Kahn RS. Brain volume abnormalities in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Hum Brain Mapp. 2009;30:3719–3735. doi: 10.1002/hbm.20801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, Sanacora G, Blumberg H, Anand A, Charney DS, Marek G, et al. Glutamate and GABA systems as targets for novel antidepressants and mood stabilizing treatments. Mol Psychiatry. 2002;7:S71–S80. doi: 10.1038/sj.mp.4001021. [DOI] [PubMed] [Google Scholar]
- Kubo Y, Tateyama M. Towards a view of functioning dimeric metabotropic receptors. Curr Opin Neurobiol. 2005;15:289–295. doi: 10.1016/j.conb.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Kuhn R. The treatment of depressive states with G22355 (imipramine hydrochloride) Am J Psychiatry. 1958;115:459–464. doi: 10.1176/ajp.115.5.459. [DOI] [PubMed] [Google Scholar]
- Ladera C, del Carmen Godino M, Cabanero M, Torres M, Watanabe M, Lujan R, et al. Presynaptic GABA receptors inhibit glutamate release through GIRK channels in rat cerebral cortex. J Neurochem. 2008;107:1506–1517. doi: 10.1111/j.1471-4159.2008.05712.x. [DOI] [PubMed] [Google Scholar]
- Lal R, Sukbuntherng J, Tai EH, Upadhyay S, Yao F, Warren MS, et al. Arbaclofen placarbil, a novel R-baclofen prodrug: improved absorption, distribution, metabolism, and elimination properties compared with R-baclofen. J Pharmacol Exp Ther. 2009;330:911–921. doi: 10.1124/jpet.108.149773. [DOI] [PubMed] [Google Scholar]
- Lesage A, Steckler T. Metabotropic glutamate mGlu(1) receptor stimulation and blockade: therapeutic opportunities in psychiatric illness. Eur J Pharmacol. 2010;639:2–16. doi: 10.1016/j.ejphar.2009.12.043. [DOI] [PubMed] [Google Scholar]
- Levinson AJ, Fitzgerald PB, Favalli G, Blumberger DM, Daigle M, Daskalakis ZJ. Evidence of cortical inhibitory deficits in major depressive disorder. Biol Psychiatry. 2010;210:458–464. doi: 10.1016/j.biopsych.2009.09.025. [DOI] [PubMed] [Google Scholar]
- Lloyd G, Thuret F, Pilc A. Upregulation of gamma-aminobutyric acid (GABA) B binding sites in rat frontal cortex: a common action of repeated administration of different classes of antidepressants and electroshock. J Pharmacol Exp Ther. 1985;235:191–199. [PubMed] [Google Scholar]
- Lucey JV, Butcher G, O'Flynn K, Clare AW, Dinan G. The growth hormone response to baclofen in obsessive compulsive disorder: does the GABA-B receptor mediate obsessive anxiety? Pharmacopsychiatry. 1994;27:23–26. doi: 10.1055/s-2007-1014269. [DOI] [PubMed] [Google Scholar]
- McCarson KE, Enna SJ. Nociceptive regulation of GABAB receptor gene expression in rat spinal cord. Neuropharmacology. 1999;38:1767–1773. doi: 10.1016/s0028-3908(99)00121-5. [DOI] [PubMed] [Google Scholar]
- McCarson KE, Krause JE. NK-1 and NK-3 type tachykinin receptor mRNA expression in the rat spinal cord dorsal horn is increased during adjuvant or formalin-induced nociception. J Neurosci. 1994;14:712–720. doi: 10.1523/JNEUROSCI.14-02-00712.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarson KE, Ralya A, Reisman SA, Enna SJ. Amitriptyline prevents thermal hyperalgesia and modifications in the rat spinal cord GABAB receptor expression and function in an animal model of neuropathic pain. Biochem Pharmacol. 2005;71:196–202. doi: 10.1016/j.bcp.2005.10.026. [DOI] [PubMed] [Google Scholar]
- McCarson KE, Duric V, Reisman SA, Winter M, Enna SJ. GABAB receptor function and subunit expression in the rat spinal cord as indicators of stress and the antinociceptive response to antidepressants. Brain Res. 2006;1068:109–117. doi: 10.1016/j.brainres.2005.11.015. [DOI] [PubMed] [Google Scholar]
- Maciag D, Hughes J, O'Dwyer G, Pride Y, Stockmeier CA, Sanacora G, et al. Reduced density of calbindin immunoreactive GABAergic neurons in the occipital cortex in major depression: relevance to neuroimaging studies. Biol Psychiatry. 2009;67:465–470. doi: 10.1016/j.biopsych.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci. 2000;20:9104–9110. doi: 10.1523/JNEUROSCI.20-24-09104.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannoury la Cour C, Herbelles C, Pasteau P, deNanteuil G, Millan MJ. Influence of positive allosteric modulators on GABAB receptor coupling in rat brain: a scintillation proximity assay characterization of G protein subtypes. J Neurochem. 2008;105:308–323. doi: 10.1111/j.1471-4159.2007.05131.x. [DOI] [PubMed] [Google Scholar]
- Marchesi C, Chiodera P, De Ferri A, De Risio C, Dasso L, Menozzi P, et al. Reduction of GH response to the GABA-B agonist baclofen in patients with major depression. Psychoneuroendocrinology. 1991;16:465–479. doi: 10.1016/0306-4530(91)90031-n. [DOI] [PubMed] [Google Scholar]
- Martin P, Pichat P, Massol J, Soubrie P, Lloyd KG, Puech J. Decreased GABAB receptors in helpless rats: reversal by tricyclic antidepressants. Neuropsychobiology. 1989;22:220–224. doi: 10.1159/000118620. [DOI] [PubMed] [Google Scholar]
- Mayberg HS. Modulating dysfunctional limbic-cortical circuits in depression: towards development of brain-based algorithms for diagnosis and optimized treatment. Br Med Bull. 2003;65:193–207. doi: 10.1093/bmb/65.1.193. [DOI] [PubMed] [Google Scholar]
- Merlo D, Mollinari C, Inaba Y, Cardinale A, Rinaldi AM, D'Antuono M, et al. Reduced GABAB receptor subunit expression and paired-pulse depression in a genetic model of absence seizures. Neurobiol Dis. 2007;25:631–641. doi: 10.1016/j.nbd.2006.11.005. [DOI] [PubMed] [Google Scholar]
- Miller BH, Schultz LE, Gulati A, Comeron MD, Pletcher MT. Genetic regulation of behavioral and neuronal responses to fluoxetine. Neuropsychopharmacology. 2008;33:1312–1322. doi: 10.1038/sj.npp.1301497. [DOI] [PubMed] [Google Scholar]
- Mizukami K, Ishikawa M, Hidaka S, Iwakiri M, Sasaki M, Iritani S. Immunohistochmical localization of GABAB receptor in the entorhinal cortex and inferior temporal cortex of schizophrenic brain. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26:393–396. doi: 10.1016/s0278-5846(01)00247-0. [DOI] [PubMed] [Google Scholar]
- Mombereau C, Kaupmann K, Froestl W, Sansig G, van der Putten H, Cryan JF. Genetic and pharmacological evidence of a role for GABAB receptors in the modulation of anxiety and antidepressant-like behavior. Neuropsychopharmacology. 2004;29:1050–1062. doi: 10.1038/sj.npp.1300413. [DOI] [PubMed] [Google Scholar]
- Mombereau C, Kaupmann K, Gassmann M, Bettler B, van der Putten H, Cryan JF. Altered anxiety and depression-related behavior in mice lacking GABAB(2) receptor subunits. Neuroreport. 2005;16:307–310. doi: 10.1097/00001756-200502280-00021. [DOI] [PubMed] [Google Scholar]
- Mutneja M, Berton F, Suen KF, Luscher C, Slesinger PA. Endogeneous RGS proteins enhance acute desensitization of GABA(B) receptor-activated GIRK currents in HEK-293T cells. Pflugers Arch. 2005;450:61–73. doi: 10.1007/s00424-004-1367-1. [DOI] [PubMed] [Google Scholar]
- Nakagawa Y, Ishima T. Possible involvement of GABAB receptors in action of antidepressants. Nihon Shinkei Seishin Yakurigaku Zasshi. 2003;23:83–89. [PubMed] [Google Scholar]
- Nakagawa Y, Ishima T, Ishibashi Y, Tsuji M, Takashima T. Involvement of GABAB receptor systems in experimental depression: baclofen but not bicuculline exacerbates helplessness in rats. Brain Res. 1996;741:240–245. doi: 10.1016/s0006-8993(96)00929-8. [DOI] [PubMed] [Google Scholar]
- Nakagawa Y, Sasaki A, Takashima T. The GABAB receptor antagonist CGP36742 improves learned helplessness in rats. Eur J Pharmacol. 1999;381:1–7. doi: 10.1016/s0014-2999(99)00567-1. [DOI] [PubMed] [Google Scholar]
- Nandan D, Reiner NE. TGF beta attenuates the class II transactivator and reveals an accessory pathway of IFN-gamma action. J Immunol. 1997;158:1095–1011. [PubMed] [Google Scholar]
- Nestler EJ, Hyman SE, Malenka RC. Molecular Pharmacology: A Foundation for Clinical Neuroscience. New York: McGraw-Hill; 2001. pp. 327–354. Chapter 15. [Google Scholar]
- Nowak G, Partyka A, Palucha A, Szewczyk B, Wieronska JM, Dybala M, et al. Antidepressant-like activity of CGP 36742 and CGP 51176, selective GABAB receptor antagonists, in rodents. Br J Pharmacol. 2006;149:581–590. doi: 10.1038/sj.bjp.0706845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Flynn K, Dinan TG. Baclofen-growth hormone release in major depression: relationship to dexamethasone suppression test. Am J Psychiatry. 1993;150:1728–1730. doi: 10.1176/ajp.150.11.1728. [DOI] [PubMed] [Google Scholar]
- Paterson NE, Hanania T. The modified Geller-Seifter test in rats was insensitive to GABAB receptor positive modulaton or blockade, or 5-HT1A receptor activation. Behav Brain Res. 2010;208:258–264. doi: 10.1016/j.bbr.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Petty F. GABA and mood disorders: a brief review of hypothesis. J Affect Disord. 1995;34:275–281. doi: 10.1016/0165-0327(95)00025-i. [DOI] [PubMed] [Google Scholar]
- Pilc A, Lloyd G. Chronic antidepressants and GABA ‘B’ receptors: a GABA hypothesis of antidepressant drug action. Life Sci. 1984;35:2149–2154. doi: 10.1016/0024-3205(84)90515-0. [DOI] [PubMed] [Google Scholar]
- Pinard A, Seddik R, Bettler B. GABAB receptors: physiological functions and mechanisms of diversity. In: Blackburn TP, editor. GABAB Receptor Pharmacology: A Tribute to Norman Bowery. Advances in Pharmacology. Vol. 58. New York: Academic Press; 2010. pp. 231–255. [DOI] [PubMed] [Google Scholar]
- Pittenger C, Duman RS. Stress, depression, and neuroplasticity: a convergence of mechanisms. Neuropsychopharmacology. 2008;33:88–109. doi: 10.1038/sj.npp.1301574. [DOI] [PubMed] [Google Scholar]
- Post RM, Ketter TA, Joffe RT, Kramlinger KL. Lack of beneficial effects of l-baclofen in affective disorder. Int Clin Psychopharmacol. 1991;6:197–207. doi: 10.1097/00004850-199100640-00001. [DOI] [PubMed] [Google Scholar]
- Pratt GD, Bowery NG. Repeated administration of desipramine and a GABAB receptor antagonist, CGP 36742, discretely up-regulates GABAB receptor binding sites in rat frontal cortex. Br J Pharmacol. 1993;110:724–735. doi: 10.1111/j.1476-5381.1993.tb13872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Drevets WC. Neurocircuitry of mood disorders. Neuropsychopharmacology. 2010;35:192–216. doi: 10.1038/npp.2009.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Princivalle AP, Duncan JS, Thom M, Bowery NG. GABAB1a, GABAB1b, and GABAB2 mRNA variants expression in hippocampus resected from patients with temporal lobe epilepsy. Neuroscience. 2003;122:975–984. doi: 10.1016/j.neuroscience.2003.08.044. [DOI] [PubMed] [Google Scholar]
- Rajkowska G. Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol Psychiatry. 2000;48:766–777. doi: 10.1016/s0006-3223(00)00950-1. [DOI] [PubMed] [Google Scholar]
- Rajkowska G, Miguel-Hidalgo JJ, Wei J, Dilley G, Pittman SD, Meltzer HY, et al. Morphomethric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol Psychiatry. 1999;45:1085–1098. doi: 10.1016/s0006-3223(99)00041-4. [DOI] [PubMed] [Google Scholar]
- Rajkowska G, O'Dwyer G, Teleki Z, Stockmeier CA, Miguel-Hidalgo JJ. GABAergic neurons immunoreactive for calcium binding proteins are reduced in the prefrontal cortex in major depression. Neuropsychopharmacology. 2007;32:471–482. doi: 10.1038/sj.npp.1301234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Mason GF, Krystal JH. Impairment of GABAergic transmission in depression: new insights from neuroimaging studies. Crit Rev Neurobiol. 2000;14:23–45. doi: 10.1615/critrevneurobiol.v14.i1.20. [DOI] [PubMed] [Google Scholar]
- Sands SA, McCarson KE, Enna SJ. Differential regulation of GABAB receptor subunit expression and function. J Pharmacol Exp Ther. 2003a;305:191–196. doi: 10.1124/jpet.102.046342. [DOI] [PubMed] [Google Scholar]
- Sands SA, Reisman SA, Enna SJ. Effects of stress and tranylcypromime on amphetamine-induced locomotor activity and GABAB receptor function in rat brain. Life Sci. 2003b;72:1085–1092. doi: 10.1016/s0024-3205(02)02360-3. [DOI] [PubMed] [Google Scholar]
- Sands SA, Reisman SA, Enna SJ. Effect of antidepressants on GABAB receptor function and subunit expression in the rat hippocampus. Biochem Pharmacol. 2004;68:1489–1495. doi: 10.1016/j.bcp.2004.07.027. [DOI] [PubMed] [Google Scholar]
- Schwenk J, Metz M, Zolles G, Turecek R, Fritzius T, Bildl W, et al. Native GABAB receptors are heteromultimers with a family of auxiliary subunits. Nature. 2010;465:231–235. doi: 10.1038/nature08964. [DOI] [PubMed] [Google Scholar]
- Seminowicz DA, Mayberg HS, McIntosh AR, Goldapple K, Kennedy S, Segal Z, et al. Limbic-frontal circuitry in major depression: a path modeling metanalysis. Neuroimage. 2004;22:409–418. doi: 10.1016/j.neuroimage.2004.01.015. [DOI] [PubMed] [Google Scholar]
- Sequeira A, Mamdani F, Ernst C, Vawter MP, Bunney WE, Lebel V, et al. Global brain gene expression analysis links glutamatergic and GABAergic alterations to suicide and major depression. PLoS One. 2009;4:e6585. doi: 10.1371/journal.pone.0006585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline YI. Hippocampal atrophy in major depression: a result of depression-induced neurotoxicity? Mol Psychiatry. 1996;1:298–299. [PubMed] [Google Scholar]
- Sheline YI, Sanghavi M, Mintun MA, Gada MH. Depression duration but not age predicts hippocampal volume loss in medically healthy women with recurrent major depression. J Neurosci. 1999;19:5034–5043. doi: 10.1523/JNEUROSCI.19-12-05034.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazaki T, Yoshimizu T, Chaki S. Melanin-concentrating hormone MCH1 receptor antagonists: a potential new approach to the treatment of depression and anxiety disorders. CNS Drugs. 2006;20:801–811. doi: 10.2165/00023210-200620100-00002. [DOI] [PubMed] [Google Scholar]
- Slattery DA, Desrayaud S, Cryan JF. GABAB receptor antagonist-mediated antidepressant-like behavior is serotonin-dependent. J Pharmacol Exp Ther. 2005;312:290–296. doi: 10.1124/jpet.104.073536. [DOI] [PubMed] [Google Scholar]
- Stan A, Ghose S, Gao XM, Roberts RC, Lewis-Amezcua K, Hatanpaa K, et al. Human postmortem tissue: what quality markers matter? Brain Res. 2006;1123:1–11. doi: 10.1016/j.brainres.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzdak PD, Gianutsos G. Effect of chronic imipramine or baclofen on GABA-B binding and cyclic AMP production in cerebral cortex. Eur J Pharmacol. 1986;131:129–133. doi: 10.1016/0014-2999(86)90526-1. [DOI] [PubMed] [Google Scholar]
- Tanti A, Belzung C. Open questions in current models of antidepressant action. Br J Pharmacol. 2010;159:1187–1200. doi: 10.1111/j.1476-5381.2009.00585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuault SJ, Brown JT, Sheardown SA, Jourdain S, Farifax B, Spencer JP, et al. The GABA(B2) subunit is critical for trafficking and function of native GABA(B) receptors. Biochem Pharmacol. 2004;68:1655–1666. doi: 10.1016/j.bcp.2004.07.032. [DOI] [PubMed] [Google Scholar]
- Towers S, Princivalle A, Billinton A, Edmunds M, Bettler B, Urban L, et al. GABAB receptor protein and mRNA distribution in rat spinal cord and dorsal root ganglia. Eur J Neurosci. 2001;12:3201–3210. doi: 10.1046/j.1460-9568.2000.00237.x. [DOI] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006;9:519–525. doi: 10.1038/nn1659. [DOI] [PubMed] [Google Scholar]
- Valdez GR. CRF receptors as a potential target in the development of novel pharmacotherapies for depression. Curr Pharm Des. 2009;15:1587–1594. doi: 10.2174/138161209788168083. [DOI] [PubMed] [Google Scholar]
- Vigot R, Barbieri S, Brauner-Osborne H, Turecek R, Shigemoto R, Zhang YP, et al. Differential compartmentalization of distinct functions of GABAB receptor variants. Neuron. 2006;18:589–601. doi: 10.1016/j.neuron.2006.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmeier PC, Wicki P, Feldtrauer JJ, Mickel SJ, Bittiger H, Baumann PA. GABA and glutamate release affected by GABAB receptor antagonists with similar potency: no evidence for pharmacologically different presynaptic receptors. Br J Pharmacol. 1994;113:1515–1521. doi: 10.1111/j.1476-5381.1994.tb17168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldvogel HJ, Billinton A, White JH, Emson PC, Faull RL. Comparative cellular distribution of GABAA and GABAB receptors in the human basal ganglia: immunohistochemical colocalization of the alpha 1 subunit of the GABAA receptor, and the GABABR1 and GABABR2 receptor subunits. J Comp Neurol. 2004;470:339–356. doi: 10.1002/cne.20005. [DOI] [PubMed] [Google Scholar]
- White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, et al. Heterodimerization is required for the formation of a functional GABA(B) receptor. Nature. 1998;396:679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]
- Yucel K, McKinnon M, Chahal R, Taylor V, Macdonald K, Joffe R, et al. Increased subgenual prefrontal cortex size in remitted patients with major depressive disorder. Psychiatry Res. 2009;173:71–76. doi: 10.1016/j.pscychresns.2008.07.013. [DOI] [PubMed] [Google Scholar]