

Abstract
BACKGROUND AND PURPOSE
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors, and three subtypes (α, β and γ) have been identified. PPAR activation has been reported to decrease renal injury and markers of glomerular dysfunction in models of renal ischemia/reperfusion (I/R). However, both the I/R effects and the effects of PPAR agonists on podocytes, an integral cellular part of the glomerular filtration barrier, remain to be established.
EXPERIMENTAL APPROACH
By using oxygen/glucose deprivation-reoxygenation as an in vitro model that mimics in vivo I/R, the effects of PPAR agonists on podocyte death were compared. Human immortalized podocytes were treated with gemfibrozil, GW0742, pioglitazone or rosiglitazone, as a single or repeated challenge. Cell loss, necrotic and apoptotic cell death were measured.
KEY RESULTS
Only the repeated treatment with each PPAR agonist significantly prevented cell death, mainly by decreasing apoptosis. In comparison, in a model of serum deprivation-induced apoptosis, both treatments were effective, although the repeated treatment achieved the more pronounced effect. Finally, our results showed that preservation of Bcl-2, Bax and nephrin expression accompanied the anti-apoptotic effects exerted by PPAR agonists in human podocytes.
CONCLUSION AND IMPLICATIONS
These findings contribute to clarification of the pathophysiological role of renal PPARs and suggest that selective PPARα, PPARβ or PPARγ agonists may exert similar protective effects on podocytes by decreasing apoptotic cell death.
Keywords: ischemia-reperfusion, podocytes, cell death, in vitro studies
Introduction
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors of the nuclear receptor superfamily. Three PPAR subtypes (α, β, and γ) have been characterized both in rodents and humans (Berger and Moller, 2002; nomenclature follows Alexander et al., 2009). All PPAR subtypes are expressed in the kidney (Ruan et al., 2008), and it has been suggested that these transcription factors contribute to renal pathophysiology (Boulanger et al., 2006; Ruan et al., 2008). However, renal PPARs still require further characterization and definition of their roles.
Experimental findings have revealed that PPAR activity influences ischemia/reperfusion (I/R) injury in several organs, including the kidney (Chatterjee, 2007; Di Paola and Cuzzocrea, 2007; Collino et al., 2008). Worse organ damage and dysfunction have been reported in PPARα- or PPARβ-null mice after renal I/R when compared with wild-type control animals (Portilla et al., 2000; Letavernier et al., 2005). Conversely, structurally different PPARα, PPARβ or PPARγ agonists have been shown to attenuate renal I/R injury in rats (Portilla et al., 2000; Sivarajah et al., 2002; 2003; Collino et al., 2005; Letavernier et al., 2005). The effects of PPAR agonists on renal I/R injury have been mainly investigated in animal models, whereas few studies have focused on specific renal cell types.
Renal I/R is known to compromise glomerular integrity. In rats, mild renal I/R has been reported to cause a decrease in glomerular filtration barrier (GFB) charge and size selectivity, proteinuria and podocyte effacement (Rippe et al., 2006; Andersson et al., 2007; Wagner et al., 2008). More prolonged insults, typically associated with organ preservation before transplantation, can cause severe glomerular lesions including podocyte loss and glomerulosclerosis (Lambert et al., 1986; Pippin et al., 2009). In patients, marked proteinuria has been reported during the initial hours after renal transplantation (Stefanidis et al., 1996). Moreover, proteinuria associated with glomerulosclerosis is increasingly common late after transplantation (Nankivell et al., 2003; 2004;). Proteinuria and glomerulosclerosis are clinical signs of several glomerulopathies, including focal segmental glomerulosclerosis, membranous nephropathy, minimal change disease and diabetic nephropathy (Shankland, 2006), where injury to podocytes is a common and determining factor (Wiggins, 2007). In contrast, the role of podocytes in renal I/R injury has been less well investigated.
In this study, by using oxygen/glucose deprivation (OGD)-reoxygenation as an in vitro model that mimics an in vivo I/R insult, we have evaluated the effects of I/R on the death of human podocytes. The OGD-reoxygenation model allows the elucidation of the putative roles of specific stimuli implicated in pathological events in vivo. It has been widely adopted to study the effects of I/R on distinct renal cell types (Russ et al., 2007). However, although a significant number of studies have been published utilising kidney tubular and endothelial cells (Molitoris and Sutton, 2004; Russ et al., 2007), relatively less is known about the effects of I/R on podocytes. In particular, the effects of I/R on podocyte death, which is known to alter GFB integrity and has been postulated to be the ‘committed’ lesion in the development of glomerulosclerosis (Shankland, 2006; Wiggins, 2007) remains largely unexplored. Therefore, we decided to characterize the PPAR subtypes expressed by human podocytes, and to study the role of these transcription factors in protecting podocytes against I/R injury. To this purpose, we have evaluated the effects of structurally different PPAR agonists – gemfibrozil, GW0742, pioglitazone and rosiglitazone – on OGD-reoxygenation-induced podocyte death.
Methods
Cultures of human podocytes
In this study we have used lines of immortalized human podocytes obtained by infection of cultures of renal cells with a hybrid Adeno5/SV40 virus (Conaldi et al., 1997). Podocytes were characterized as previously reported (Doublier et al., 2001) and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with fetal bovine serum (FBS) 10%, penicillin (100 IU·mL−1), streptomycin (100 µg·mL−1) and L-glutamine (2 mM) (standard conditions). The day before the experiment, cells were plated on 6-well culture plates (300 × 103 cells per well).
In vitro models of podocyte injury and drug treatments
OGD-reoxygenation was performed as previously described (Miglio et al., 2004) and experimental conditions were optimized following the recommendations of Russ et al. (2007). Briefly, during OGD the culture medium was replaced with 1 mL (∼100 µL·cm−2) of a bicarbonate-buffered balanced salt solution (BSS; in mM: 134, NaCl; 15.7, NaHCO3; 3.1, KCl; 1.2, CaCl2; 1.2, MgSO4; 0.25, KH2PO4; pH 7.2), equilibrated with an hypoxic gas mixture N2 (95%)/CO2 (5%), and cultures were placed in an anaerobic chamber (Oxoid, Hampshire, UK) filled with a hypoxic gas mixture (hypoxic conditions) at 37°C. Control samples received glucose (10 mM)-supplemented BSS and were maintained at 37°C in fully humidified air (95%)/CO2 (5%) incubator. Reoxygenation was started at the designated time points (see results) by returning cell cultures to standard culture conditions. Similar experiments were performed to evaluate the contribution of glucose deprivation (GD) and oxygen deprivation (OD)-reoxygenation to the OGD-reoxygenation-induced podocyte injury. In the GD experiments, the culture medium was exchanged with the BSS and cultures were maintained in fully humidified air (95%)/CO2 (5%) incubator. In the OD experiments, the culture medium was exchanged with the glucose (10 mM)-supplemented BSS and cultures were maintained under hypoxic conditions. At the designated time points (see results), GD and OD were interrupted by returning cultures to standard culture conditions.
Serum deprivation (SD) was obtained by culturing cells in a serum-deprived culture medium (DMEM supplemented with FBS 0.1%) for increasing periods of time (24–72 h; Tejada et al., 2008).
Cells were treated with either PPAR agonists or vehicle alone according to the four experimental protocols shown in Figure 1 of the supplementary materials. For the OGD-reoxygenation, single treatment (Protocol 1), cells were treated for 19 h with the PPAR agonists and then exposed to OGD-reoxygenation. For the OGD-reoxygenation, repeated treatment (Protocol 2) cells were treated for 72 h with the PPAR agonists and then exposed to OGD-reoxygenation. For the SD, single treatment (Protocol 3), cells were treated with the PPAR agonists simultaneously with SD. Finally, for the SD, repeated treatment (Protocol 4) cells were pretreated for 72 h with the PPAR agonists and then exposed to SD. Drugs were replaced at every medium change.
Figure 1.
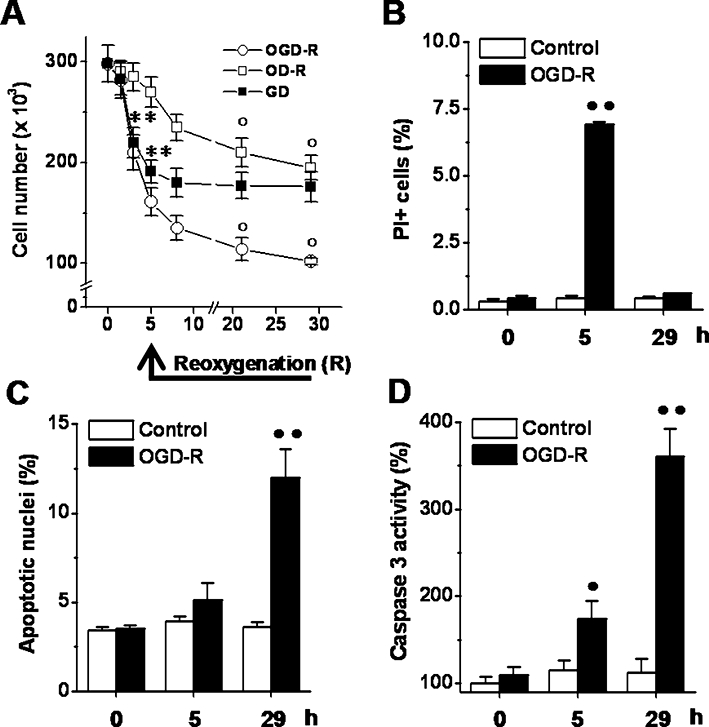
Effects of OGD-reoxygenation on podocyte death. (A) Cells were exposed to OGD (1.5–5 h)-reoxygenation (3–24 h), GD (1.5–5 h) or OD (1.5–5 h)-reoxygenation (3–24 h) and the cell number were determined at the indicated time points. (B–D) Cells were exposed to OGD (5 h)-reoxygenation (24 h) and the parcentage of PI+ cells (B), that of apoptotic nuclei (C) and the caspase 3 activity (D) were determined at the indicated time points. Results are expressed as mean ± SEmean of four experiments run in triplicate. **P < 0.01 versus basal levels; °P < 0.01 versus 5 h-OGD, -GD or -OD; •P < 0.05; ••P < 0.01 versus control.
Evaluation of cell loss, necrotic, and apoptotic cell death
Evaluation of cell loss was performed, without knowledge of the treatments, by determining the number of viable cells in a haemacytometer, using the Trypan blue exclusion test.
Necrosis and apoptosis were evaluated by detecting characteristic hallmarks of the necrotic (loss of plasma membrane integrity) and apoptotic (nuclear pyknosis and/or fragmentation) cell death in the cell cultures (Aras et al., 2008; Galluzzi et al., 2009; Kroemer et al., 2009). Necrotic cells were detected after staining with fluorescein diacetate (FDA)/propidium iodide (PI). Cells were washed twice with phosphate-buffered saline (PBS) and stained with the mixture FDA (4.0 µM)/PI (0.4 µM). After washing, cells were examined using a fluorescence microscope (Leica Microsystems Wetzlar, Germany). Green or red cells (PI+ cells) were scored as healthy/viable and dead respectively (Aras et al., 2008). Apoptotic cells were detected after cell staining with 4′,6-diamidino-2-phenylindole (DAPI). Cells were washed twice with ice-cold PBS, fixed and permeabilized with methanol (4°C; 5 min), and stained with DAPI (0.3 µM). After washing, cells were examined using a fluorescent microscope. Pyknotic and/or fragmented nuclei were scored as apoptotic nuclei (Aras et al., 2008). Between 80 and 120 cells (or nuclei) per field were counted, without knowledge of the treatments and for each experimental condition ∼5000 cells (or nuclei) were examined.
Evaluation of caspase 3 activation
Increase in caspase 3 activity was evaluated as marker of apoptotic cell death. Using a commercial kit (Biovision Research Products, Mountain View, CA, USA), caspase 3 activity was determined by measuring the ability of cell lysates to cleave the P-nitroaniline coupled caspase 3 substrate DEVD, according to the manufacturer's instructions.
Western blot analysis
Western blot analyses were performed as previously described (Miglio et al., 2009). PPARα, PPARβ, PPARγ, Bcl-2, Bax and synaptopodin were detected following incubation with rabbit polyclonal antibodies (Santa Cruz Biotechnology, Sta. Cruz, CA, USA). Nephrin was detected following incubation with a guinea pig polyclonal antibody (GP-N1; Progen Biotechnik, Germany), according to the manufacturer's instructions. To confirm the homogeneity of the proteins loaded, the membranes were stripped and incubated with an anti-β-actin monoclonal antibody (Santa Cruz). The membranes were overlaid with Western Lightning Chemiluminescence Reagent Plus (Perkin Elmer Life Science, Norwalk, CT, USA) and then exposed to Hyperfilm ECL film (Amersham Biosciences, Piscataway, NJ, USA). Protein bands were quantified on film by densitometry using the software Image J 1.41.
Quantitative real-time PCR analysis
Total RNA was extracted with OMNIzol reagent (Euroclone, Milan Italy) according to the manufacturer's instructions. First-strand cDNA was synthesized from 0.5 µg of total RNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA, USA). Real-time PCR experiments were performed in 25 µL reaction mixtures containing 10 ng of cDNA template, the Power SYBR® Green PCR Master Mix and the AmpliTaq Gold® DNA Polymerase LD (Applied Biosystems). The sequence-specific oligonucleotide primers (purchased from Sigma-Genosys, Milan, Italy) and thermal cycling conditions are listed in Supporting Information Table S1. Relative quantization of the products was performed using a 48-well StepOne™ Real Time System (Applied Biosystems). For all real-time PCR analyses, β-actin mRNA was used to normalize RNA inputs. Fold-change expression with respect to control was calculated for all samples.
Confocal microscopy
Indirect immunofluorescence was performed on podocytes cultured on chamber slides (Nalgen Nunc International, Rochester, NY, USA) fixed in 4% paraformaldehyde containing 2% sucrose. Subconfluent cells were stained with a polyclonal anti-nephrin (GP-N1; Progen) or an anti-synaptopodin (Santa Cruz) antibody. An immunologically irrelevant guinea pig serum was used as a control where appropriate. Alexa Fluor-488 anti guinea pig polyclonal antibody (Molecular Probes, Leiden, the Netherlands) was used as a secondary antibody. Confocal microscopy analysis was performed using a Zeiss LSM 5 Pascal Model Confocal Microscope (Carl Zeiss International, Jena, Germany). Hoechst 33258 was added for nuclear staining.
Data and statistical analysis
Results are expressed as mean ± SEmean of at least four experiments. Drug effect percent was calculated as follow:
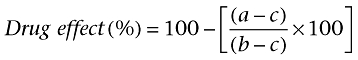 |
where a, b and c were the cell number, the percentage of PI+ cells, the percentage of apoptotic nuclei or the caspase 3 activity measured in cells: unexposed to the insult (c); drug-treated and exposed to the insult (a); drug-untreated and exposed to the insults (b). Statistical significance was evaluated by one-way anova followed by a post hoc Dunnett's test. Differences were considered statistically significant when P < 0.05.
Materials
Pioglitazone and rosiglitazone were from Alexis (Vinci, Italy). GW0742, gemfibrozil, and all other reagents were from Sigma-Aldrich (Milan, Italy). PPAR agonists were dissolved in dimethylsulfoxide and the final drug concentrations were obtained by dilution of stock solutions in the experimental buffers. The final concentration of organic solvent was less than 0.1%, which had no effect on cell viability.
Results
Effects of OGD-reoxygenation on human podocyte death
To study whether renal I/R may cause podocyte death, the effects of OGD-reoxygenation on cell loss was first measured. OGD (1.5–5 h)-reoxygenation (3–24 h) significantly decreased the cell number by about 50% after 5 h of OGD, with a further fall in numbers after 24 h of reoxygenation (Figure 1A). GD and OD-reoxygenation affected cell number differently. In GD cultures, cell numbers decreased after 5 h of GD, but did not fall further during the following 24 h (Figure 1A). In OD cultures, cell numbers did not change during the 5 h of OD, but did fall significantly during the 24 h of reoxygenation (Figure 1A). Hence, GD and OD-reoxygenation contributed to the cell loss associated with OGD and reoxygenation respectively.
To investigate how necrotic and apoptotic cell death contributed to the OGD-reoxygenation-induced cell loss, the percentage of PI+ cells, apoptotic nuclei and caspase 3 activity were measured. The percentage of PI+ cells increased from the basal level after 5 h of OGD and returned to near the basal values after 24 h of reoxygenation (Figure 1B). By contrast, the percentage of apoptotic nuclei increased from the basal level after 5 h of OGD and was further elevated after 24 h of reoxygenation (Figure 1C). Caspase 3 activity also increased from the basal level after 5 h of OGD and after 24 h of reoxygenation (Figure 1D). Compared with the basal level no significant change in the evaluated parameters was measured in control cells (Figure 1B–D). The significant and temporally distinct increase in the percentage of PI+ cells, apoptotic nuclei and in the caspase 3 activity suggests that necrotic and apoptotic cell death contribute to the cell loss associated with OGD and reoxygenation respectively.
Expression and transcriptional activity of PPARs in human podocytes
To characterize our cellular model, first we studied the expression of PPAR subtypes at both mRNA and protein level by quantitative real-time PCR and Western blot analysis respectively. Cells constitutively express genes encoding for all PPAR subtypes and cell treatment with gemfibrozil, GW0742 or pioglitazone, increased PPARA, PPARB, and PPARG expression in a time- and concentration-dependent manner (P < 0.05 vs. control; Figure 2A). Western blot analyses confirmed the findings of the PCR experiments, showing that the PPAR agonists (72 h) up-regulated PPARα, PPARβ and PPARγ in a concentration-dependent manner (Figure 2B). Then, to assess the PPAR transcriptional activity we evaluated whether PPAR agonists up-regulate expression of PPAR-target genes. As shown in Figure 2C, cell exposure (72 h) to gemfibrozil (10 µM), GW0742 (0.1 µM) or pioglitazone (1 µM) significantly up-regulated PPARGC1A, and UCP2, known PPAR target genes (Hondares et al., 2006; Villarroya et al., 2007). Together, these results indicate that our cultures of human podocytes expressed all PPAR subtypes, which were pharmacologically responsive to known agonists and transcriptionally active.
Figure 2.
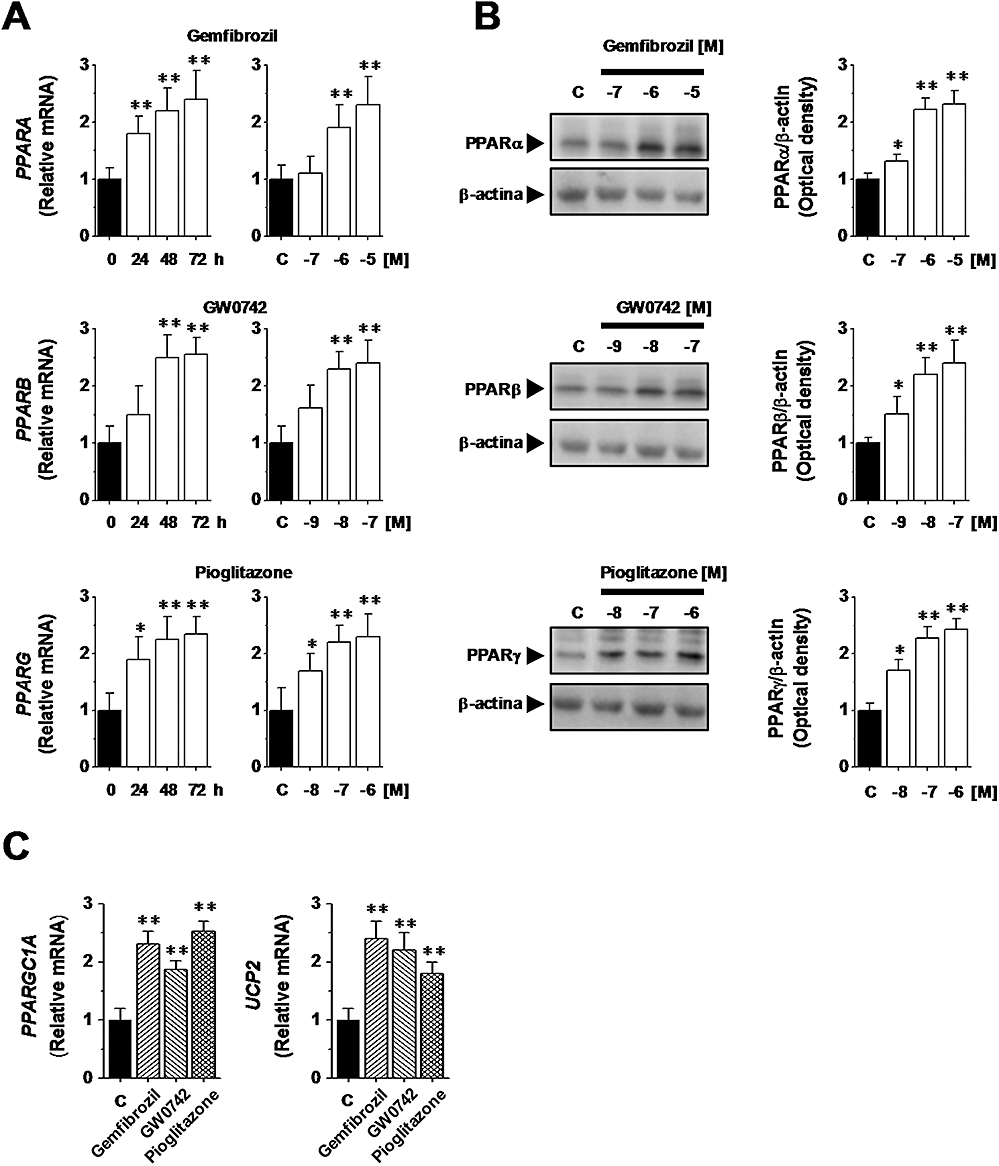
Peroxisome proliferator-activated receptor (PPAR) expression and transcriptional activity in human podocytes. (A) Cells were treated with gemfibrozil (10 µM), GW0742 (0.1 µM) or pioglitazone (1 µM) for increasing times (24–72 h) or with gemfibrozil (0.1–10 µM), GW0742 (1–100 nM) or pioglitazone (0.01–1 µM) for 72 h. PPARA, PPARB and PPARG expression was evaluated by quantitative real-time PCR analysis. (B) Cells were treated with gemfibrozil (0.1–10 µM), GW0742 (1 nM–100 nM) or to pioglitazone (0.01–1 µM) for 72 h and PPARα, PPARβ and PPARγ expression was evaluated by Western blot analysis. β-actin was used as an internal control. The figures are representative of four independent experiments. (C) Cells were treated with gemfibrozil (10 µM), GW0742 (0.1 µM) or pioglitazone (1 µM) for 72 h and PPARGC1A, and UCP2 expression was evaluated by quantitative real-time PCR analysis. Results are expressed as mean ± SEmean of four experiments. *P < 0.05; **P < 0.01 versus untreated cells (c).
Effects of PPAR agonists on OGD-reoxygenation-induced podocyte loss
Cells were treated with gemfibrozil (10 µM), GW0742 (0.1 µM), pioglitazone (1 µM) or rosiglitazone (0.1 µM) as a single (Protocol 1) or repeated treatment (Protocol 2) and were then exposed to OGD (5 h)-reoxygenation (24 h). None of the PPAR agonists decreased the OGD-associated cell loss when added as a single treatment and they had minimal effects (not significant) on the reoxygenation-associated cell loss (Figure 3). However, when added as a repeated treatment, they all decreased both the OGD-associated cell loss and the reoxygenation-associated cell loss (Figure 3). Consequently, all PPAR agonists significantly decreased the overall cell loss (by 33 ± 6%, 29 ± 6%, 36 ± 5% and 37 ± 6%, respectively; P < 0.01 vs. vehicle alone) when used as a repeated treatment but not as a single treatment. Neither longer periods of cell treatment nor higher drug concentrations increased the magnitude of these effects (data not shown).
Figure 3.
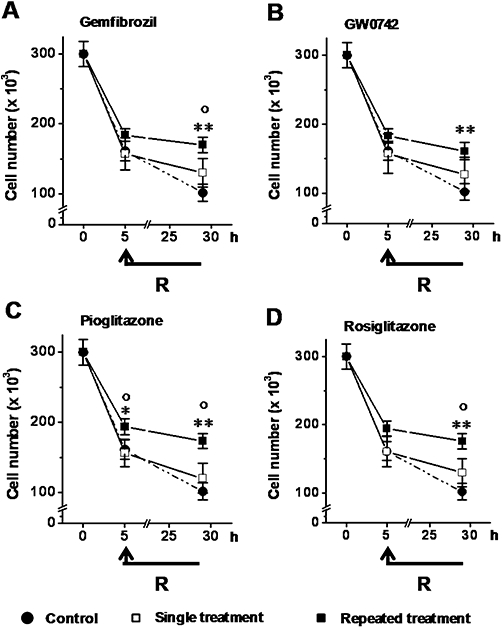
Effects of peroxisome proliferator-activated receptor (PPAR) agonists on OGD-reoxygenation-induced podocyte loss. Cells were treated with gemfibrozil (10 µM; A), GW0742 (0.1 µM; B), pioglitazone (1 µM; C) or rosiglitazone (0.1 µM; D), as a single or repeated treatment and then exposed to OGD (5 h)-reoxygenation (24 h). Results are expressed as mean ± SEmean of five experiments run in triplicate. *P < 0.05; **P < 0.01 versus control (vehicle alone). °P < 0.01 versus single treatment.
Effects of PPAR agonists on OGD-reoxygenation-induced necrosis and apoptosis
None of the drugs affected the OGD-induced increase in the percentage of PI+ cells when added as a single treatment (Figure 4A). However, they significantly prevented (with the exception of GW0742) the increase in the percentage of PI+ cells (P < 0.05 vs. vehicle alone) when added as a repeated treatment (Figure 4A). When compared with the single treatment, the repeated treatment significantly (P < 0.01) reduced the OGD-induced increase in the percentage of PI+ cells (Figure 4A). All drugs significantly decreased the reoxygenation-associated increase in the percentage of apoptotic nuclei (P < 0.05 vs. vehicle alone) when added as single treatment (Figure 4B). Similarly, when added as a repeated treatment, they all prevented the reoxygenation-associated increase in the percentage of apoptotic nuclei (P < 0.01 vs. vehicle alone) (Figure 4B). In comparison with the single treatment, the repeated treatment decreased the reoxygenation-associated increase in the percentage of apoptotic nuclei significantly only for GW0742 (P < 0.01). PPAR agonists also significantly decreased the reoxygenation-associated increase in caspase 3 activity when added as a single treatment or as repeated treatment (Figure 4C). In comparison with the single treatment, the repeated treatment decreased the reoxygenation-associated increase in the percentage of apoptotic nuclei, again significantly only for GW0742 (P < 0.01).
Figure 4.
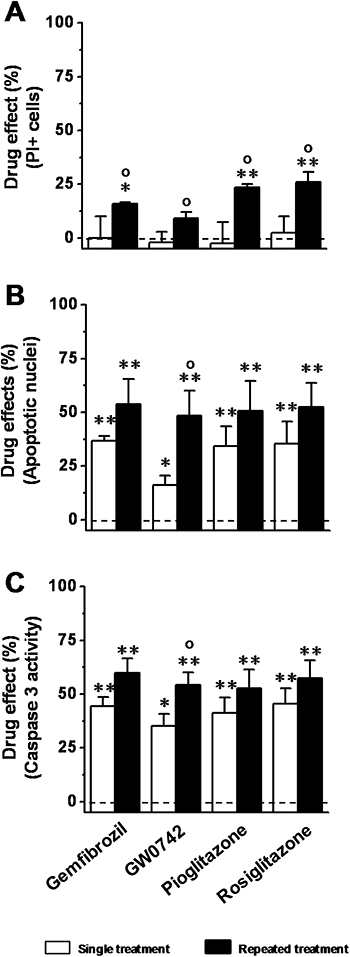
Effects of peroxisome proliferator-activated receptor (PPAR) agonists on OGD-reoxygenation-induced podocyte necrosis and apoptosis. Cells were treated with gemfibrozil (10 µM), GW0742 (0.1 µM), pioglitazone (1 µM) or rosiglitazone (0.1 µM) as a single or repeated treatment, and then exposed to OGD (5 h)-reoxygenation (24 h). (A) Percentage of PI+ cells at the end of 5 h-OGD. (B) Percentage of apoptotic nuclei at the end of 24 h-reoxygenation. (C) Caspase 3 activity at the end of 24 h-reoxygenation. Results are expressed as mean ± SEmean of four experiments run in triplicate. *P < 0.05; **P < 0.01 versus control (vehicle alone). °P < 0.05 versus single treatment. Broken line indicates cells treated with vehicle alone.
Effects of SD on podocyte death
In order to assess the anti-apoptotic effects of PPAR agonists, we evaluated the effects of gemfibrozil, GW0742, pioglitazone, and rosiglitazone on SD-induced podocyte death, which is known to be mainly due to apoptosis (Foster et al., 2005; Tejada et al., 2008). SD (24–72 h) induced significant cell loss at each time of assay (24, 48, and 72 h) (Figure 5A). The percentage of apoptotic nuclei (Figure 5B) and caspase 3 activity (Figure 5C) also increased over the 72 h of SD. In comparison with the basal level no significant changes in the evaluated parameters was measured in control cells. As the cell loss caused by 48 h of SD was similar to that measured in the OGD (5 h)-reoxygenation (24 h) experiments, and at this time point the SD-induced increase in the percentage of apoptotic nuclei and of caspase 3 activity achieved a constant value, we decided to perform the subsequent experiments using these experimental conditions.
Figure 5.
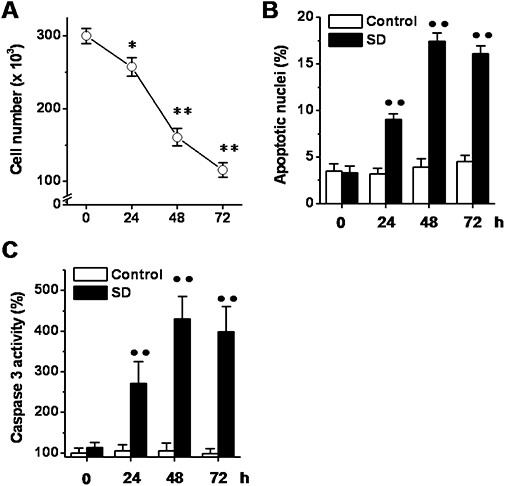
Effects of serum deprivation (SD) on podocyte death. Cells were exposed to SD (24–72 h), and at the indicated time points, the cell number (A), the percentage of apoptotic nuclei (B) and the caspase 3 activity (C) were determined. Results are expressed as mean ± SEmean of four experiments run in triplicate. *P < 0.05; **P < 0.01 versus basal level; ••P < 0.01 versus control.
Effects of PPAR agonists on SD-induced podocyte apoptosis
Cells were treated with increasing concentrations of gemfibrozil (1 nM–30 µM), GW0742 (0.01–300 nM), pioglitazone (0.3 nM–10 µM) or rosiglitazone (0.03 nM–3 µM) as a single (Protocol 3) or repeated treatment (Protocol 4), and were then exposed to SD. When added as a single treatment, all PPAR agonists decreased the SD-induced increase in the percentage of apoptotic nuclei in a concentration-dependent manner; the EC50s (Table 1) showed the following rank order: GW0742 < rosiglitazone < pioglitazone < gemfibrozil. Pioglitazone and rosiglitazone were the most effective (maximal effects were 83.0 ± 7.8% and 85.2 ± 4.4%, at 10 µM and 1 µM, respectively; Figure 6C,D), whereas gemfibrozil and GW0742 were partially effective (maximal effects were 75.2 ± 5.0% and 62.9 ± 5.2%, at 30 µM and at 0.1 nM, respectively; Figure 6A,B). When added as repeated treatment, all the drugs significantly prevented the SD-induced increase in the percentage of apoptotic nuclei. Moreover, repeated treatment caused a leftward shift of the concentration-response curves and the EC50s (Table 1) were decreased by >1 Log unit. Moreover, repeated treatment also increased the maximal effects to 85.1 ± 1.9%, 79.8 ± 3.0%, 98.5 ± 3.5%, and 99.8 ± 2.4%, for gemfibrozil (1 µM), GW0742 (0.1 µM), pioglitazone (0.1 µM), and rosiglitazone (0.1 µM) respectively (Figure 6A–D). Comparable results were obtained when cell loss or caspase 3 activity were measured (Table 1 and Supporting Information Figure S2).
Table 1.
EC50 values (M) of the selective PPAR agonists
| Decrease of apoptosis | Decrease of cell loss | Decrease of caspase 3 activation | ||||
|---|---|---|---|---|---|---|
| Agonist | Single treatment | Repeated treatment | Single treatment | Repeated treatment | Single treatment | Repeated treatment |
| Gemfibrozil | 3.6 ± 0.5 × 10−6 | 7.4 ± 0.1 × 10−8 | 3.5 ± 0.2 × 10−6 | 5.7 ± 1.2 × 10−8 | 4.4 ± 0.4 × 10−6 | 6.3 ± 0.7 × 10−8 |
| GW0742 | 1.1 ± 0.2 × 10−8 | 9.5 ± 1.4 × 10−10 | 7.5 ± 0.6 × 10−9 | 4.1 ± 0.8 × 10−10 | 1.6 ± 0.2 × 10−8 | 6.9 ± 0.1 × 10−10 |
| Pioglitazone | 2.4 ± 0.4 × 10−7 | 8.6 ± 1.0 × 10−9 | 3.0 ± 0.2 × 10−7 | 4.2 ± 0.4 × 10−9 | 1.9 ± 0.1 × 10−7 | 6.1 ± 0.1 × 10−9 |
| Rosiglitazone | 5.6 ± 1.2 × 10−8 | 1.7 ± 0.1 × 10−9 | 7.5 ± 0.7 × 10−8 | 1.1 ± 0.1 × 10−9 | 7.7 ± 0.8 × 10−8 | 2.7 ± 0.2 × 10−9 |
Figure 6.
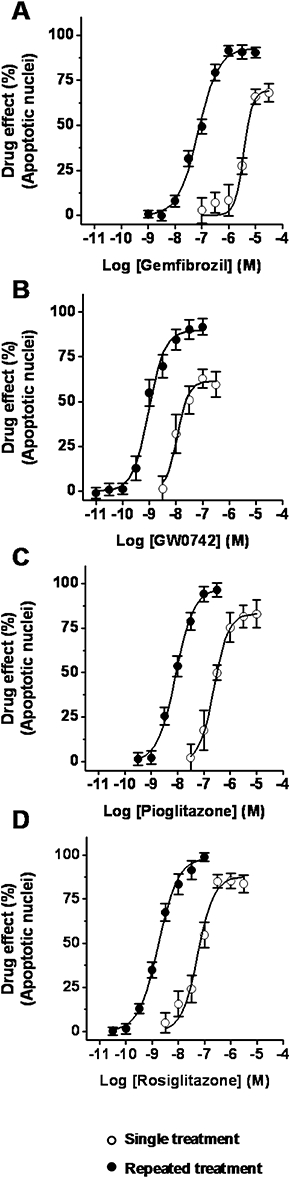
Effects of selective peroxisome proliferator-activated receptor (PPAR) agonists on serum deprivation (SD)-induced podocyte apoptosis. Cells were treated with either gemfibrozil (1 nM–30 µM, A), GW0742 (0.01 nM–300 nM, B), pioglitazone (0.3 nM–10 µM, C) or rosiglitazone (0.03nM–3 µM, D) as single or repeated treatment, and then exposed to SD (48 h). Results are expressed as mean ± SEmean of five experiments run in triplicate.
Effects of PPAR agonists on Bcl-2 and Bax expression
In comparison with the control, SD significantly decreased expression of the anti-apoptotic protein Bcl-2, whereas it increased that of the pro-apoptotic Bax. These changes caused a significant decrease in the Bcl-2-to-Bax ratio (Figure 7). Pioglitazone (1 µM), rosiglitazone (0.1 µM), and gemfibrozil (10 µM) restored Bcl-2 and Bax expression, and the Bcl-2-to-Bax ratio values approximated to that in control cells (Figure 7). GW0742 (0.1 µM) only partially reversed the effects of SD on Bcl-2-to-Bax ratio (Figure 7).
Figure 7.
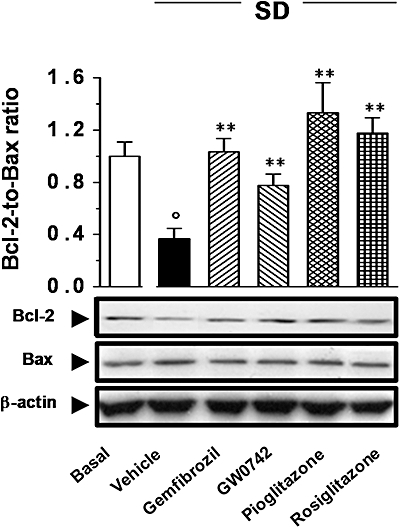
Effects of peroxisome proliferator-activated receptor (PPAR) agonists on the expression of Bcl-2 or Bax. Cells were treated with gemfibrozil (10 µM), GW0742 (0.1 µM), pioglitazone (1 µM) or rosiglitazone (0.1 µM) following Protocol 3 and exposed to serum deprivation (SD; 48 h). (A) Bcl-2 and Bax expression was determined by Western blot analysis. Results are expressed as mean ± SEmean of four experiments run in triplicate. **P < 0.01 versus vehicle alone; °P < 0.01 versus basal.
Effects of PPAR agonists on nephrin and synaptopodin expression
Finally, in order to further evaluate the protective effects of PPAR agonists on human podocytes, we determined whether gemfibrozil, GW0742, pioglitazone and rosiglitazone could affect the expression of nephrin and synaptopodin, which are functionally relevant podocyte proteins (Shankland, 2006; Wiggins, 2007). SD blunted nephrin gene (NPHS1) expression (by 88 ± 9%; P < 0.01 vs. basal; Figure 8A) and this effect was accompanied by a significant decrease in the protein level (83 ± 8%; P < 0.01; Figure 8B). Gemfibrozil (10 µM), GW0742 (0.1 µM), pioglitazone (1 µM) and rosiglitazone (0.1 µM) restored the nephrin expression, as measured at both the mRNA and protein levels. This result was confirmed by confocal microscopy. As shown in Figure 8C, the expression of nephrin was reduced by SD and restored by the PPAR agonists. SD generated a loss of the fine punctuate pattern of nephrin and several cells became negative. Under treatment with PPAR agonists, the intensity of nephrin expression was increased and displayed a coarse granular pattern. Neither SD nor PPAR agonists affected synaptopodin (Figure 8A–C).
Figure 8.
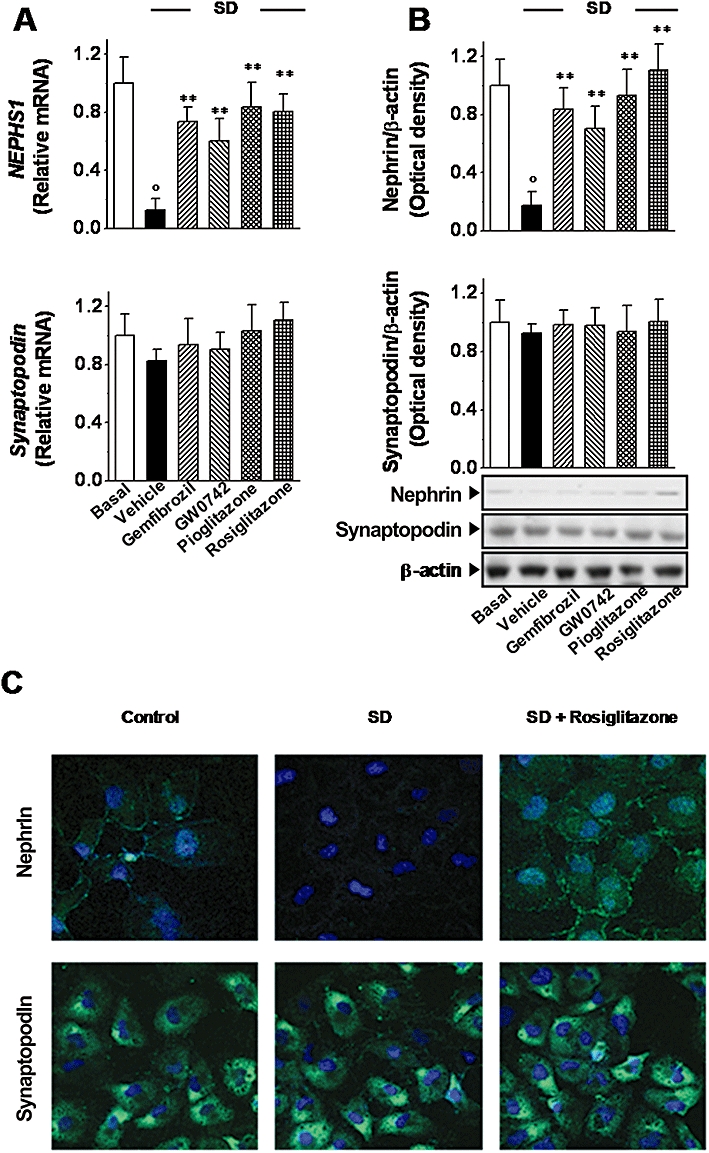
Effects of peroxisome proliferator-activated receptor (PPAR) agonists on the expression of nephrin and synaptopodin. Cells were treated with gemfibrozil (10 µM), GW0742 (0.1 µM), pioglitazone (1 µM) or rosiglitazone (0.1 µM) following Protocol 3 and exposed to serum deprivation (SD; 48 h). (A) Nephrin and synaptopodin expression was evaluated at both mRNA (A) and protein level (B) by real-time PCR (A), Western blot (B) respectively. (C) Representative micrographs of nephrin and synaptopodin expression detected by immunofluorescence in podocytes cultured with serum (control) exposed to serum deprivation (SD) and treated with rosiglitazone (original magnification: 630×). Results are expressed as mean ± SEmean of four experiments run in triplicate. **P < 0.01 versus vehicle alone; °P < 0.01 versus basal.
Discussion
The role of podocyte death in renal I/R injury remains to be elucidated. By using an OGD-reoxygenation model we have evaluated the contribution of specific stimuli (implicated in I/R-induced cell injury) to podocyte death. In our experiments, OGD-reoxygenation caused podocyte loss by triggering necrotic and apoptotic cell death. In addition, both GD and OD-reoxygenation contributed to the overall cell loss. In rats, renal I/R significantly affects podocytes, causing effacement (Wagner et al., 2008), cytoplasmic oedema, detachment (Lambert et al., 1986), and cell loss (Pippin et al., 2009). In addition, OGD, GD and OD (all without reoxygenation) have been reported to cause death of murine podocytes (Brukamp et al., 2007). Our results suggest that I/R may also cause podocyte death, which, together with the previously reported effects, may contribute to the glomerular injury associated with renal I/R.
PPAR activation has been reported to attenuate renal I/R injury, but the role of these transcription factors in glomerular cells has been largely unexplored to date. The expression of PPARs in podocytes has been previously reported both in vivo and in cultured cells (Ren et al., 2005; Yang et al., 2006; Kanjanabuch et al., 2007). Our results extend these findings showing that our cultures of human podocytes express all PPAR subtypes, which were transcriptionally active and induced by PPAR agonists.
By using two different models of podocyte injury, four structurally different PPAR agonists and two regimes of cell treatment, we showed that PPAR stimulation may decrease podocyte death. This experimental approach represented the mainstay of our studies and allowed us to compare the cyto-protective effects exerted by selective PPAR agonists. In our experiments, PPAR agonists decreased both OGD-reoxygenation- and SD-induced cell loss, mainly through an anti-apoptotic effect. The anti-apoptotic effects of PPAR agonists have been reported in many cell types exposed to different insults (Fuenzalida et al., 2007; Jung et al., 2007; Zanetti et al., 2008). Kanjanabuch et al. (2007) have shown that pioglitazone decreased puromycin-induced death of murine podocytes. Here we extend these findings showing that gemfibrozil, GW0742, pioglitazone and rosiglitazone decreased OGD-reoxygenation- or SD-induced death of human podocytes. Together, these results suggest that PPAR agonists may exert anti-apoptotic effects in different cell types, including podocytes of different origin and regardless of the nature of the initial insult. In addition, they indicated that, in human podocytes, the three PPAR subtypes might mediate equivalent anti-apoptotic effects, thus suggesting a possible redundancy between these transcription factors.
Through molecular mechanism involving both the direct activation of PPAR target genes and the interaction with other signalling pathways, such as those of the mitogen-activated protein kinases (MAPKs) and nuclear factor-κB (NF-κB), the PPAR subtypes mediate both distinct and similar actions (Berger and Moller, 2002). In our experiments, all PPAR agonists prevented the SD-induced podocyte apoptosis and these effects were accompanied by the preservation of the Bcl-2 and Bax levels, as well as by the attenuation of caspase 3 activation. Increased expression of the cell cycle inhibitor p27 and the anti-apoptotic protein Bcl-xL, and inhibition of caspase 3 activation have been previously postulated to mediate the anti-apoptotic effects exerted by pioglitazone in murine podocytes (Kanjanabuch et al., 2007). Moreover, PPARα and PPARγ agonists have been reported to modulate Bcl-2 and Bax expression in different cell types (Fuenzalida et al., 2007; Jung et al., 2007; Zanetti et al., 2008). All these findings suggest that multiple events might mediate the anti-apoptotic effects exerted by PPAR agonists. However, whether these events result from a cross-talk between PPAR and other signalling pathways and/or to binding to a PPAR response element (PPRE) in the promoter regions of PPAR target genes [as reported for the bcl-2 gene (Butts et al., 2004)] remain to be established.
Our studies reveal that the experimental cell treatment regime may influence both the nature and the magnitude of the drug effects. In our experiments all PPAR agonists significantly decreased apoptosis, but not necrosis, when added as a single treatment. Pioglitazone and rosiglitazone abolished the SD-induced apoptosis, while gemfibrozil and GW0742 were partially effective. Notably, the EC50s were similar to those reported in transactivation assays (Willson et al., 2000; Mukherjee et al., 2002; Sznaidman et al., 2003). All drugs decreased apoptotic cell loss and, albeit marginally, necrotic cell loss when added as repeated treatments. They abolished the SD-induced apoptosis, and in comparison with a single treatment, the EC50s were decreased by >1 Log unit. Taken together, these results indicate that: (i) PPAR subtypes expressed by human podocytes are pharmacologically similar to those used in transactivation assays; (ii) PPAR agonists decrease podocyte apoptosis in a receptor-dependent manner; (iii) repeated treatment may also decrease necrotic cell death; and (iv) repeated exposure sensitizes cells to the effects of the drug. The agonist-induced receptor up-regulation is a known physiological cell response to PPAR agonists and a regulatory mechanism of the PPAR activity. It is has been reported in many cell types (Fu et al., 2003; Kim et al., 2003; Miglio et al., 2009), including murine podocytes (Kanjanabuch et al., 2007). Our results suggest that such a mechanism exists in human podocytes and that our observation of the greater effects exerted by PPAR agonists after repeated cell treatment may be explained by an increased receptor abundance.
In our experiments all PPAR agonists prevented the SD-induced decrease nephrin down-regulation. Nephrin is a transmembrane protein located at the slit diaphragm complex. Nephrin down-regulation results in proteinuria and may cause podocyte effacement (Shankland, 2006; Wiggins, 2007). Thus, preservation of nephrin expression may be relevant for the maintenance of podocyte function. In this regard, we have recently shown that rosiglitazone prevents stretch-induced nephrin down-regulation in human podocytes (Miceli et al., 2010). Previous research has shown that PPARα and PPARγ agonists induce nephrin expression (Ren et al., 2005; Benigni et al., 2006). In addition, Benigni et al. (2006) have identified PPREs in the promoter region of the nephrin gene, thus indicating NPHS1 as a putative PPAR target gene and our results are consistent with these findings. Interestingly, it has been shown that nephrin actively exerts pro-survival effects in podocytes (Foster et al., 2005; Huber and Benzing, 2005). Therefore, as we have shown that preservation of nephrin expression accompanies the anti-apoptotic effects exerted by the PPAR agonists, we speculate that the preservation of nephrin expression may underlie the anti-apoptotic effects exerted by PPAR agonists
In conclusion, we have shown that podocyte death may be involved in the I/R-induced loss of glomerular integrity. Human podocytes express all PPAR subtypes, which are functionally responsive to selective agonists, pharmacologically similar to those used in transactivation assays and transcriptionally active. Activation of each PPAR subtype by selective agonist may exert similar protective effects by decreasing apoptotic cell death. Together these findings contribute to the characterization of renal PPARs and to the elucidation of the protective effects of PPAR agonists against I/R injury.
Acknowledgments
This work was funded by the Italian Ministry of Education, University, and Research (Programmi di ricerca scientifica di rilevanza nazionale, PRIN 2007), the Regione Piemonte (2008 and 2009) and the University of Turin.
Glossary
Abbreviations
- BBS
bicarbonate-buffered balanced salt solution
- DAPI
4′,6-diamidino-2-phenylindole
- DMEM
Dulbecco's modified Eagle's medium
- FBS
fetal bovine serum
- FDA
fluorescein diacetate
- GD
glucose deprivation
- GFB
glomerular filtration barrier
- I/R
ischemia/reperfusion
- OD
oxygen deprivation
- OGD
oxygen-glucose deprivation
- PBS
phosphate-buffered saline
- PI
propidium iodide
- PPARs
peroxisome proliferator-activated receptors
- PPRE
PPAR response element
- SD
serum deprivation
Conflicts of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 For the OGD-reoxygenation, single treatment (Protocol 1), cells were treated for 19 h with the PPAR agonists and then exposed to OGD-reoxygenation. For the OGD-reoxygenation, repeated treatment (Protocol 2) cells were treated for 72 h with the PPAR agonists and then exposed to OGD-reoxygenation. For the SD, single treatment (Protocol 3), cells were treated with the PPAR agonists simultaneously with SD. Finally, for the SD, repeated treatment (Protocol 4) cells were pretreated for 72 h with the PPAR agonists and then exposed to SD.
Figure S2 Effects of selective PPAR agonists on SD-induced podocyte loss and caspase 3 activation. Cells were treated with increasing concentration of either gemfibrozil (A and E), GW0742 (B and F), pioglitazone (C and G) or rosiglitazone (D and H) as single or repeated treatment, and then exposed to SD (48 h). Data are means ± SEM of five experiments run in triplicate. Broken line indicates cells treated with vehicle alone.
Table S1 Oligonucleotides and PCR conditions used in this study
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158:S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson M, Nilsson U, Hjalmarsson C, Haraldsson B, Nyström JS. Mild renal ischemia-reperfusion reduces charge and size selectivity of the glomerular barrier. Am J Physiol Renal Physiol. 2007;292:F1802–F1809. doi: 10.1152/ajprenal.00152.2006. [DOI] [PubMed] [Google Scholar]
- Aras MA, Hartnett KA, Aizenman E. Assessment of cell viability in primary neuronal cultures. Curr Protoc Neurosci. 2008;44:7.18.1–7.18.15. doi: 10.1002/0471142301.ns0718s44. [DOI] [PubMed] [Google Scholar]
- Benigni A, Zoja C, Tomasoni S, Campana M, Corna D, Zanchi C, et al. Transcriptional regulation of nephrin gene by peroxisome proliferator-activated receptor-gamma agonist: molecular mechanism of the antiproteinuric effect of pioglitazone. J Am Soc Nephrol. 2006;17:1624–1632. doi: 10.1681/ASN.2005090983. [DOI] [PubMed] [Google Scholar]
- Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- Boulanger H, Mansouri R, Gautier JF, Glotz D. Are peroxisome proliferator-activated receptors new therapeutic targets in diabetic and non-diabetic nephropathies? Nephrol Dial Transplant. 2006;21:2696–2702. doi: 10.1093/ndt/gfl448. [DOI] [PubMed] [Google Scholar]
- Brukamp K, Jim B, Moeller MJ, Haase VH. Hypoxia and podocyte-specific Vhlh deletion confer risk of glomerular disease. Am J Physiol Renal Physiol. 2007;293:F1397–F1407. doi: 10.1152/ajprenal.00133.2007. [DOI] [PubMed] [Google Scholar]
- Butts BD, Tran NL, Briehl MM. Identification of a functional peroxisome proliferator activated receptor response element in the 3′ untranslated region of the human bcl-2 gene. Int J Oncol. 2004;24:1305–1310. [PubMed] [Google Scholar]
- Chatterjee PK. Novel pharmacological approaches to the treatment of renal ischemia-reperfusion injury: a comprehensive review. Naunyn Schmiedebergs Arch Pharmacol. 2007;376:1–43. doi: 10.1007/s00210-007-0183-5. [DOI] [PubMed] [Google Scholar]
- Collino M, Patel NS, Lawrence KM, Collin M, Latchman DS, Yaqoob MM, et al. The selective PPARγ antagonist GW9662 reverses the protection of LPS in a model of renal ischemia-reperfusion. Kidney Int. 2005;68:529–536. doi: 10.1111/j.1523-1755.2005.00430.x. [DOI] [PubMed] [Google Scholar]
- Collino M, Patel NS, Thiemermann C. PPARs as new therapeutic targets for the treatment of cerebral ischemia/reperfusion injury. Ther Adv Cardiovasc Dis. 2008;2:179–197. doi: 10.1177/1753944708090924. [DOI] [PubMed] [Google Scholar]
- Conaldi PG, Biancone L, Bottelli A, De Martino A, Camussi G, Toniolo A. Distinct pathogenic effects of group B coxsackieviruses on human glomerular and tubular kidney cells. J Virol. 1997;71:9180–9187. doi: 10.1128/jvi.71.12.9180-9187.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Paola R, Cuzzocrea S. Peroxisome proliferator-activated receptors ligands and ischemia-reperfusion injury. Naunyn Schmiedebergs Arch Pharmacol. 2007;375:157–175. doi: 10.1007/s00210-007-0141-2. [DOI] [PubMed] [Google Scholar]
- Doublier S, Ruotsalainen V, Salvidio G, Lupia E, Biancone L, Conaldi PG, et al. Nephrin redistribution on podocytes is a potential mechanism for proteinuria in patients with primary acquired nephritic syndrome. Am J Pathol. 2001;158:1723–1731. doi: 10.1016/S0002-9440(10)64128-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster RR, Saleem MA, Mathieson PW, Bates DO, Harper SJ. Vascular endothelial growth factor and nephrin interact and reduce apoptosis in human podocytes. Am J Physiol Renal Physiol. 2005;288:F48–F57. doi: 10.1152/ajprenal.00146.2004. [DOI] [PubMed] [Google Scholar]
- Fu J, Gaetani S, Oveisi F, Lo Verme J, Serrano A, Rodríguez De Fonseca F, et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-alpha. Nature. 2003;425:90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- Fuenzalida K, Quintanilla R, Ramos P, Piderit D, Fuentealba RA, Martinez G, et al. Peroxisome proliferator-activated receptor gamma up-regulates the Bcl-2 anti-apoptotic protein in neurons and induces mitochondrial stabilization and protection against oxidative stress and apoptosis. J Biol Chem. 2007;282:37006–37015. doi: 10.1074/jbc.M700447200. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, et al. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ. 2009;16:1093–1107. doi: 10.1038/cdd.2009.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondares E, Mora O, Yubero P, Rodriguez de la Concepción M, Iglesias R, Giralt M, et al. Thiazolidinediones and rexinoids induce peroxisome proliferator-activated receptor-coactivator (PGC)-1alpha gene transcription: an autoregulatory loop controls PGC-1alpha expression in adipocytes via peroxisome proliferator-activated receptor-gamma coactivation. Endocrinology. 2006;147:2829–2838. doi: 10.1210/en.2006-0070. [DOI] [PubMed] [Google Scholar]
- Huber TB, Benzing T. The slit diaphragm: a signalling platform to regulate podocyte function. Curr Opin Nephrol Hypertens. 2005;14:211–216. doi: 10.1097/01.mnh.0000165885.85803.a8. [DOI] [PubMed] [Google Scholar]
- Jung TW, Lee JY, Shim WS, Kang ES, Kim SK, Ahn CW, et al. Rosiglitazone protects human neuroblastoma SH-SY5Y cells against MPP+ induced cytotoxicity via inhibition of mitochondrial dysfunction and ROS production. J Neurol Sci. 2007;253:53–60. doi: 10.1016/j.jns.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Kanjanabuch T, Ma LJ, Chen J, Pozzi A, Guan Y, Mundel P. PPAR-γ agonist protects podocytes from injury. Kidney Int. 2007;71:1232–1239. doi: 10.1038/sj.ki.5002248. [DOI] [PubMed] [Google Scholar]
- Kim EJ, Park KS, Chung SY, Sheen YY, Moon DC, Song YS, et al. Peroxisome proliferator-activated receptor-γ activator 15-deoxy-Delta12,14-prostaglandin J2 inhibits neuroblastoma cell growth through induction of apoptosis: association with extracellular signal-regulated kinase signal pathway. J Pharmacol Exp Ther. 2003;307:505–517. doi: 10.1124/jpet.103.053876. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert R, Henry M, Howden B, Jablonski P, Rae D, Tavanlis G, et al. Glomerular damage after kidney preservation. Transplantation. 1986;42:125–130. doi: 10.1097/00007890-198608000-00004. [DOI] [PubMed] [Google Scholar]
- Letavernier E, Perez J, Joye E, Bellocq A, Fouqueray B, Haymann JP, et al. Peroxisome proliferator-activated receptor β/δ exerts a strong protection from ischemic acute renal failure. J Am Soc Nephrol. 2005;16:2395–2402. doi: 10.1681/ASN.2004090802. [DOI] [PubMed] [Google Scholar]
- Miceli I, Burt D, Tarabra E, Camussi G, Perin PC, Gruden G. Stretch reduces nephrin expression via an Angiotensin II-AT1-dependent mechanism in human podocytes: effect of rosiglitazone. Am J Physiol Renal Physiol. 2010;298:F381–F390. doi: 10.1152/ajprenal.90423.2008. [DOI] [PubMed] [Google Scholar]
- Miglio G, Rosa AC, Rattazzi L, Collino M, Lombardi G, Fantozzi R. PPARγ stimulation promotes mitochondrial biogenesis and prevents glucose deprivation-induced neuronal cell loss. Neurochem Int. 2009;55:496–504. doi: 10.1016/j.neuint.2009.05.001. [DOI] [PubMed] [Google Scholar]
- Miglio G, Varsaldi F, Francioli E, Battaglia A, Canonico PL, Lombardi G. Cabergoline protects SH-SY5Y neuronal cells in an in vitro model of ischemia. Eur J Pharmacol. 2004;489:157–165. doi: 10.1016/j.ejphar.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Molitoris BA, Sutton TA. Endothelial injury and dysfunction: role in the extension phase of acute renal failure. Kidney Int. 2004;66:496–499. doi: 10.1111/j.1523-1755.2004.761_5.x. [DOI] [PubMed] [Google Scholar]
- Mukherjee R, Sun S, Santomenna L, Miao B, Walton H, Liao B. Ligand and coactivator recruitment preferences of peroxisome proliferator activated receptor. J Steroid Biochem Mol Biol. 2002;81:217–225. doi: 10.1016/s0960-0760(02)00066-3. [DOI] [PubMed] [Google Scholar]
- Nankivell BJ, Borrows RJ, Fung CL, O'Connell PJ, Allen RD, Chapman JR. The natural history of chronic allograft nephropathy. N Engl J Med. 2003;349:2326–2333. doi: 10.1056/NEJMoa020009. [DOI] [PubMed] [Google Scholar]
- Nankivell BJ, Borrows RJ, Fung CL, O'Connell PJ, Allen RD, Chapman JR. Evolution and pathophysiology of renal-transplant glomerulosclerosis. Transplantation. 2004;78:461–468. doi: 10.1097/01.tp.0000128612.75163.26. [DOI] [PubMed] [Google Scholar]
- Pippin J, Kumar V, Stein A, Jablonski P, Shankland SJ, Davis CL. The contribution of podocytes to chronic allograft nephropathy. Nephron Exp Nephrol. 2009;111:e1–e10. doi: 10.1159/000178762. [DOI] [PubMed] [Google Scholar]
- Portilla D, Dai G, Peters JM, Gonzalez FJ, Crew MD, Proia AD. Etomoxir-induced PPARα-modulated enzymes protect during acute renal failure. Am J Physiol Renal Physiol. 2000;278:F667–F675. doi: 10.1152/ajprenal.2000.278.4.F667. [DOI] [PubMed] [Google Scholar]
- Ren S, Xin C, Beck KF, Saleem MA, Mathieson P, Pavenstädt H, et al. PPARα activation upregulates nephrin expression in human embryonic kidney epithelial cells and podocytes by a dual mechanism. Biochem Biophys Res Commun. 2005;338:1818–1824. doi: 10.1016/j.bbrc.2005.10.158. [DOI] [PubMed] [Google Scholar]
- Rippe C, Rippe A, Larsson A, Asgeirsson D, Rippe B. Nature of glomerular capillary permeability changes following acute renal ischemia-reperfusion injury in rats. Am J Physiol Renal Physiol. 2006;291:F1362–F1368. doi: 10.1152/ajprenal.00123.2006. [DOI] [PubMed] [Google Scholar]
- Ruan X, Zheng F, Guan Y. PPARs and the kidney in metabolic syndrome. Am J Physiol Renal Physiol. 2008;294:F1032–F1047. doi: 10.1152/ajprenal.00152.2007. [DOI] [PubMed] [Google Scholar]
- Russ AL, Haberstroh KM, Rundell AE. Experimental strategies to improve in vitro models of renal ischemia. Exp Mol Pathol. 2007;83:143–159. doi: 10.1016/j.yexmp.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Shankland SJ. The podocyte's response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 2006;69:2131–2147. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- Sivarajah A, Chatterjee PK, Hattori Y, Brown PA, Stewart KN, Todorovic Z, et al. Agonists of peroxisome-proliferator activated receptor-alpha (clofibrate and WY14643) reduce renal ischemia/reperfusion injury in the rat. Med Sci Monit. 2002;8:BR532–BR539. [PubMed] [Google Scholar]
- Sivarajah A, Chatterjee PK, Patel NS, Todorovic Z, Hattori Y, Brown PA, et al. Agonists of peroxisome-proliferator activated receptor-gamma reduce renal ischemia/reperfusion injury. Am J Nephrol. 2003;23:267–276. doi: 10.1159/000072088. [DOI] [PubMed] [Google Scholar]
- Stefanidis I, Heintz B, Stöcker G, Mrowka C, Sieberth HG, Haubeck HD. Association between heparan sulfate proteoglycan excretion and proteinuria after renal transplantation. J Am Soc Nephrol. 1996;7:2670–2676. doi: 10.1681/ASN.V7122670. [DOI] [PubMed] [Google Scholar]
- Sznaidman ML, Haffner CD, Maloney PR, Fivush A, Chao E, Goreham D, et al. Novel selective small molecule agonists for peroxisome proliferator-activated receptor δ (PPARδ)-synthesis and biological activity. Bioorg Med Chem Lett. 2003;13:1517–1521. doi: 10.1016/s0960-894x(03)00207-5. [DOI] [PubMed] [Google Scholar]
- Tejada T, Catanuto P, Ijaz A, Santos JV, Xia X, Sanchez P, et al. Failure to phosphorylate AKT in podocytes from mice with early diabetic nephropathy promotes cell death. Kidney Int. 2008;73:1385–1393. doi: 10.1038/ki.2008.109. [DOI] [PubMed] [Google Scholar]
- Villarroya F, Iglesias R, Giralt M. PPARs in the control of uncoupling proteins gene expression. PPAR Res. 2007;2007:74364. doi: 10.1155/2007/74364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner MC, Rhodes G, Wang E, Pruthi V, Arif E, Saleem MA, et al. Ischemic injury to kidney induces glomerular podocyte effacement and dissociation of slit diaphragm proteins Neph1 and ZO-1. J Biol Chem. 2008;283:35579–35589. doi: 10.1074/jbc.M805507200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 2007;71:1205–1214. doi: 10.1038/sj.ki.5002222. [DOI] [PubMed] [Google Scholar]
- Willson TM, Brown PJ, Sternbach DD, Henke BR. The PPARs: from orphan receptors to drug discovery. J Med Chem. 2000;43:527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- Yang HC, Ma LJ, Ma J, Fogo AB. Peroxisome proliferator-activated receptor-γ agonist is protective in podocyte injury-associated sclerosis. Kidney Int. 2006;69:1756–1764. doi: 10.1038/sj.ki.5000336. [DOI] [PubMed] [Google Scholar]
- Zanetti M, Stocca A, Dapas B, Farra R, Uxa L, Bosutti A, et al. Inhibitory effects of fenofibrate on apoptosis and cell proliferation in human endothelial cells in high glucose. J Mol Med. 2008;86:185–195. doi: 10.1007/s00109-007-0257-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.