

Abstract
BACKGROUND AND PURPOSE
Introducing the calcineurin inhibitors cyclosporin (CsA) and tacrolimus (Tac) has improved the outcome of organ transplants, but complications such as new onset diabetes mellitus after transplantation (NODAT) decrease survival rates.
EXPERIMENTAL APPROACH
We sought, in a beta-cell culture model, to elucidate the pathogenic mechanisms behind NODAT and the relative contribution of the calcineurin inhibitors. INS-1E cells were incubated at basal and stimulatory glucose concentrations, while exposed to pharmacologically relevant doses of CsA, Tac and vehicle for 6 or 24 h.
RESULTS
Tac inhibited basal (P < 0.05), but not glucose-stimulated insulin secretion (GSIS) after 6 h of exposure. After 24 h, both agents inhibited basal and GSIS (P < 0.05). Calcineurin phosphatase activity was decreased by both drugs during all conditions. Apoptosis was only seen with CsA treatment, which also induced a slight suppression of calcineurin and insulin mRNA, as well as increased levels of the sterol receptor element binding protein (SREBP)-1c, a transcription factor thought to suppress genes essential for beta-cell function and induce insulin resistance. Expression levels of nuclear factor of activated T-cells (NFAT)-c1, -c2, -c3 and -c4 were not decreased notably by either drug.
CONCLUSIONS AND IMPLICATIONS
Tac had acute inhibitory effects on basal insulin secretion, but prolonged exposure (24 h) to Tac or CsA revealed similar suppression of insulin secretion. These prolonged effects were mirrored by a total inhibition of calcineurin activity in beta-cells. CsA showed greater inhibition of beta-cell survival and transcriptional markers, essential for beta-cell function.
Keywords: beta-cells, calcineurin, cyclosporin A, insulin, tacrolimus, NFAT, SREBP diabetes transplantation
Introduction
Introducing the calcineurin inhibitors, cyclosporin (CsA) and tacrolimus (Tac), into the field of transplantation has improved the outcome of organ transplants; however, their widespread therapeutic use is restricted by a number of side effects shared by both drugs. Among these, the condition of new onset diabetes mellitus after transplantation (NODAT) is particularly important because of its frequent occurrence (Heisel et al., 2004; Vincenti et al., 2007) and its associations with increased risk of cardiovascular diseases as well as impaired survival rates (Cosio et al., 2002; 2005;). Previous in vitro studies on purified islets and insulin-producing beta-cells have proposed several diabetogenic actions of CsA and Tac. Both drugs have been shown to impair insulin secretion (Nielsen et al., 1986; Robertson, 1986; Paty et al., 2002; Polastri et al., 2002), decrease insulin content of the beta-cell (Redmon et al., 1996; Uchizono et al., 2004) and impair insulin transcription (Lawrence et al., 2001; 2002; Oetjen et al., 2003), although the primary mechanisms remain unexplained.
In insulin-secreting cells, calcineurin is involved in the stimulation of insulin gene transcription through the activation of the transcription factor nuclear factor of activated T-cells (NFAT; Lawrence et al., 2002). Indeed, mice deficient in calcineurin B1 develop diabetes mellitus during aging due to insufficient insulin production, while transgenic expression of constitutively active NFAT protects against diabetes mellitus (Heit et al., 2006). Other studies have suggested that calcineurin may support anti- as well as pro-apoptotic events in the cell (Drachenberg et al., 1999; Lotem et al., 1999; Lawrence et al., 2005; Ranta et al., 2008). Furthermore, opposing results of transient versus sustained calcineurin inhibition in the beta-cell have also been reported (Ebihara et al., 1996; Lester et al., 2001) and, collectively, these studies point out that calcineurin controls a number of beta-cell functions subjected to tight regulation. Further to these preclinical findings, clinical research has yielded similar conflicting results regarding the impact of calcineurin inhibitors on beta-cell function and glucose metabolism. Hence, both insulin resistance (Ekstrand et al., 1992; Midtvedt et al., 2004) and impaired insulin secretion (Ekstrand et al., 1992; Duijnhoven et al., 2001; Hjelmesaeth et al., 2007) have been put forward as elements in the pathogenesis of NODAT, which is more frequently related to treatment with Tac than with CsA (Heisel et al., 2004; Vincenti et al., 2007). Nevertheless, the degree and comparability of the calcineurin inhibitors in impairing beta-cell function is yet to be established. Furthermore, the underlying molecular mechanisms of their diabetogenic actions are still unresolved.
The present study was designed to elucidate and compare the direct effects of calcineurin inhibition by CsA and Tac in insulin-secreting cells. In addition, we sought to discover whether calcineurin signalling controls the expression of several essential beta-cell factors. To this end, beta-cells were exposed to varying concentrations of CsA and Tac, at basal and stimulatory glucose concentrations for 6 and 24 h. Insulin secretion, insulin content and mRNA were measured along with calcineurin phosphatase (CaN) activity and calcineurin mRNA, expression levels NFAT-c1–c4 and the lipogenic transcription factor sterol receptor element binding protein (SREBP)-1c as well as apoptosis-related markers.
Methods
INS-1E cells
INS-1E cells were cultured in a 5% CO2 atmosphere in complete RPMI 1640 supplemented with 11 mmol·L−1 glucose, 10% (v/v) heat-inactivated fetal bovine serum, 50 µmol·L−1β-mercaptoethanol, 2 mmol·L−1 L-glutamine, 100 U·mL−1 penicillin and 100 mg·mL−1 streptomycin. For insulin assays, cells were plated (3.0 × 105 cells per well) into 24-well plates (NUNC, Roskilde, Denmark) at 11 mmol·L−1 glucose and cultured for 3 days. For PCR and calcineurin activity assays, cells were plated (1.4 × 106 cells/well and 15 × 106 cells per well, respectively) into 6-well plates. Then the cells were cultured at basal (3.3 and 6.6 mmol·L−1) and stimulatory (16.7 mmol·L−1) glucose concentrations with varying drug solutions: CsA (0.10 and 2.5–10 µmol·L−1, corresponding to 120 and 3000–12.000 µg·L−1) and Tac (0.01 and 0.05–0.2 µmol·L−1, corresponding to 8 and 40–160 µg·L−1) and vehicle for 6 and 24 h.
Insulin measurements
INS-1E cells were incubated for 2 h in a Krebs-Ringer bicarbonate HEPES buffer (at pH 7.4) containing 115 mmol·L−1 NaCl, 4.7 mmol·L−1 KCl, 1.2 mmol·L−1 MgSO4, 2.6 mmol·L−1 CaCl2, 1.2 mmol·L−1 KH2PO4, 20 mmol·L−1 HEPES, 5 mmol·L−1 NaHCO3, 0.1% (v/v) human serum albumin (Sigma, Brøndby, Denmark) for insulin release determination. The incubation medium was collected for insulin analysis and insulin release was measured. Afterwards, the cells were washed in Earle's basal medium (Invitrogen, Taastrup, Denmark) at room temperature before adding 1 mL ice-cold Earle's basal medium in which the cells were scraped off with a rubber policeman. After centrifugation, the medium was discarded and the intact cells re-suspended partly in a buffer containing 0.75% (v/v) glycine and 0.25% (v/v) BSA adjusted to pH 8.8 for insulin determination after sonication and centrifugation at 30 000×g for 30 min at 4°C, and partly in 0.1 mol·L−1 NaOH for protein determination. Total protein was determined using BCA Protein Assay Reagent Kit from PIERCE, US (Bie & Berntsen A/S, Rødovre, Denmark). Samples of the incubation medium were immediately frozen for insulin analysis. Insulin content was determined using an ultrasentive Rat Insulin Elisa Kit from DRG Diagnostics (VWR, Herlev, Denmark). Results are expressed as insulin secretion µg·mg−1 protein normalised to insulin content µg·mg−1 protein.
CaN activity
Lysed INS-1E cells were used to determine CaN activity. The activity of the enzyme was measured as described by Fruman et al. (1992), with modifications made by Koefoed-Nielsen et al. (2004). Briefly, CaN activity was measured as the release of radiolabelled phosphate from a previously phosphorylated 19 amino acid peptide, which mimics NFAT, the natural substrate of CaN. Radioactivity was quantitated by liquid scintillation spectrometry, in counts per minute, and results are expressed as units CaN/107 beta-cells, utilizing a calibration curve made with bovine CaN.
Quantification of mRNA by real-time PCR
INS-1E RNA was extracted using Qiagen RNeasy Mini Kit (VWR) and treated with DNase (VWR). The RNA quality was controlled on a 1% (v/v) agarose gel stained with ethidium bromide. Total RNA (500 ng) was reversely transcribed using ImProm-II™ Reverse Transcription System (Promega, Nacka, Sweden) and oligo dT18 primers (TAC, Copenhagen, Denmark). The cDNA was checked for genomic DNA contamination by PCR analysis using Qiagen HotStarTaq Master Mix Kit (VWR) with an intron-spanning primer-set of β-actin (TAC). The PCR product was analysed by ethidiumbromide staining after electrophoresis in a 1% agarose gel. Quantitative real-time PCR was performed in duplicate with IQ Sybr Green supermix (Bio-Rad, Copenhagen, Denmark) in a MyiQ SingleColor Real-Time PCR Detection System (Bio-Rad). For all reactions, a melting curve was included. The results were analysed with iQ™5 Optical System Software, Version 2.0 (Copenhagen, Denmark). Starting quantities were calculated from a standard curve. Values were normalized to the geomean of three housekeeping genes.
PCR primers used for real-time PCR: heat shock protein (HSPcb): F: 5′GAT TGA CAT CAT CCC CAA CC 3′, R:5′CTG CTC ATC ATC GTT GTG CT 3′; Clathrin (Cltc): F: 5′AAG GAG GCG AAA CTC ACA GA 3′, R:5′GAG CAG TCA ACA TCC AGC AA 3′; Insulin (INS): F: 5′CGC TTC CTG CCC CTG CTG GC 3′, R: 5′CGG GCC TCC ACC CAG CTC CA 3′; Bax F: 5′GTG AGC GGC TGC TTG TCT 3′, R: 5′GTG GGG GTC CCG AAG TAG 3′; Bcl-2 F: 5′CGA CTT TGC AGA GAT GTC CA 3′, R: 5′ATG CCG GTC AGG TAC TCA G 3′; Calcineurin A beta (CaN): F: 5′ACA GGG ATG TTG CCT AGT GG 3′, R: 5′TCA GTG GTA TGT GCG GTG TT 3′;NFATc1: F: 5′ TGA GCA GTA TCT GTC GGT GC 3′, R: 5′ CCT GTG GTG AGA CTT GGG TT 3′; NFATc2: F: 5′GTG TTT GCT AGC TTC TGG GC3′, R: 5′ACA TTG TGC TCA ACA GGC AG 3′; NFATc3: F: 5′ TCC CAC TTG AAC CGT CCT AC 3′, R: 5′AGA GCC ACC TGG AGA AGT CA 3′; NFATc4: F: 5′AGA TGG CTG GCA TGG ATT AC 3′, R: 5′AGCCTA GGA GCT TGA CCA CA 3′; SREBF-1c: F: 5′ATG GGC AAG TAC ACA GGA GG 3′, R: 5′ATC CAC GAA GAA ACG GTG AC 3′. The intron-spanning primers of β-actin: F: 5′CTA CAA TGA GCT GCG TGT GGC 3′, R: 5′ GTC CAG ACG CAG GAT GGC ATG 3′. cDNA gives a band of 269 base pairs, and genomic DNA gives a band of 732 base pairs. All PCR sequences were confirmed by sequencing with Thermo Sequenase™ Dye Terminator Cycle Sequencing Premix Kit (Amersham Bioscience, Brøndby, Denmark).
Cell death elisa assay
Fragmentation of histone-associated-DNA after cell death induced by glucose, CsA or Tac was determined by photometric enzyme immunoassay (Cell Death Detection ELISAPLUS, Roche Applied Science, Hvidovre, Denmark). INS-1E cells were plated on 24-well plate with 200 000 cells per well and stimulated 36 h later either with 6.6 mM or 16.7 mM glucose ± 2.5 µmol·L−1 CsA or 0.5 µmol·L−1 Tac for 24 h. Experiments were performed in quadruplicate. Cells were scraped off with a rubber policeman and centrifuged at 200×g for 10 min. Supernatants containing DNA from necrotic cells were removed and stored at 4°C for further analysis. Cell pellets containing DNA fragments were lysed and centrifuged at 200×g for 10 min. The supernatant containing the cytoplasmic fraction and the supernatant containing the DNA from necrotic cells were transferred into streptavidin-coated microtiter plate in duplicate and incubated with anti-histone-biotin. The amount of fragmented DNA bound to anti-DNA-peroxidase was measured by ABTS (2,2′-azino-bis(3-ethylbenzthiazoline-6-sulphonic acid) at 405 and 490 nm as reference wavelength.
Data analysis
Statistical analyses were conducted using STATA 10.0 software (College Station, TX, USA). Results in the Figures and Tables are presented as medians with range as data were non-normally distributed. Comparisons between groups in insulin secretion studies, CaN activity measurements, RT-PCR experiments and DNA fragmentation analyses were made by Kruskall–Wallis and Mann–Whitney U-tests. P-value < 0.05 was considered statistically significant.
Materials
CsA and Tac were supplied by Sigma; the INS-1E cells were provided by Prof CB Wollheim, Geneva, Switzerland; the CaN substrate peptide was from Schäfer-N (Copenhagen, Denmark) and the bovine CaN standard from Biomol (Hamburg, Germany). Drug and molecular target nomenclature follows Alexander et al. (2009).
Results
Effects of calcineurin inhibition on insulin release and insulin mRNA
We cultured INS-1E cells for 6 and 24 h at drug concentrations ranging from therapeutic blood levels to toxic amounts in order to study the effects on basal and glucose-stimulated insulin secretion (GSIS). Tac had acute (6 h incubation) inhibitory effects on basal insulin output, which was decreased by 61–87% (median values; P= 0.01), over the concentration range. Neither of the drugs showed any short-term inhibitory effects on GSIS (Figure 1A). After prolonged incubation (24 h), Tac suppressed basal insulin release by 48–62% (P= 0.03) as well as GSIS by 70–88% (P= 0.01). Long-term inhibitory effects were also observed with CsA exposure, which decreased basal insulin secretion by 58–69% (P= 0.01) and GSIS by 19–74% (P= 0.01) (Figure 1B). A separate study of long-term effects with lower doses of Tac (10 nmol·L−1) and CsA (100 nmol·L−1) revealed that neither basal insulin release nor GSIS was inhibited, which suggests that the inhibitory capacities of the drugs are dose-dependent (Figure 1C). Both drugs mainly affected the secreted amount of insulin while the insulin content of the beta-cells remained unaltered. Insulin mRNA levels were only marginally decreased with 24 h of CsA exposure in glucose-stimulated cells, otherwise insulin mRNA levels were comparable between cells exposed to calcineurin inhibitors and controls (Figure 2A,B).
Figure 1.
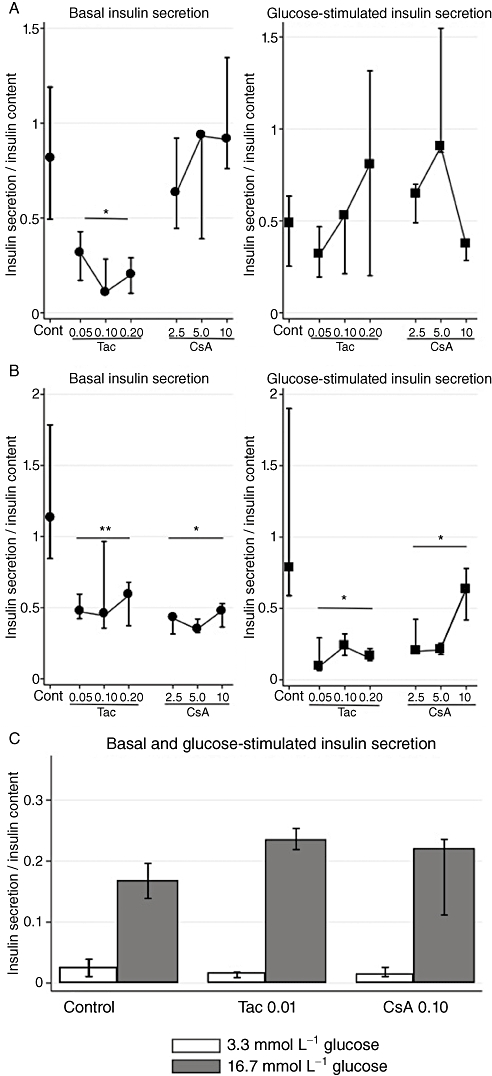
Effects of tacrolimus (Tac; µmol·L−1) and cyclosporin (CsA; µmol·L−1) on insulin secretion normalized to insulin content in INS-1E cells. (A) Six hours of incubation at basal (6.6 mmol·L−1) and stimulatory (16.7 mmol·L−1) glucose concentrations. Data are medians with range. n= 6 for controls and n= 3 for each drug concentration. (B) Twenty-four hours of incubation at basal (6.6 mmol·L−1) and stimulatory (16.7 mmol·L−1) glucose concentrations. Data are medians with range. n= 6 for controls and n= 3 for each drug concentration. (C) Twenty-four hours of incubation at basal (3.3 mmol·L−1) and stimulatory (16.7 mmol·L−1) glucose concentrations. Data are medians with range. n= 6 for controls and n= 4 for each drug concentration. *P= 0.01, **P= 0.03 versus control, Kruskal–Wallis test.
Figure 2.
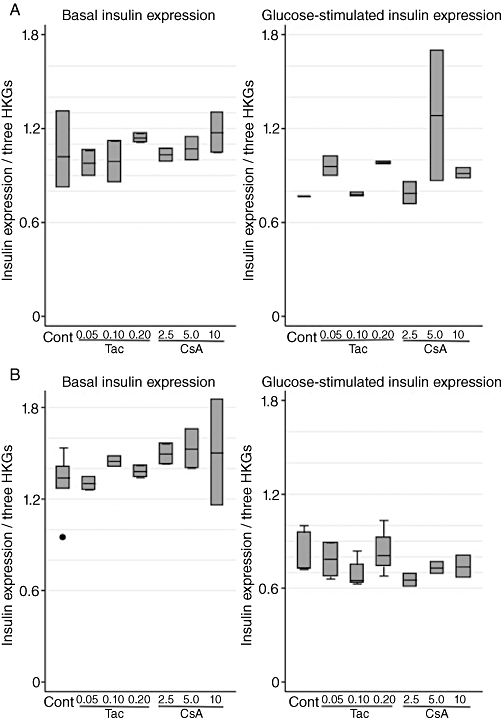
Effects of tacrolimus (Tac; µmol·L−1) and cyclosporin (CsA; µmol·L−1) on relative gene expression of insulin (normalized to β-actin, clathrin and HSP) in INS-1E cells. (A) Six hours of incubation at basal (6.6 mmol·L−1) and stimulatory (16.7 mmol·L−1) glucose concentrations. Data are medians with range. n= 3 for controls and n= 2 for each drug concentration. (B) Twenty-four hours of incubation at basal (6.6 mmol·L−1) and stimulatory (16.7 mmol·L−1) glucose concentrations. Data are medians with range. n= 3 for controls and n= 2 for each drug concentration. HKG, housekeeping gene.
Does calcineurin regulate insulin release?
After studying the effects induced by calcineurin inhibitors on insulin secretion, we addressed the involvement of calcineurin in regulating this pathway, by measuring CaN activity, as well as calcineurin mRNA levels. High glucose concentrations (16.7 mmol·L−1) in controls stimulated CaN activity approximately 20% during short-term incubations (6 h). This glucose stimulatory effect was, however, not seen after 24 h of incubation. Tac and CsA equally suppressed CaN activity completely after 6 and 24 h (Figure 3A,B), ultimately matching the similarly inhibited insulin release after 24 h, yet not explaining the partially maintained insulin output after 6 h of exposure. Calcineurin mRNA levels were decreased after prolonged CsA exposure (P= 0.06) in glucose stimulated cells, but were unaffected during all other conditions (Table 1A–D).
Figure 3.
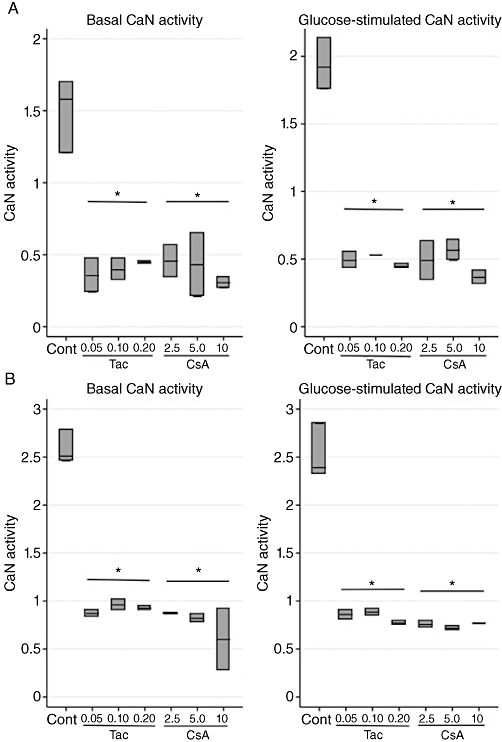
Effects of tacrolimus (Tac; µmol·L−1) and cyclosporin (CsA; µmol·L−1) on calcineurin phosphatase (CaN) activity (units/107 cells) in INS-1E cells. (A) Six hours of incubation at basal (6.6 mmol·L−1) and stimulatory (16.7 mmol·L−1) glucose concentrations. Data are medians with range. n= 3 for controls and n= 2 for each drug concentration. (B) Twenty-four hours of incubation at basal (6.6 mmol·L−1) and stimulatory (16.7 mmol·L−1) glucose concentrations. Data are medians with range. n= 3 for controls and n= 2 for each drug concentration. *P= 0.02 versus control, Kruskal–Wallis test.
Table 1.
Effects of tacrolimus and cyclosporin on relative gene expression levels (normalized to the housekeeping genes β-actin, clathrin and HSP) in INS-1E cells
| Tacrolimus (µmol·L−1) | Cyclosporin (µmol·L−1) | ||||||
|---|---|---|---|---|---|---|---|
| Control | 0.05 | 0.10 | 0.20 | 2.5 | 5.0 | 10 | |
| (A)Six hours incubation at basal (6.6 mmol·L−1) glucose concentration | |||||||
| Calcineurin | 0.85 (0.84–0.85) | 0.76 (0.75–0.83) | 0.93 (0.89–0.97) | 0.78 (0.77–0.79) | 0.8 (0.8–0.8) | 0.83 (0.73–0.93) | 0.74 (0.71–0.77) |
| Bax/Bcl-2 | 0.71 (0.51–0.75) | 0.74 (0.69–0.79) | 0.77 (0.75–0.78) | 0.82 (0.81–0.82) | 0.72 (0.66–0.78) | 0.76 (0.7–0.83) | 0.92 (0.86–0.99) |
| NFATc1 | 1.03 (0.96–1.13) | 0.99 (0.89–1.09) | 1.06 (0.94–1.18) | 1.02 (0.95–1.09) | 1.17 (1.12–1.22) | 1.11 (1.07–1.14) | 1.09 (1.04–1.13) |
| NFATc2 | 12.76 (4.29–13.7) | 7.77 (5.65–9.9) | 6.88 (5.11–8.64) | 3.77 (3.64–3.89) | 2.38 (2–2.76) | 5.12 (0.93–9.32) | 5.07 (2.15–7.98) |
| NFATc3 | 1.11 (1.04–1.34) | 0.91 (0.88–0.94) | 1.09 (0.96–1.22) | 1.23 (1.17–1.28) | 1.14 (1.11–1.17) | 1.11 (1.04–1.19) | 1.29 (1.05–1.54) |
| NFATc4 | 0.35 (0.28–0.36) | 0.37 (0.35–0.39) | 0.39 (0.32–0.45) | 0.36 (0.31–0.41) | 0.38 (0.32–0.44) | 0.41 (0.36–0.46) | 0.37 (0.36–0.38) |
| (B)Six hours incubation at stimulatory (16.7 mmol·L−1) glucose concentration | |||||||
| Calcineurin | 0.83 (0.76–0.96) | 0.89 (0.76–0.96) | 0.8 (0.78–0.83) | 0.94 (0.84–1.04) | 0.86 (0.83–0.9) | 0.82 (0.74–0.9) | 0.72 (0.69–0.75) |
| Bax/Bcl-2 | 0.9 (0.83–1.05) | 0.89 (0.84–0.93) | 0.78 (0.66–0.92) | 0.98 (0.74–1.21) | 0.98 (0.96–0.99) | 1.15 (1.1–1.2) | 1.34 (1.22–1.46) |
| NFATc1 | 0.75 (0.71–0.77) | 0.83 (0.73–0.93) | 0.83 (0.78–0.87) | 0.8 (0.64–0.97) | 0.75 (0.72–0.77) | 0.82 (0.76–0.88) | 0.75 (0.71–0.78) |
| NFATc2 | 4.12 (0.97–9.72) | 24.16 (2.6–45.7) | 4.53 (3.17–5.9) | 4.19 (2.87–5.51) | 1.56 (0.95–2.17) | 4.42 (1.31–7.52) | 26.27 (8.34–44) |
| NFATc3 | 1.11 (0.98–1.47) | 1.06 (1.02–1.09) | 1.06 (0.9–1.23) | 1.13 (1.12–1.14) | 1.14 (1.13–1.15) | 1.16 (1.09–1.22) | 1.34 (1.32–1.36) |
| NFATc4 | 0.21 (0.18–0.22) | 0.19 (0.18–0.19) | 0.19 (0.18–0.21) | 0.19 (0.19–0.19) | 0.19 (0.18–0.19) | 0.18 (0.17–0.18) | 0.20 (0.19–0.21) |
| (C)Twenty-four hours of incubation at basal (6.6 mmol·L−1) glucose concentration | |||||||
| Calcineurin | 0.93 (0.83–1.08) | 1.01 (0.98–1.05) | 0.91 (0.9–0.93) | 1.00 (0.95–1.05) | 1.22 (1.0–1.44) | 0.95 (0.93–0.96) | 1.06 (0.93–1.2) |
| Bax/Bcl-2 | 0.74 (0.66–0.89) | 0.66 (0.65–0.68) | 0.79 (0.75–0.82) | 0.67 (0.61–0.73) | 0.74 (0.68–0.79) | 0.59 (0.56–0.62) | 0.71 (0.65–0.76) |
| NFATc1 | 0.88 (0.78–0.95) | 0.7 (0.69–0.71) | 0.77 (0.73–0.8) | 0.8 (0.73–0.86) | 1.25 (1.23–1.26) | 1.11 (1.07–1.15) | 1.08 (1.04–1.12) |
| NFATc2 | 17.75 (4.76–60.9) | 19.26 (16.8–21.7) | 18.13 (15.7–20.6) | 12.93 (4.58–21.3) | 42.11 (15.3–68.9) | 5.18 (5.03–5.34) | 17.13 (15.3–19) |
| NFATc3 | 1.47 (1.29–1.75) | 1.57 (1.48–1.67) | 1.82 (1.73–1.91) | 2.1 (1.91–2.28) | 1.59 (1.34–1.84) | 1.5 (1.38–1.62) | 1.52 (1.44–1.61) |
| NFATc4 | 0.81 (0.62–0.88) | 0.9 (0.85–0.96) | 0.94 (0.86–1.02) | 0.99 (0.95–1.04) | 1.15 (1.05–1.24) | 1.1 (1.02–1.18) | 1.12 (1.05–1.19) |
| (D)Twenty-four hours of incubation at stimulatory (16.7 mmol·L−1) glucose concentrations | |||||||
| Calcineurin | 1.0 (0.92–1.29) | 0.94 (0.83–1.15) | 0.95 (0.89–1.28) | 0.93 (0.91–0.99) | 1.03 (0.93–1.14) | 0.84 (0.76–0.92) | 0.74 (0.62–0.85) |
| Bax/Bcl-2 | 1.08 (0.83–1.53) | 0.98 (0.85–1.23) | 0.99 (0.89–1.41) | 1.14 (1.02–1.37) | 1.12 (1.03–1.22) | 1.18 (1.15–1.22) | 1.42 (1.14–1.71) |
| NFATc1 | 0.64 (0.53–0.71) | 0.6 (0.51–0.78) | 0.61 (0.5–0.75) | 0.61 (0.56–0.68) | 0.65 (0.6–0.7) | 0.64 (0.6–0.68) | 0.59 (0.58–0.6) |
| NFATc2 | 20.05 (16.5–44.2) | 23.04 (12.5–29.9) | 46.97 (12.9–498) | 53.23 (11.2–124) | 16.31 (9.61–23) | 8.22 (3.06–13.4) | 36.0 (12.9–59.1) |
| NFATc3 | 1.66 (1.33–1.79) | 1.53 (1.31–1.79) | 1.62 (1.27–1.89) | 1.54 (1.36–1.8) | 1.75 (1.68–1.82) | 1.44 (1.38–1.49) | 1.48 (1.29–1.68) |
| NFATc4 | 0.26 (0.25–0.36) | 0.29 (0.23–0.33) | 0.34 (0.33–0.65) | 0.45 (0.31–0.65) | 0.29 (0.22–0.36) | 0.28 (0.26–0.29) | 0.28 (0.22–0.35) |
Data are medians with range. n= 3 for controls and n= 2 for each drug concentration.
Bax, Bcl-2 associated X protein; Bcl-2, B-cell leukaemia/lymphoma 2; HSP, heat shock protein; NFATc, nuclear factor of activated T cells cytoplasmic.
Is NFATc expression controlled by the calcineurin/NFAT signalling pathway?
The calcineurin/NFAT pathway has emerged as an essential component of normal endocrine pancreas function and development (Lawrence et al., 2001; 2002; Heit et al., 2006). In order to explore which of the NFATc transcription factors were modulated by blocking calcineurin, we measured gene expression levels of NFAT-c1–c4. Neither short- nor long-term cell cultures with Tac or CsA suppressed levels of NFATc. On the contrary, NFATc1 and NFATc4 levels were up-regulated after 24 h of CsA exposure with basal glucose concentrations (Table 1C). Tac had similar effects on NFATc4, which was up-regulated in glucose-stimulated cells after prolonged incubation (Table 1D). Neither NFAT-c1, -c2 nor -c3 levels differed between controls and cells treated with the calcineurin inhibitors.
Calcineurin inhibitor-induced cell death and insulin resistance
To further unveil the molecular basis of the beta-cell dysfunction induced by calcineurin inhibitors, we studied the regulation of SREBP-1c, a lipogenic transcription factor known to suppress genes essential for beta-cell function and beta-cell mass (for the insulin receptor substrate 2 and the pancreatic and duodenal homebox factor 1), next to disrupting the secretory machinery of the beta-cell as well as promoting hepatic insulin resistance (Takahashi et al., 2005; Kato et al., 2006; Shimano et al., 2007). CsA significantly up-regulated expression levels of SREBP-1c (P= 0.03); meanwhile, prolonged Tac treatment interestingly down-regulated expression levels in glucose-stimulated cells (P= 0.03) (Figure 4A,B). Finally, cell death, as a possible consequence of treatment with calcineurin inhibitors, was investigated. (Drachenberg et al., 1999; Lotem et al., 1999; Wang et al., 1999; Shibasaki et al., 2002; Lawrence et al., 2005; Ranta et al., 2008). Initially, cell cultures were evaluated microscopically, where only very high doses of Tac (200 nmol·L−1) and CsA (10 µmol·L−1) resulted in beta-cell death in approximately 75 and 50% of cells, respectively, after 24 h. Gene expression analysis showed that high glucose concentrations alone mediated up-regulation of the Bax/Bcl2 ratio. A tendency towards an increased Bax/Bcl2 ratio was seen after 6 and 24 h of incubation with CsA (Table 1A–D) and analysis of DNA fragmentation confirmed cell death by apoptosis (P= 0.03) and necrosis (Figure 5A,B).
Figure 4.
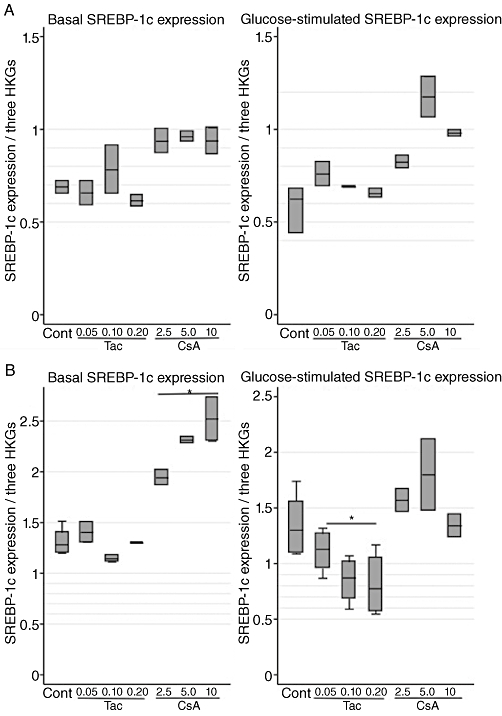
Effects of tacrolimus (Tac; µmol·L−1) and cyclosporin (CsA; µmol·L−1) on relative gene expression of SREBP-1c (normalized to β-actin, clathrin and HSP) in INS-1E cells. (A) Six hours of incubation at basal (6.6 mmol·L−1) and stimulatory (16.7 mmol·L−1) glucose concentrations. Data are medians with range. n= 3 for controls and n= 2 for each drug concentration. (B) Twenty-four hours of incubation at basal (6.6 mmol·L−1) and stimulatory (16.7 mmol·L−1) glucose concentrations. Data are medians with range. n= 3 for controls and n= 2 for each drug concentration. *P= 0.03 versus control, Kruskal–Wallis test. HKG, housekeeping gene.
Figure 5.
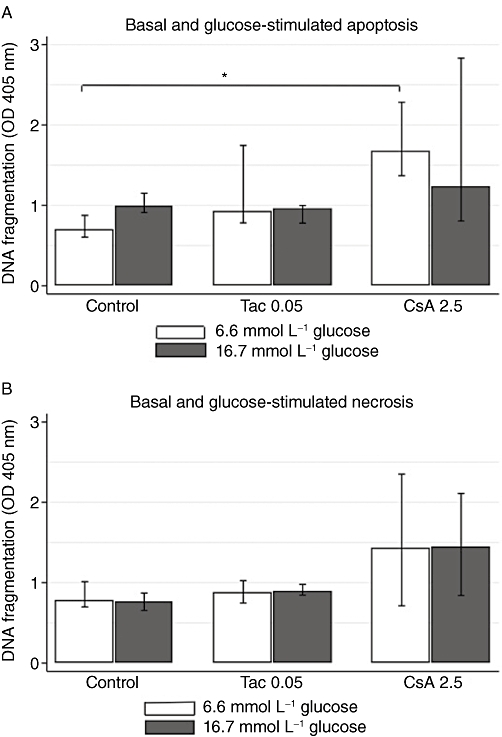
Effects of tacrolimus (Tac; µmol·L−1) and cyclosporin (CsA; µmol·L−1) on apoptosis and necrosis in INS-1E cells. (A) Intracellular DNA fragments (apoptosis) and (B) DNA fragments in medium (necrosis) after 24 h of incubation at basal (6.6 mmol·L−1) and stimulatory (16.7 mmol·L−1) glucose. Data are medians with range. n= 4 for controls and n= 4 for each drug concentration. *P= 0.03, Mann–Whitney U-test.
Discussion and conclusions
Chronic treatment with anti-calcineurinic immunosuppressants of organ transplanted patients secondarily induces a diabetic state relatively frequently and with unfavourable outcomes including graft failure and death (Cosio et al., 2002; 2005;). Recent evidence points towards a pivotal role of calcineurin for beta-cell function and survival (Heit et al., 2006); however, the relative diabetogenicity of anti-calcineurinic immunosuppressants and the molecular background of their diabetogenic actions have not been fully investigated. We herein report the effects of CsA and Tac on beta-cell function in a rat insulinoma cell line (INS-1E cells). The major findings of our study are firstly that both drugs equally impair basal as well as GSIS, after prolonged exposure. Secondly, such impairment was mirrored by an equal and total inhibition of CaN activity in the beta-cells. Finally, we present a novel connection between CsA treatment and activation of SREBP-1c, a lipogenic transcription factor promoting insulin resistance and beta-cell dysfunction.
A large body of preclinical literature has demonstrated that CsA and Tac exert deleterious effects on pancreatic beta-cells. Many previous studies are confounded by the use of very high doses of the drugs or difficulties in accounting for the insulin content of the beta-cells (Robertson, 1986; Ebihara et al., 1996; Dufer et al., 2001; Lawrence et al., 2001; 2002; 2005; Paty et al., 2002; Polastri et al., 2002). The present study demonstrates that doses high in the pharmacological range of both drugs primarily impair basal and GSIS after prolonged exposure, while insulin content and mRNA remained unaltered. Differential effects of the two drugs were however present, as Tac singularly displayed acute inhibitory effects on basal insulin secretion, and CsA paradoxically increased GSIS after short-term exposure. Similar time-dependent, opposing effects of calcineurin inhibitors on both insulin secretion and insulin content have been reported previously (Ebihara et al., 1996; Fuhrer et al., 2001; Lester et al., 2001; Johnson et al., 2009; Rauch et al., 2009), and collectively these data support clinical evidence of a more rapid development of diabetes during Tac treatment, than during treatment with CsA (van Duijnhoven et al., 2002; Hoitsma and Hilbrands, 2006). The natural time course of the beta-cell toxicity induced by calcineurin inhibitors may thus be impaired insulin secretion, followed by a decreased insulin production, as indicated by other long-term reports (Redmon et al., 1996; Drachenberg et al., 1999; Uchizono et al., 2004; Hernandez-Fisac et al., 2007). Thus, an explanation for the lack of effects on insulin content and mRNA, in our study, might be that the exposure time was too short. Apart from these time-dependent effects, the diabetogenic actions of the calcineurin inhibitors also appear to be dose dependent (Redmon et al., 1996; Drachenberg et al., 1999; Fuhrer et al., 2001; Paty et al., 2002; Uchizono et al., 2004). In the present study, beta-cell toxicity was highly and equally present for CsA and Tac during long-term incubations with increasing drug doses, yet our separate study using very low doses of Tac (0.01 µmol·L−1) and CsA (0.1 µmol·L−1), revealed no reductions in insulin output. This partly corresponds to a recent study on low-dose Tac-treated human islets, albeit some inhibitions of GSIS with low doses were observed (Johnson et al., 2009). These observations highlight the need to investigate newer low-dose regimens and their relation to NODAT in organ transplant recipients. Nonetheless, studying the in vitro effects of high calcineurin inhibitor concentrations may be relevant to islet transplantation, as the degree of calcineurin inhibitor exposure in this setting may, by far, exceed recommended blood levels.
The importance of the calcineurin/NFAT signalling pathway in maintaining beta-cell function and growth was documented in a recent study by Heit et al. (2006). We observed that a total blockade of CaN activity in the beta-cells was present at all times with both calcineurin inhibitors; and although this result matched the impaired insulin output seen after 24 h, it did not explain the largely maintained insulin output during short-term incubations. Calcineurin-independent regulatory mechanisms have previously been proposed by others. Indeed, Dufer et al. (2001) showed that CsA acutely and directly impairs GSIS in rodent beta-cells and islets, by acting on mitochondrial permeability transition pores, which determine the oscillatory activity of the cell membrane potential. Fuhrer et al. (2001) speculated that Tac had acute calcineurin independent effects in rodent beta-cells, through the regulation of the ATP-K channel, although a direct channel interaction was not determined. Aside from these presumed different acute actions, CsA and Tac may also act on different targets in the long run, alongside their shared calcineurin/NFAT signalling pathway. Previous studies have argued both against (Heit et al., 2006; Hernandez-Fisac et al., 2007) and in favour (Drachenberg et al., 1999; Uchizono et al., 2004; Plaumann et al., 2008; Johnson et al., 2009) of beta-cell apoptosis as a molecular rationale for diabetes-induced by calcineurin inhibitors. Our analysis of Bax/Bcl2 gene expression and DNA fragmentation mainly support an apoptotic mechanism of action for CsA, but not for Tac. Interestingly, we also found a novel relationship between CsA treatment and activation of the lipogenic transcription factor SREBP-1c, which is known to depress beta-cell secretory function and to promote insulin resistance (Kato et al., 2006; Shimano et al., 2007). Our findings contrast with observations made by Wu et al. (1999), where SREBP1 mRNA was suppressed in CsA-treated mice. However, the use of different methodologies makes it difficult to directly compare results. A final important finding in the present study was that no transcriptional repression of the NFAT-c1–c4 isoforms was observed despite calcineurin inhibition. However, it remains to be determined whether specific NFATc proteins can be detected in the beta-cell and whether or not they have redundant or dominant roles in insulin gene transcription. It will be of interest to expand these observations and determine whether a regulation of SREBP and other beta-cell factors by calcineurin inhibitors contribute to the development of diabetic states in humans.
In conclusion, we have shown that CsA and Tac are equally diabetogenic after prolonged exposure and that these prolonged effects coincide with a total inhibition of CaN activity. Only Tac produces acute inhibitory effects on basal insulin release, while CsA has a larger negative impact on transcriptional levels of several beta-cell essential genes including SREBP-1c, suggesting that Tac and CsA exert their effects not solely through calcineurin inhibition, but with additional highly individual pathways. Characterizing these potential targets of calcineurin inhibitors will contribute to the discovery of novel therapeutic modalities for the treatment of pancreatic and beta-cell diseases, and furthermore will help in tailoring immunosuppressive regimens for transplant recipients.
Acknowledgments
In memory of Professor Ole Schmitz, who contributed in the most inspiring way to this study. This work was supported in part by grants from The Danish Strategic Research Council (grant # 09-067009), The Research Foundation of the Danish Kidney Association, The Danish Society of Nephrology Research Foundation and The AP Moeller Foundation. We thank Karen Skjoedt Soerensen and Elin Carstensen, Department of Pharmacology, Aarhus University, Denmark, and Ilse Rasmussen and Birgitte Kildevaeld Sahl, C-Laboratory, Department of Nephrology, Aarhus University Hospital Skejby, Denmark for their expert technical assistance.
Glossary
Abbreviations
- Bax
Bcl-2 associated X protein
- Bcl-2
B-cell leukaemia/lymphoma 2
- CaN
calcineurin phosphatase
- GSIS
glucose-stimulated insulin secretion
- IRS-2
insulin receptor substrate 2
- NFATc
nuclear factor of activated T-cells cytoplasmic
- NODAT
new onset diabetes mellitus after transplantation
- PDX1
pancreatic and duodenal homebox factor 1
- SREBP-1c
sterol receptor element binding protein 1c
Conflict of interests
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosio FG, Pesavento TE, Kim S, Osei K, Henry M, Ferguson RM. Patient survival after renal transplantation: IV. Impact of post-transplant diabetes. Kidney Int. 2002;62:1440–1446. doi: 10.1111/j.1523-1755.2002.kid582.x. 10.1111/j.1523-1755.2002.kid582.x. [DOI] [PubMed] [Google Scholar]
- Cosio FG, Kudva Y, van der Velde M, Larson TS, Textor SC, Griffin MD, et al. New onset hyperglycemia and diabetes are associated with increased cardiovascular risk after kidney transplantation. Kidney Int. 2005;67:2415–2421. doi: 10.1111/j.1523-1755.2005.00349.x. [DOI] [PubMed] [Google Scholar]
- Drachenberg CB, Klassen DK, Weir MR, Wiland A, Fink JC, Bartlett ST, et al. Islet cell damage associated with tacrolimusrolimus and cyclosporine: morphological features in pancreas allograft biopsies and clinical correlation. Transplantation. 1999;68:396–402. doi: 10.1097/00007890-199908150-00012. [DOI] [PubMed] [Google Scholar]
- Dufer M, Krippeit-Drews P, Lembert N, Idahl LA, Drews G. Diabetogenic effect of cyclosporin A is mediated by interference with mitochondrial function of pancreatic B-cells. Mol Pharmacol. 2001;60:873–879. [PubMed] [Google Scholar]
- Duijnhoven EM, Boots JM, Christiaans MH, Wolffenbuttel BH, Van Hooff JP. Influence of tacrolimusrolimus on glucose metabolism before and after renal transplantation: a prospective study. J Am Soc Nephrol. 2001;12:583–588. doi: 10.1681/ASN.V123583. [DOI] [PubMed] [Google Scholar]
- van Duijnhoven EM, Christiaans MH, Boots JM, Nieman FH, Wolffenbuttel BH, van Hooff JP. Glucose metabolism in the first 3 years after renal transplantation in patients receiving tacrolimusrolimus versus cyclosporine-based immunosuppression. J Am Soc Nephrol. 2002;13:213–220. doi: 10.1681/ASN.V131213. [DOI] [PubMed] [Google Scholar]
- Ebihara K, Fukunaga K, Matsumoto K, Shichiri M, Miyamoto E. Cyclosporin A stimulation of glucose-induced insulin secretion in MIN6 cells. Endocrinology. 1996;137:5255–5263. doi: 10.1210/endo.137.12.8940343. [DOI] [PubMed] [Google Scholar]
- Ekstrand AV, Eriksson JG, Gronhagenriska C, Ahonen PJ, Groop LC. Insulin resistance and insulin deficiency in the pathogenesis of posttransplantation diabetes in man. Transplantation. 1992;53:563–568. doi: 10.1097/00007890-199203000-00014. [DOI] [PubMed] [Google Scholar]
- Fruman DA, Klee CB, Bierer BE, Burakoff SJ. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc Natl Acad Sci USA. 1992;89:3686–3690. doi: 10.1073/pnas.89.9.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrer DK, Kobayashi M, Jiang H. Insulin release and suppression by tacrolimusrolimus, rapamycin and cyclosporin A are through regulation of the ATP-sensitive potassium channel. Diabetes Obes Metab. 2001;3:393–402. doi: 10.1046/j.1463-1326.2001.00150.x. [DOI] [PubMed] [Google Scholar]
- Heisel O, Heisel R, Balshaw R, Keown P. New onset diabetes mellitus in patients receiving calcineurin inhibitors: a systematic review and meta-analysis. Am J Transplant. 2004;4:583–595. doi: 10.1046/j.1600-6143.2003.00372.x. [DOI] [PubMed] [Google Scholar]
- Heit JJ, Apelqvist AA, Gu X, Winslow MM, Neilson JR, Crabtree GR, et al. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature. 2006;443:345–349. doi: 10.1038/nature05097. 10.1038/nature05097. [DOI] [PubMed] [Google Scholar]
- Hernandez-Fisac I, Pizarro-Delgado J, Calle C, Marques M, Sanchez A, Barrientos A, et al. Tacrolimusrolimus-induced diabetes in rats courses with suppressed insulin gene expression in pancreatic islets. Am J Transplant. 2007;7:2455–2462. doi: 10.1111/j.1600-6143.2007.01946.x. 10.1111/j.1600-6143.2007.01946.x. [DOI] [PubMed] [Google Scholar]
- Hjelmesaeth J, Hagen LT, Asberg A, Midtvedt K, Storset O, Halvorsen CE, et al. The impact of short-term ciclosporin A treatment on insulin secretion and insulin sensitivity in man. Nephrol Dial Transplant. 2007;22:1743–1749. doi: 10.1093/ndt/gfl820. [DOI] [PubMed] [Google Scholar]
- Hoitsma AJ, Hilbrands LB. Relative risk of new-onset diabetes during the first year after renal transplantation in patients receiving tacrolimusrolimus or cyclosporine immunosuppression. Clin Transplant. 2006;20:659–664. doi: 10.1111/j.1399-0012.2006.00535.x. [DOI] [PubMed] [Google Scholar]
- Johnson JD, Ao Z, Ao P, Li H, Dai LJ, He Z, et al. Different effects of FK506, rapamycin, and mycophenolate mofetil on glucose-stimulated insulin release and apoptosis in human islets. Cell Transplant. 2009;18:833–845. doi: 10.3727/096368909X471198. 10.3727/096368909X471198. [DOI] [PubMed] [Google Scholar]
- Kato T, Shimano H, Yamamoto T, Yokoo T, Endo Y, Ishikawa M, et al. Granuphilin is activated by SREBP-1c and involved in impaired insulin secretion in diabetic mice. Cell Metab. 2006;4:143–154. doi: 10.1016/j.cmet.2006.06.009. 10.1016/j.cmet.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Koefoed-Nielsen PB, Karamperis N, Jorgensen KA. Validation of the calcineurin phosphatase assay. Clin Chem. 2004;50:2331–2337. doi: 10.1373/clinchem.2004.034066. 10.1373/clinchem.2004.034066. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Bhatt HS, Watterson JM, Easom RA. Regulation of insulin gene transcription by a Ca2+- responsive pathway involving calcineurin and nuclear factor of activated T cells. Mol Endocrinol. 2001;15:1758–1767. doi: 10.1210/mend.15.10.0702. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Bhatt HS, Easom RA. NFAT regulates insulin gene promoter activity in response to synergistic pathways induced by glucose and glucagon-like peptide-1. Diabetes. 2002;51:691–698. doi: 10.2337/diabetes.51.3.691. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, McGlynn K, Park BH, Cobb MH. ERK1/2-dependent activation of transcription factors required for acute and chronic effects of glucose on the insulin gene promoter. J Biol Chem. 2005;280:26751–26759. doi: 10.1074/jbc.M503158200. 10.1074/jbc.M503158200. [DOI] [PubMed] [Google Scholar]
- Lester LB, Faux MC, Nauert JB, Scott JD. Targeted protein kinase A and PP-2B regulate insulin secretion through reversible phosphorylation. Endocrinology. 2001;142:1218–1227. doi: 10.1210/endo.142.3.8023. [DOI] [PubMed] [Google Scholar]
- Lotem J, Kama R, Sachs L. Suppression or induction of apoptosis by opposing pathways downstream from calcium-activated calcineurin. Proc Natl Acad Sci USA. 1999;96:12016–12020. doi: 10.1073/pnas.96.21.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midtvedt K, Hjelmesaeth J, Hartmann A, Lund K, Paulsen D, Egeland T, et al. Insulin resistance after renal transplantation: the effect of steroid dose reduction and withdrawal. J Am Soc Nephrol. 2004;15:3233–3239. doi: 10.1097/01.ASN.0000145435.80005.1E. [DOI] [PubMed] [Google Scholar]
- Nielsen JH, Mandrup-Poulsen T, Nerup J. Direct effects of cyclosporin A on human pancreatic beta-cells. Diabetes. 1986;35:1049–1052. doi: 10.2337/diab.35.9.1049. [DOI] [PubMed] [Google Scholar]
- Oetjen E, Baun D, Beimesche S, Krause D, Cierny I, Blume R, et al. Inhibition of human insulin gene transcription by the immunosuppressive drugs cyclosporin A and tacrolimusrolimus in primary, mature islets of transgenic mice. Mol Pharmacol. 2003;63:1289–1295. doi: 10.1124/mol.63.6.1289. [DOI] [PubMed] [Google Scholar]
- Paty BW, Harmon JS, Marsh CL, Robertson RP. Inhibitory effects of immunosuppressive drugs on insulin secretion from HIT-T15 cells and wistar rat islets. Transplantation. 2002;73:353–357. doi: 10.1097/00007890-200202150-00007. [DOI] [PubMed] [Google Scholar]
- Plaumann S, Blume R, Borchers S, Steinfelder HJ, Knepel W, Oetjen E. Activation of the dual-leucine-zipper-bearing kinase and induction of beta-cell apoptosis by the immunosuppressive drug cyclosporin A. Mol Pharmacol. 2008;73:652–659. doi: 10.1124/mol.107.040782. [DOI] [PubMed] [Google Scholar]
- Polastri L, Galbiati F, Bertuzzi F, Fiorina P, Nano R, Gregori S, et al. Secretory defects induced by immunosuppressive agents on human pancreatic beta-cells. Acta Diabetol. 2002;39:229–233. doi: 10.1007/s005920200039. [DOI] [PubMed] [Google Scholar]
- Ranta F, Dufer M, Stork B, Wesselborg S, Drews G, Haring HU, et al. Regulation of calcineurin activity in insulin-secreting cells: stimulation by Hsp90 during glucocorticoid-induced apoptosis. Cell Signal. 2008;20:1780–1786. doi: 10.1016/j.cellsig.2008.06.003. 10.1016/j.cellsig.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Rauch MC, San Martin A, Ojeda D, Quezada C, Salas M, Carcamo JG, et al. Tacrolimusrolimus causes a blockage of protein secretion which reinforces its immunosuppressive activity and also explains some of its toxic side-effects. Transpl Immunol. 2009;22:72–81. doi: 10.1016/j.trim.2009.07.001. 10.1016/j.trim.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Redmon JB, Olson LK, Armstrong MB, Greene MJ, Robertson RP. Effects of tacrolimusrolimus (FK506) on human insulin gene expression, insulin mRNA levels, and insulin secretion in HIT-T15 cells. J Clin Invest. 1996;98:2786–2793. doi: 10.1172/JCI119105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson RP. Cyclosporin-induced inhibition of insulin secretion in isolated rat islets and HIT cells. Diabetes. 1986;35:1016–1019. doi: 10.2337/diab.35.9.1016. [DOI] [PubMed] [Google Scholar]
- Shibasaki F, Hallin U, Uchino H. Calcineurin as a multifunctional regulator. J Biochem. 2002;131:1–15. doi: 10.1093/oxfordjournals.jbchem.a003063. [DOI] [PubMed] [Google Scholar]
- Shimano H, Amemiya-Kudo M, Amemiya-Kudo M, Takahashi A, Takahashi A, Kato T, et al. Sterol regulatory element-binding protein-1c and pancreatic beta-cell dysfunction. Diabetes Obes Metab. 2007;9(Suppl 2):133–139. doi: 10.1111/j.1463-1326.2007.00779.x. [DOI] [PubMed] [Google Scholar]
- Takahashi A, Motomura K, Kato T, Yoshikawa T, Nakagawa Y, Yahagi N, et al. Transgenic mice overexpressing nuclear SREBP-1c in pancreatic beta-cells. Diabetes. 2005;54:492–499. doi: 10.2337/diabetes.54.2.492. [DOI] [PubMed] [Google Scholar]
- Uchizono Y, Iwase M, Nakamura U, Sasaki N, Goto D, Iida M. Tacrolimusrolimus impairment of insulin secretion in isolated rat islets occurs at multiple distal sites in stimulus-secretion coupling. Endocrinology. 2004;145:2264–2272. doi: 10.1210/en.2003-1152. [DOI] [PubMed] [Google Scholar]
- Vincenti F, Friman S, Scheuermann E, Rostaing L, Jenssen T, Campistol JM, et al. Results of an international, randomized trial comparing glucose metabolism disorders and outcome with cyclosporine versus tacrolimusrolimus. Am J Transplant. 2007;7:1506–1514. doi: 10.1111/j.1600-6143.2007.01749.x. [DOI] [PubMed] [Google Scholar]
- Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, et al. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–343. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- Wu J, Zhu YH, Patel SB. Cyclosporin-induced dyslipoproteinemia is associated with selective activation of SREBP-2. Am J Physiol. 1999;277:E1087–E1094. doi: 10.1152/ajpendo.1999.277.6.E1087. [DOI] [PubMed] [Google Scholar]