

Abstract
BACKGROUND AND PURPOSE
Frankincense, the gum resin derived from Boswellia species, showed anti-inflammatory efficacy in animal models and in pilot clinical studies. Boswellic acids (BAs) are assumed to be responsible for these effects but their anti-inflammatory efficacy in vivo and their molecular modes of action are incompletely understood.
EXPERIMENTAL APPROACH
A protein fishing approach using immobilized BA and surface plasmon resonance (SPR) spectroscopy were used to reveal microsomal prostaglandin E2 synthase-1 (mPGES1) as a BA-interacting protein. Cell-free and cell-based assays were applied to confirm the functional interference of BAs with mPGES1. Carrageenan-induced mouse paw oedema and rat pleurisy models were utilized to demonstrate the efficacy of defined BAs in vivo.
KEY RESULTS
Human mPGES1 from A549 cells or in vitro-translated human enzyme selectively bound to BA affinity matrices and SPR spectroscopy confirmed these interactions. BAs reversibly suppressed the transformation of prostaglandin (PG)H2 to PGE2 mediated by mPGES1 (IC50 = 3–10 µM). Also, in intact A549 cells, BAs selectively inhibited PGE2 generation and, in human whole blood, β-BA reduced lipopolysaccharide-induced PGE2 biosynthesis without affecting formation of the COX-derived metabolites 6-keto PGF1α and thromboxane B2. Intraperitoneal or oral administration of β-BA (1 mg·kg−1) suppressed rat pleurisy, accompanied by impaired levels of PGE2 and β-BA (1 mg·kg−1, given i.p.) also reduced mouse paw oedema, both induced by carrageenan.
CONCLUSIONS AND IMPLICATIONS
Suppression of PGE2 formation by BAs via interference with mPGES1 contribute to the anti-inflammatory effectiveness of BAs and of frankincense, and may constitute a biochemical basis for their anti-inflammatory properties.
Keywords: inflammation, prostaglandin, boswellic acid, microsomal prostaglandin E2 synthase, arachidonic acid
Introduction
Extracts of the gum resins from Boswellia species (frankincense) are commonly used in folk medicine to treat various inflammatory disorders. The pentacyclic triterpenes boswellic acids (BAs; Figure 1) are assumed to be the pharmacological active principles of frankincense extracts, and based on data obtained from experiments using cellular and animal models (Poeckel and Werz, 2006), there is accumulating evidence for anti-inflammatory and anti-tumorigenic effects of BAs. Pilot clinical studies have suggested some efficacy of frankincense preparations in the treatment of osteoarthritis (OA), rheumatoid arthritis (RA), inflammatory bowel diseases, asthma and cancer (see Ammon, 2006; Poeckel and Werz, 2006). Molecular mechanisms responsible for these therapeutics effects have been mainly attributed to the interference of 3-O-acetyl-11-keto-β-BA (AKBA) with signalling pathways including the nuclear factor-κB route (Syrovets et al., 2005), mitogen-activated protein kinase pathway and Ca2+ signalling (Poeckel et al., 2006a) as well as targeting human leukocyte elastase (HLE) (Safayhi et al., 1997), 5-lipoxygenase (5-LOX) (Safayhi et al., 1992), platelet-type 12-lipoxygenase (12S-LOX) (Poeckel et al., 2006b) and COX-1 (Siemoneit et al., 2008). However, in vivo studies confirming the pharmacological relevance of these proposed target interactions are still missing, and the efficacy of defined BAs in in vivo models of inflammation remains to be assessed. Recently, we showed that the serine protease cathepsin G is a high affinity and pharmacologically relevant target of BAs (Tausch et al., 2009).
Figure 1.
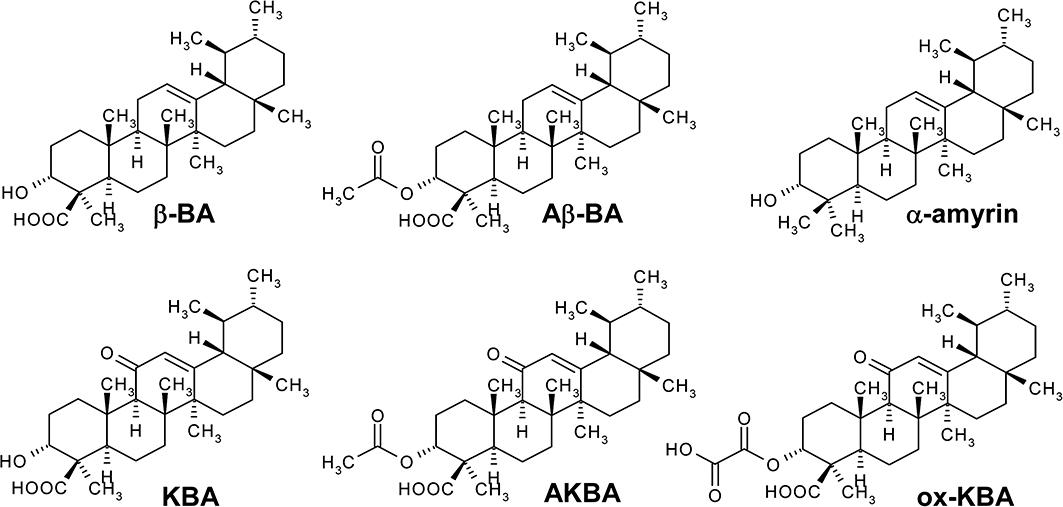
Chemical structures of boswellic acids (BAs) and α-amyrin. 3-O-Acetyl-β-boswellic acid (Aβ-BA); 3-O-acetyl-11-keto-β-boswellic acid (AKBA); β-boswellic acid (β-BA); 11-keto-β-boswellic acid (KBA); 3-O-oxaloyl-11-β-keto-boswellic acid (ox-KBA).
Prostaglandins (PGs) are important lipid mediators derived from arachidonic acid (AA) that control not only numerous physiological events such as blood pressure, blood clotting and sleep, but also inflammation (Funk, 2001). PGE2 is a key player in pyresis, pain and inflammatory responses (Smith, 1989), and the beneficial therapeutic effects of non-steroidal anti-inflammatory drugs (NSAIDs) are essentially attributed to the suppression of PGE2 (Funk, 2001). The biosynthetic pathway to PGE2 includes the release of AA from membrane phospholipids by phospholipases A2 followed by conversion via COX-1 and −2 to PGH2 and its subsequent isomerization by PGE2 synthases (PGES). mPGES1 is induced by pro-inflammatory stimuli such as interleukin-1β (IL-1β) or lipopolysaccharide (LPS), and receives PGH2 preferentially from COX-2 (Murakami et al., 2002). Thus, inflammation, pain, fever and different types of cancer are closely linked to the increased PGE2 formation originating from up-regulated mPGES1 (Samuelsson et al., 2007). Data from studies using mPGES1-deficient mice indicate that suppression of mPGES1 may provide an efficient pharmacological approach for the treatment of inflammatory diseases (Trebino et al., 2003), avoiding effects on the formation of physiologically important and homeostatic PGs.
Here, we have demonstrated that BAs were direct inhibitors of mPGES1, and particularly β-BA (the most abundant BA in frankincense) was highly effective in vitro and in vivo. Moreover, we analysed and compared for the first time the anti-inflammatory efficacy of the four major β-configured BAs in vivo. As pathophysiological PGE2 formation is mainly related to mPGES1 (Samuelsson et al., 2007), our findings may provide a molecular mechanism contributing to the anti-inflammatory efficacy of BAs or frankincense preparations.
Methods
Animals
All animal care and experimental procedures complied with Italian regulations on the protection of animals used for experimental and other scientific purpose (Ministerial Decree 116192), and with the European Economic Community regulations (Official Journal of E.C. L 358/1 12/18/1986). Male adult CD1 mice (25–35 g, Harlan, Milan, Italy) and Wistar Han rats (200–220 g, Harlan) were housed in a controlled environment and provided with standard rodent chow and water.
Cells and cell viability assay
A549 cells were cultured as described (Koeberle et al., 2008) and cell viability was measured using the colorimetric MTT dye reduction assay in a 96-well format using a multi-well scanning spectrophotometer (Victor3 plate reader, PerkinElmer, Rodgau-Juegesheim, Germany) as recently reported (Koeberle et al., 2008). Neither α-amyrin nor any of the five BAs (30 µM each) significantly reduced cell viability within 5 h, compared with the effects of dimethyl sulphoxide (DMSO) as vehicle (data not shown), excluding possible acute cytotoxic effects of the compounds in the cellular assays.
Cell-free expression of human mPGES1
Human mPGES1 was obtained by the continuous-exchange cell-free expression system according to Schwarz et al. (2007). This system comprises a reaction mixture that contains Escherichia coli S30 extract (derived from the A19 strain), T7 polymerase, tRNAs, pyruvate kinase and the template DNA for human mPGES1 (cloned in the pBH4 vector derived from pET19b, Novagen, Gibbstown, NJ, USA). The reaction mixture was dialysed against the feeding mixture that supplies amino acids, energy equivalents acetyl phosphate, and phosphoenol pyruvate as well as nucleotides. Reactions are incubated at 30°C for up to 20 h. Protein synthesis takes place in the reaction mixture and up to 1.5 mg of mPGES1 per mL are obtained in the precipitate. mPGES1 was resuspended in 50 mM potassium phosphate buffer pH 7.4, 1 mM GSH, 10% glycerol and 2% (w/v) LysoFos12 choline (Anatrace, Maumee, OH, USA) for 2 h at 30°C, and insoluble parts were removed by centrifugation (10 000×g 10 min, 10°C).
Protein pull-down assays using immobilized BAs
For immobilization of BAs, β-BA or KBA were linked to EAH Sepharose 4B beads via the C3-OH group using glutaric acid as linker as described previously (Poeckel et al., 2006b). For protein pull-down experiments, 1 × 107 A549 cells were lysed in 375 µL lysis buffer (50 mM HEPES pH 7.4, 200 mM NaCl, 1 mM EDTA, 1% Triton X-100, 2 mM phenylmethanesulfonyl fluoride, 10 µg·mL−1 leupeptin and 120 µg·mL−1 soybean trypsin inhibitor). After sonification (3 × 8 s) and centrifugation (12 000×g, 10 min, 4°C), 125 µL of the Sepharose slurries (50%, v/v) were added to the lysates and incubated at 4°C overnight, with continuous rotation. For pull-down experiments with isolated mPGES1, 200 ng of the purified enzyme was diluted into 500 µL lysis buffer containing 1000-fold excess of E. coli (BL21 strain) protein, and 100 µL of the Sepharose slurries (50%, v/v) were added. Beads were extensively washed three times with 10 volumes of binding buffer (HEPES pH 7.4, 200 mM NaCl, 1 mM EDTA) and precipitated proteins were denatured by the addition of sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) sample loading buffer (20 mM Tris-HCl, pH 8, 2 mM EDTA, 5% (m/v) SDS, 10% β-mercaptoethanol). After boiling (95°C, 6 min), proteins were separated by SDS-PAGE and analysed by Western blotting, using specific antibodies against COX-2 (Biomol Intern., Hamburg, Germany) and mPGES1 (Cayman Chemical, Ann Arbor, MI, USA). Ponceau S-staining of the membranes after blotting assured equal protein loading of bound proteins to the beads.
Surface plasmon resonance spectroscopy
A BIAcore X device (GE Healthcare Bio-Sciences, Freiburg, Germany) was used. In vitro-translated mPGES1 (100 µg·mL−1) in 10 mM Na-acetate pH 6.0 was coupled to a carboxymethylated dextran surface (CM-5 chip, GE Healthcare) using standard amine coupling procedure according to the manufacturer's instructions. Flow cell 1 on the chip was not altered (reference) whereas on flow cell 2, mPGES1 (236 fmol·mm−2) was immobilized corresponding to 4700 resonance units (RU) and equilibrated by a continuous flow of assay buffer (10 mM HEPES, 150 mM NaCl, 0.01% surfactant P20, and 1% DMSO, pH 7.4) The stock solution of 3-O-oxaloyl-KBA (in DMSO) was diluted into assay buffer. Measurements were performed at 25°C and at a flow rate of 30 µL·min−1. After recording association, the liquid phase was replaced by assay buffer, and the dissociation was monitored. The binding profiles were obtained after subtracting the response signal of the untreated reference cell 1, and sensograms were processed by using automatic correction for non-specific bulk refractive index effects using BIAEVALUATION Version 3.1 software (GE Healthcare Bio-Sciences).
To obtain dissociation constants from the equilibrium binding data, two different fitting models were adopted. First, the change in the equilibrium amount of compound bound as a function of the concentration of compound was fitted to the equation (Eqn 1) for a simple 1:1 binding model:
| (1) |
where Req is the equilibrium response, Rmax is the maximum response and KD is the dissociation constant. A Scatchard analysis was also used to determine KD (represented by the negative reciprocal of the slope). Analysis employing the BIAEVALUATION software version 3.1 was performed to determine kinetics. The integrated rate equation describing a 1:1 Langmuir interaction was fitted simultaneously to the entire concentration range for 3-O-oxaloyl-KBA. This fit yielded the association rate ka, the dissociation rate kd and the dissociation constant KD (Roden and Myszka, 1996; Karlsson and Falt, 1997). The quality of the fit was determined by the χ2-values as well as the magnitude and distribution of the residuals.
Stimulation of A549 cells and isolation of microsomes
Preparation of A549 cells was performed as described (Koeberle et al., 2009). In brief, cells were incubated for 16 h at 37°C and 5% CO2, and after changing the medium, mPGES1 expression was induced by IL-1β (1 ng·mL−1). After 72 h, cells were frozen in liquid nitrogen, ice-cold homogenization buffer (0.1 M potassium phosphate buffer pH 7.4, 1 mM phenylmethanesulfonyl fluoride, 60 µg·mL−1 soybean trypsin inhibitor, 1 µg·mL−1 leupeptin, 2.5 mM GSH and 250 mM sucrose) was added, and after 15 min, cells were resuspended and sonicated on ice (3 × 20 s). The homogenate was subjected to differential centrifugation at 10 000×g for 10 min and at 174 000×g for 1 h at 4°C. The pellet (microsomal fraction) was resuspended in 1 mL homogenization buffer and the protein concentration was determined by the Coomassie protein assay.
Determination of PGES1 activity in microsomes of A549 cells
Microsomal membranes of A549 cells were diluted in potassium phosphate buffer (0.1 M, pH 7.4) containing 2.5 mM GSH (100 µL total volume), and PGE2 formation was initiated by the addition of PGH2 (20 µM, final concentration). After 1 min at 4°C, the reaction was terminated with 100 µL of stop solution (40 mM FeCl2, 80 mM citric acid and 10 µM of 11β-PGE2); PGE2 was separated by solid phase extraction and analysed by reversed phase-high performance liquid chromatography (RP-HPLC) as described (Koeberle et al., 2009).
Determination of PGE2 and 6-keto PGF1α formation in intact A549 cells
The expression of mPGES1 in A549 cells was induced as described previously. After trypsinization, cells were washed twice with phosphate-buffered saline (PBS), resuspended in PBS (4 × 106 mL−1) containing CaCl2 (1 mM) and pre-incubated with the indicated compounds at 37°C for 10 min. Prostanoid formation was started by the addition of ionophore A23187 (2.5 µM), AA (1 µM) and [3H]AA (18.4 kBq). The reaction was stopped after 15 min at 37°C, and the samples were put on ice. For quantification of radiolabelled PGE2, samples were extracted, fractionated by HPLC and then analysed by liquid scintillation counting (Koeberle et al., 2008). 6-Keto PGF1α was determined by High Sensitivity EIA Kits (Assay Designs, Ann Arbor, MI, USA) according to the manufacturer's protocols.
Determination of prostanoid formation in human whole blood
Human venous blood from healthy adult donors, who had not received any medication for at least 2 weeks (Blood Center, University Hospital Tuebingen, Germany), was freshly withdrawn and collected in monovettes containing 16 IE heparin mL−1 (Sarstedt, Nümbrecht, Germany). For determination of PGE2 and 6-keto PGF1α, aliquots of whole blood (0.8 mL) were mixed with CV4151 (1 µM) and with aspirin (50 µM). For determination of TXB2, aliquots of whole blood (0.5 mL) were used without the addition of CV4151. A total volume of 1 mL was adjusted with sample buffer (10 mM potassium phosphate buffer pH 7.4, 3 mM KCl, 140 mM NaCl and 6 mM d-glucose). After pre-incubation with the indicated compounds for 5 min at room temperature, the samples were stimulated with LPS (10 µg·mL−1) for 5 h at 37°C. Prostanoid formation was stopped on ice, the samples were centrifuged (2300×g, 10 min, 4°C), and 6-keto PGF1α and TXB2 were quantified in the supernatant using High Sensitivity EIA Kits (Assay Designs), according to the manufacturer's protocols. PGE2 was determined as described (Koeberle et al., 2008). In brief, the supernatant was acidified with citric acid (30 µL, 2 M), and after centrifugation (2300×g, 10 min, 4°C), solid phase extraction and RP-HPLC, analysis of PGE2 was performed to isolate PGE2. The PGE2 peak (3 mL), identified by co-elution with the authentic standard, was collected, and acetonitrile was removed under a nitrogen stream. The pH was adjusted to 7.2 by the addition of 10 × PBS buffer pH 7.2 (230 µL) before PGE2 contents were quantified using a PGE2 High Sensitivity EIA Kit (Assay Designs) according to the manufacturer's protocol.
For determination of the COX product 12(S)-hydroxy-5-cis-8,10-trans-heptadecatrienoic acid (12-HHT), freshly drawn human blood (2 mL) was pre-incubated with the indicated compounds at 37°C for 10 min, and 30 µM Ca2+ ionophore A23187 was added. After 10 min, the reaction was stopped on ice, and the samples were centrifuged (600×g, 10 min, 4°C). Aliquots of the resulting plasma (500 µL) were then mixed with 2 mL of methanol, and 200 ng prostaglandin B1 was added as internal standard. The samples were cooled to −20°C for 2 h and centrifuged again (600×g, 15 min, 4°C). The supernatants were collected and diluted with 2.5 mL PBS and 75 µL HCl 1N, and formed 12-HHT was extracted and analysed by HPLC as described (Siemoneit et al., 2008).
Carrageenan-induced paw oedema
Mice were divided into groups (n = 10 for each group) and lightly anaesthetized with enflurane (4%) mixed with O2, 0.5 L·min−1, and N2O, 0.5 L·min−1. Each group of animals received subplantar administration of saline (0.05 mL) or λ-carrageenan type IV (1% w/v) (0.05 mL) in saline. The paw was marked in order to immerse it to the same extent in the measurement chamber. The volume was measured by using a hydroplethysmometer, specially modified for small volumes (Ugo Basile, Milan, Italy) immediately before subplantar injection and 2, 4 and 6 h thereafter. The assessment of paw volume was performed always under double blind conditions and by the same operator. The increase in paw volume was calculated by subtracting the initial paw volume (basal) to the paw volume measured at each time point.
Carrageenan-induced pleurisy
Rats were anaesthetized with enflurane (4%) mixed with O2, 0.5 L·min−1, N2O, 0.5 L·min−1, and submitted to a skin incision at the level of the left sixth intercostal space. The underlying muscle was dissected, and saline (0.2 mL) or λ-carrageenan type IV (1% w/v) (0.2 mL) was injected into the pleural cavity. The skin incision was closed with a suture, and the animals were allowed to recover. At 4 h after the injection of λ-carrageenan, the animals were killed by inhalation of CO2. The chest was carefully opened, and the pleural cavity was rinsed with 2 mL saline solution containing heparin (5 U·mL−1). The exudate and washing solution were removed by aspiration, and the total volume was measured using an adjustable-volume pipette (P1000, range volume 1000–200 µL or P200, range volume 200–30 µL). Any exudate that was contaminated with blood was discarded. The amount of exudate was calculated by subtracting the volume injected (2 mL) from the total volume recovered. Leukocytes in the exudate were resuspended in PBS and counted with an optical light microscope in a Burker's chamber after vital staining with Trypan blue.
The amounts of PGE2 and 6-keto PGF1α in the supernatant of centrifuged exudate (800×g, 10 min) were assayed by radioimmunoassay (PGE2) and EIA (6-keto PGF1α), respectively (Cayman Chemical), according to the manufacturer's protocol. The results are expressed as ng per rat and represent the mean ± SE of 10 rats.
Experimental design of animal experiments
For i.p. administration, the test compounds were dissolved in DMSO and diluted with saline, achieving a final DMSO concentration of 2 or 4%. For oral administration, the compounds were dissolved in water containing 0.5% (w/v) carboxymethylcellulose, 10% Tween-20 (v/v), and 1% sesame oil (v/v).
For carrageenan-induced paw oedema in the treated group of animals, β-BA (0.25 and 1 mg·kg−1) or indomethacin (5 mg·kg−1, reference compound) were given i.p. 30 min before carrageenan. The vehicle-treated group of mice received DMSO 2% (i.p.).
For carrageenan-induced pleurisy in the treated group of animals, the BAs (1 mg·kg−1, each) and indomethacin (5 mg·kg−1, reference compound) were given either i.p. or p.o. 30 min before carrageenan. The vehicle-treated group of rats received DMSO 4% (i.p.) or water containing 0.5% CMC, 10% Tween-20 and 1% sesame oil (p.o.). DMSO (4%, i.p.) itself did not affect exudate volume as compared with saline (0.30 ± 0.071 and 0.34 ± 0.02 mL respectively).
Data analysis
Data are expressed as mean ± SE. The program Graphpad Instat (Graphpad Software Inc., San Diego, CA, USA) was used for statistical comparisons of the data by one-way analyses of variance for independent or correlated samples followed by Tukey HSD post hoc tests. Where appropriate, Student's t-test for paired and correlated samples was applied. A P value of <0.05 (*) was considered significant. IC50 values of compounds are approximations determined by graphical analysis (linear interpolation between the points between 50% activity).
Materials
BAs were prepared as previously described (Jauch and Bergmann, 2003). All BAs and synthetic derivatives were at least 95% pure. Structures were confirmed by 1H-nuclear magnetic resonance (NMR) spectroscopy, 13C-NMR spectroscopy, H,H-correlated spectroscopy (COSY), heteronuclear multiple quantum coherence (HMQC), heteronuclear multiple bond correlation, HMQC-COSY, and nuclear Overhauser and exchange spectroscopy as well as electrospray ionization mass spectrometry in the negative mode. α-Amyrin was from Extrasynthèse (Genay, France); EAH-Sepharose 4B was from GE Healthcare Bio-Sciences (Freiburg, Germany); thromboxane synthase inhibitor CV4151 was a gift by Dr Stefan Laufer (Tuebingen, Germany); DMEM/High glucose (4.5 g·L−1) medium, penicillin, streptomycin, trypsin/EDTA solution, were from PAA (Coelbe, Germany); PGH2 was from Larodan (Malmö, Sweden); 11β-PGE2, MK-886, [5, 6, 8, 9, 11, 12, 14, 15-3H] AA ([3H]AA), were from BioTrend Chemicals GmbH (Cologne, Germany); Ultima Gold™ XR was from Perkin Elmer (Boston, MA, USA); λ-carrageenan type IV isolated from Gigartina aciculaire and Gigartina pistillata was from Sigma-Aldrich (Milan, Italy); [3H-PGE2], PerkinElmer Life Sciences (Milan, Italy); PGE2 antibody, Sigma-Aldrich. AA, LPS, fetal calf serum and all other chemicals were obtained from Sigma-Aldrich (Deisenhofen, Germany) unless stated otherwise. Nomenclature for the receptors and molecular targets studied here follows Alexander et al. (2009).
Results
Identification of mPGES1 as a BA-binding protein
A target fishing approach using immobilized BAs (Poeckel et al., 2006b) was applied in order to investigate whether or not BAs interact with mPGES1. Lysates of IL-1β-treated A549 cells, expressing mPGES1 (Jakobsson et al., 1999), were incubated with resins composed of β-BA or KBA, linked via the C3-OH moiety to glutaric acid and coupled to EAH Sepharose 4B beads yielding β-BA-Seph or KBA-Seph respectively. EAH Sepharose 4B beads without ligand (Seph) were used as the negative control. Beads were extensively washed and the bound proteins were detached by the addition of SDS-PAGE sample loading buffer followed by separation by SDS-PAGE and Western blot analysis, using specific antibodies against mPGES1 and COX-2. Substantial amounts of mPGES1 were bound to BA-Seph beads but hardly any was bound to Seph beads without ligand (Figure 2A). In contrast, COX-2 was not detected in these precipitated proteins. Furthermore, in vitro-translated mPGES1 (200 ng purified protein) in the presence of 1000-fold excess of E. coli protein was precipitated by both KBA-Seph and β-BA-Seph, but was minimally precipitated by Seph beads (Figure 2B).
Figure 2.
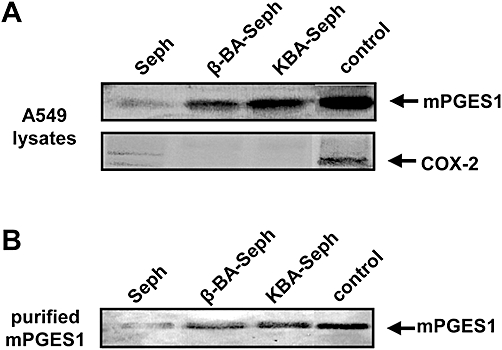
Boswellic acids (BAs) bind to microsomal prostaglandin E2 synthase 1 (mPGES1). (A) Supernatants of A549 cell lysates were incubated with β-BA-Seph, KBA-Seph or with Seph, as indicated. (B) Purified, in vitro-translated mPGES1 (200 ng) was incubated with β-BA-Seph, KBA-Seph or Seph beads. Precipitated proteins were separated by SDS-PAGE, and visualized by Western blotting, using specific antibodies against mPGES1 (A,B) or COX-2 (A). An aliquot of the supernatant was used as positive control. Similar results were obtained in three additional experiments. KBA, 11-keto-β-boswellic acid; SDS-PAGE, sodium dodecyl sulphate polyacrylamide gel electrophoresis.
To characterize the interaction of BAs with mPGES1 in more detail, surface plasmon resonance (SPR) spectroscopy studies were carried out. The progress of interaction, binding of analyte (association) and dissociation from the immobilized mPGES1 was monitored as a sensogram, expressing changes in binding responses as RU. Unfortunately, no consistent binding patterns were obtained using naturally occurring BAs as analytes, presumably due to their high lipophilicity leading to concentration-dependent aggregation and super-stoichiometric binding behaviour (Giannetti et al., 2008). Neither the addition of bovine serum albumin as carrier protein nor the variation of commercial assay buffers (with or without detergent) or changes in temperature improved the quality of the recorded sensograms. Hence, we used the more hydrophilic synthetic derivate 3-O-oxaloyl-KBA (10 µM) to obtain more valuable and reproducible sensograms, which indicated specific reversible binding to mPGES1 (Figure 3A). α-Amyrin (a pentacyclic triterpene which lacks the C4-COOH moiety of BAs; Figure 1) failed to bind mPGES1 up to concentrations of 30 µM (Figure 3A). In order to determine equilibrium-binding constants, 3-O-oxaloyl-KBA was analysed at concentrations ranging from 0.5 to 25 µM. The equilibrium response (Req) was calculated, and fitting the data to the 1:1 binding model (Eqn. 1) and Scatchard plot analysis yielded KD values of 13 and 5.2 µM respectively (Figure 3B).
Figure 3.
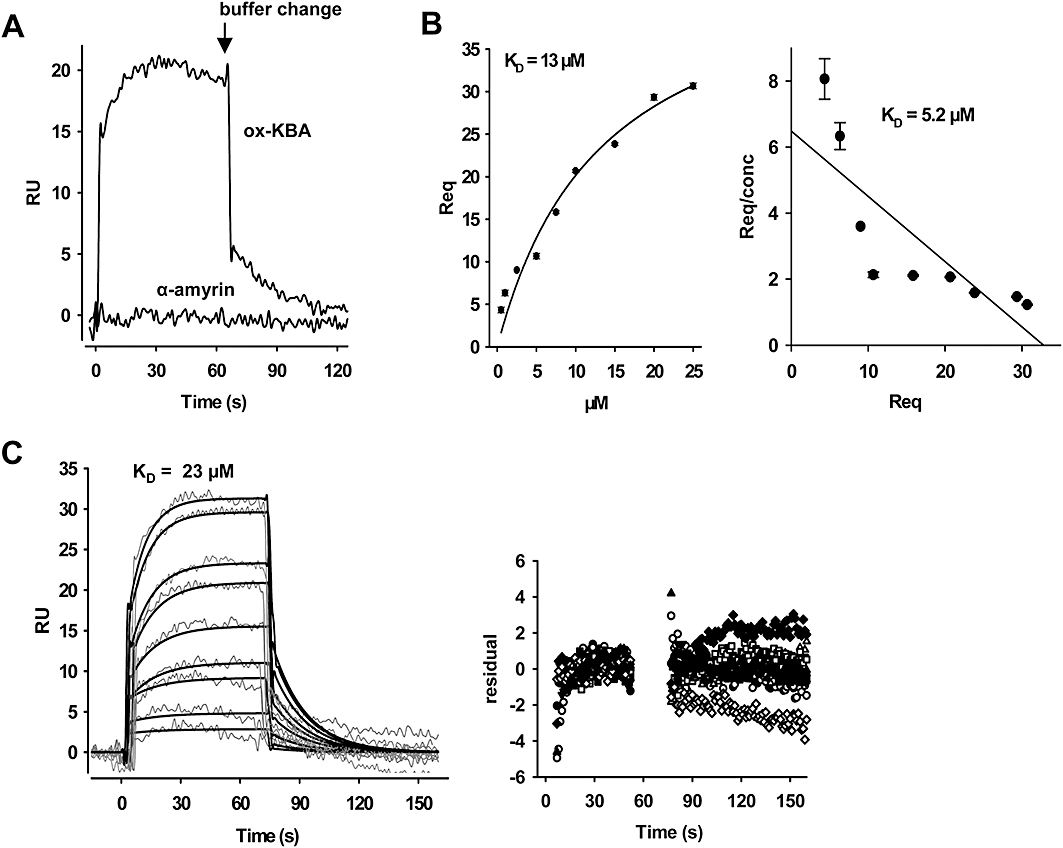
Analysis of the binding of boswellic acids to microsomal prostaglandin E2 synthase 1 (mPGES1) by surface plasmon resonance spectroscopy. In vitro-translated mPGES1 was coupled to a CM5 biosensor chip and 3-O-oxaloyl-KBA was used as analyte. Specific binding profiles were obtained after subtracting the signal [response units (RU)] from the untreated control cell. (A) Binding of 3-O-oxaloyl-KBA (ox-KBA) and α-amyrin (10 µM each) to mPGES1. (B) Binding curves for 3-O-oxaloyl-KBA. The equilibrium responses (Req) for 3-O-oxaloyl-KBA at different concentrations were plotted versus the concentration of the compound. (C) Kinetic analysis of 3-O-oxaloyl-KBA-binding to mPGES1. Representative sensograms for the injection of 0.5 µM up to 25 µM 3-O-oxaloyl-KBA are shown. A general analysis was applied to fit the data to a 1:1 binding model (bold lines), and the quality of the fit is displayed by the plots of the residuals. Results are representative for at least three independent experiments. KBA, 11-keto-β-boswellic acid.
Kinetic data were estimated using BIAEVALUATION 3.1 software. Assuming the simple relationship ka/kd = KD for 3-O-oxaloyl-KBA, a KD value of 23 µM was calculated that essentially matches the KD obtained from the equilibrium binding data (Figure 3C). Nevertheless, these kinetic parameters should be regarded as rough determinations rather than absolute values. Together, these data support a direct physical interaction between BAs and mPGES1.
BAs inhibit the catalytic activity of mPGES1 in a cell-free assay
Next, we investigated whether BAs may affect the catalytic activity of mPGES1. Isolated microsomes of IL-1β-treated A549 cells were pre-incubated with BAs, and PGE2 formation was induced by the addition of 20 µM PGH2. The mPGES1 inhibitor MK-886 was used as reference drug (Claveau et al., 2003; Koeberle et al., 2008) and blocked PGE2 formation with an IC50 = 2 µM (not shown). AKBA, β-BA and KBA concentration dependently suppressed PGE2 formation with IC50 values of 3, 5, and 10 µM respectively. As previously observed for other mPGES1 inhibitors (Koeberle et al., 2008; Koeberle and Werz, 2009), about 20–30% activity still remained even at high concentrations of BAs (100 µM, Figure 4A) or of MK-886 (30 µM, not shown), suggesting mPGES1-independent basal formation of PGE2. The synthetic derivative 3-O-oxaloyl-KBA suppressed PGE2 formation (IC50 = 5 µM, not shown), which is in good agreement with the SPR data. Aβ-BA was less potent (IC50 ≥ 30 µM) and α-amyrin was entirely inactive up to 100 µM (data not shown).
Figure 4.
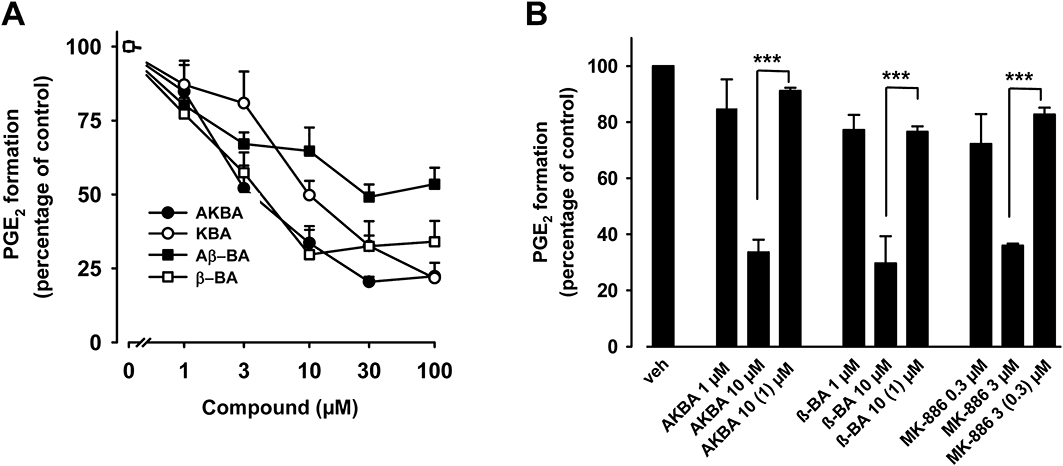
Effects of boswellic acids (BAs) on the activity of microsomal prostaglandin E2 synthase 1 (mPGES1) in a cell-free assay. (A) Concentration-response analysis. Microsomal preparations of IL-1β-stimulated A549 cells were pre-incubated with vehicle (DMSO) or BAs for 15 min at 4°C. PGH2 was added and after 1 min, the reaction was stopped and PGE2 was analysed by RP-HPLC as described. The 100% value corresponds to 944 ± 118 pmol PGE2 formed. (B) Reversibility of mPGES1 inhibition. Microsomal fractions of IL-1β-stimulated A549 cells were pre-incubated with 3 µM MK-886 or 10 µM BAs. An aliquot was diluted 10-fold to obtain an inhibitor concentration of 0.3 and 1 µM respectively. For comparison, microsomal preparations were pre-incubated with 0.3 µM MK-886, 1 µM BA or with vehicle (veh, DMSO), and then, 20 µM PGH2 was added (no dilution). After 1 min, PGE2 formation was analysed by RP-HPLC. Data are given as mean + SE, n = 3–4, ***P < 0.001 versus vehicle (DMSO) control. DMSO, dimethyl sulphoxide; RP-HPLC, reversed phase-high performance liquid chromatography.
To assess whether the inhibition of mPGES1 by BAs occurs in a reversible fashion, washout experiments were performed. Microsomal preparations of A549 cells were pre-incubated with BAs (10 µM, each), and MK-886 (3 µM) served as a control for a reversible mPGES1 inhibitor (Koeberle et al., 2008). Because all four BAs should act by a common mode, only the most active representatives (i.e. AKBA and β-BA) were tested. MK-886 at 3 µM or BAs at 10 µM efficiently blocked PGE2 formation (Figure 4B). Upon 10-fold dilution, a significant loss of potency was observed and the inhibition was comparable with the effect of BAs at 1 µM or MK-886 at 0.3 µM. Hence, BAs may inhibit mPGES1 in a reversible manner, as did MK-886.
Effects of BAs on prostanoid biosynthesis in intact cells
The treatment of A549 cells with IL-1β for 72 h results in the co-expression of COX-2 and mPGES1 (Thoren and Jakobsson, 2000), whereas COX-1 is essentially absent (Asano et al., 1996). We used this model to assess whether BAs inhibit PGE2 formation also in intact cells, and whether BAs selectively inhibit COX-2-derived PGH2 transformation to PGE2 without affecting the biosynthesis of other COX-2-derived prostanoids (i.e. 6-keto PGF1α). The formation of prostanoids was induced by the stimulation of A549 cells with 2.5 µM A23187 plus 1 µM AA and 3[H]AA (18.4 kBq). The use of A23187 and exogenous AA to induce prostanoid formation excludes effects of BAs on receptor-coupled signal transduction and/or on endogenous substrate supply for COX-2. In agreement with the effects in the cell-free mPGES1 activity assay, AKBA, KBA and β-BA (30 µM, each) significantly inhibited PGE2 synthesis to the same extent as MK-886 (30 µM), whereas Aβ-BA or α-amyrin were barely or not active (Figure 5A). More detailed analysis showed that AKBA, KBA and β-BA suppressed PGE2 synthesis in a concentration-dependent manner (IC50 approximately 20–30 µM, Figure 5B). Nevertheless, the suppression of PGE2 formation by BAs was not complete and 30 µM MK-886 also caused only 47% inhibition (Figure 5A). Importantly, the concomitant generation of 6-keto PGF1α was unaffected by all BAs, α-amyrin and by MK-886 (Figure 5A), whereas the COX-2 inhibitor celecoxib (5 µM) blocked the formation of both PGE2 and 6-keto PGF1α to the same extent, as expected. Also, celecoxib was much more efficient in the suppression of PGE2 formation, compared with BAs or MK-886 (Figure 5A).
Figure 5.
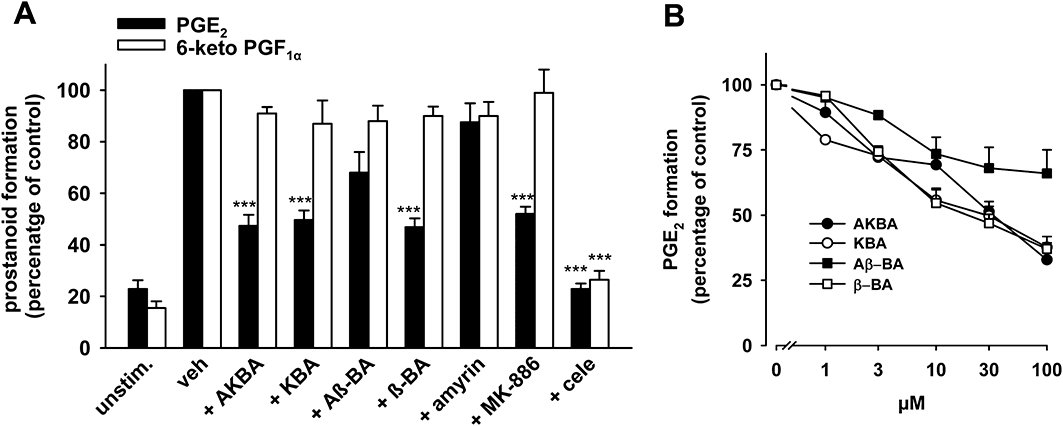
Effects of boswellic acids (BAs) on PGE2 and 6-keto PGF1α formation in intact A549 cells. IL-1β-treated A549 cells were pre-incubated (A) with vehicle (veh, DMSO), BAs (30 µM each), α-amyrin (30 µM), MK-886 (30 µM) or celecoxib (cele, 5 µM) (B) with BAs at the indicated concentrations. After 10 min at 37°C, 2.5 µM A23187 plus 1 µM AA and [3H]AA (18.4 kBq) were added (or left untreated = unstim.) and after another 15 min, formed [3H]PGE2 was analysed as described in the Methods. 6-Keto PGF1α was analysed using High Sensitivity EIA Kits; the 100% value corresponds to 87 ± 11 pg 106 cells. Data are given as mean + SE, n = 3–5. ***P < 0.001 versus vehicle (DMSO) control. AA; arachidonic acid; DMSO, dimethyl sulphoxide.
Effects of BAs on prostanoid formation in human whole blood
In order to estimate the efficacy of BAs to interfere with (COX-2/mPGES1-derived) PGE2 formation in a more complex biological test system, human whole blood assays were performed. Heparinized blood was pre-incubated with BAs for 10 min, prior to stimulation with LPS (10 µg·mL−1) for 5 h (Koeberle et al., 2008). β-BA significantly reduced PGE2 synthesis (46% inhibition) at 10 µM comparable with MK-886 at 30 µM (45% inhibition), whereas the other BAs and α-amyrin failed in this respect (Figure 6A). Concentration-response experiments revealed an IC50 value of 10 µM for β-BA (Figure 6B), but even at high concentrations (100 µM), about 40% PGE2 still remained. Indomethacin (50 µM) and celecoxib (20 µM) efficiently inhibited PGE2 formation in whole blood. In contrast, the concomitant formation of the COX-2-derived 6-keto PGF1α (Figure 6C) or TXB2 (Figure 6D) was not affected by β-BA or AKBA (and also not by Aβ-BA or KBA, not shown), implying that the suppressive effect on PGE2 synthesis is not related to reduced generation of the common precursor PGH2 (e.g. by inhibition of PLA2 or COX), but instead is due to select inhibition of PGH2 transformation to PGE2. Indomethacin and celecoxib efficiently suppressed the formation of all three prostanoids as expected. Moreover, β-BA (50 µM), in contrast to indomethacin, failed to affect COX-1-mediated generation of 12-HHT in whole blood stimulated with A23187 (Figure 6E).
Figure 6.
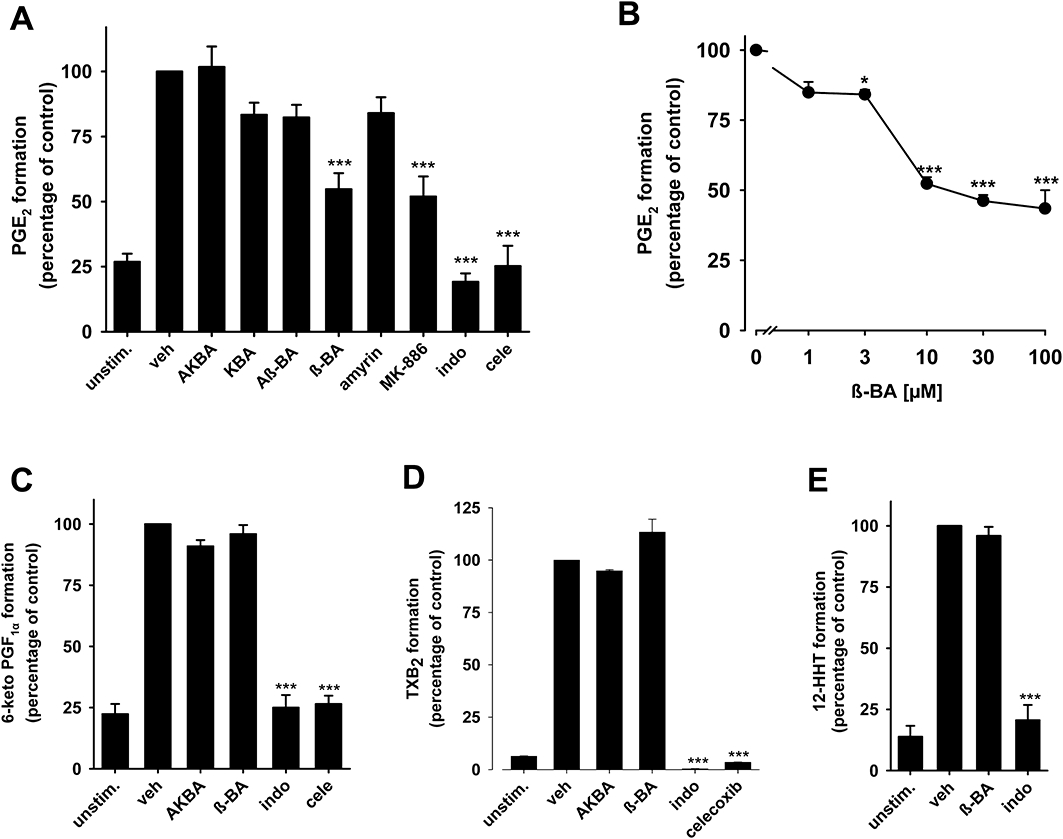
Effects of boswellic acids (BAs) on prostanoid biosynthesis in human whole blood. Heparinized human whole blood, treated with 1 µM CV4152 and 50 µM aspirin, was pre-incubated with (A) BAs and α-amyrin (10 µM, each) or vehicle (veh, DMSO) for 10 min at RT and then 10 µg·mL−1 LPS was added (or left untreated = unstim.). After 5 h at 37°C, PGE2 was separated by RP-HPLC and quantified by EIA. Controls: MK-886 (30 µM), indomethacin (indo, 50 µM), and celecoxib (cele, 20 µM). (B) Concentration-response of β-BA. (C,D) 6-keto PGF1α and TXB2 formation. 6-Keto PGF1α (C) was directly determined in blood plasma from samples above (see A) incubated with β-BA or AKBA (10 µM, each), indo (50 µM), cele (20 µM) or veh (DMSO). Inhibition of TXB2 formation (D) was assessed in heparinized human whole blood without CV4152 and aspirin. Both 6-keto PGF1α and TXB2 were measured by EIA. The 100% values corresponds to 221.8 ± 19.7 pg·mL−1 PGE2, 382.5 ± 22.3 pg·mL−1 6-keto PGF1α, and 37.9 ± 4.4 ng·mL−1 TXB2 respectively. (E) 12-HHT formation. Heparinized human blood was pre-incubated with β-BA (50 µM), indo (20 µM) or veh (DMSO) for 10 min, and A23187 (30 µM) was added (or left untreated = unstim.). After 10 min at 37°C, 12-HHT was analysed by HPLC. The 100% value corresponds to 148.8 ± 16.7 ng·mL−1 12-HHT. Data are given as mean + SE, n = 4–5; *P < 0.05; ***P < 0.001 versus vehicle (0.1% DMSO) control. β-BA, β-boswellic acid; AKBA, 3-O-acetyl-11-keto-β-boswellic acid; DMSO, dimethyl sulphoxide; RP-HPLC, reversed phase-high performance liquid chromatography.
Effects of BAs on carrageenan-induced rat pleurisy and mouse paw oedema
Although the anti-inflammatory efficacy of undefined mixtures of BAs in animal models are well documented (Poeckel and Werz, 2006; Singh et al., 2007), it is still unclear which of the BAs is responsible for the beneficial actions in vivo. Therefore, we assessed the effects of the four major β-configured BAs in the well-recognized carrageenan-induced rat pleurisy model. An injection of carrageenan into the pleural cavity of rats (DMSO 4% group) elicited an acute inflammatory response within 4 h characterized by the accumulation of exudate that contained large numbers of inflammatory cells (Figure 7A). β-BA (1 mg·kg−1) given i.p. 30 min prior to carrageenan potently inhibited the inflammatory response, as demonstrated by the significant attenuation of exudate formation (75%) and cell infiltration (64%), being as effective as the reference drug indomethacin (5 mg·kg−1, i.p.) (Figure 7A). Aβ-BA and KBA (1 mg·kg−1 i.p., each) were markedly less efficient and AKBA (1 mg·kg−1 i.p.) caused no significant effects. For β-BA, the PGE2 levels in the exudates were reduced by 51%, whereas the amount of 6-keto PGF1α was only slightly, and non-significantly, reduced. The other BAs hardly reduced PGE2 levels (<28%). Nevertheless, KBA and Aβ-BA significantly inhibited the generation of 6-keto PGF1α by 35 and 40%. Indomethacin almost completely suppressed formation of PGE2 as well as 6-keto PGF1α (Figure 7A).
Figure 7.
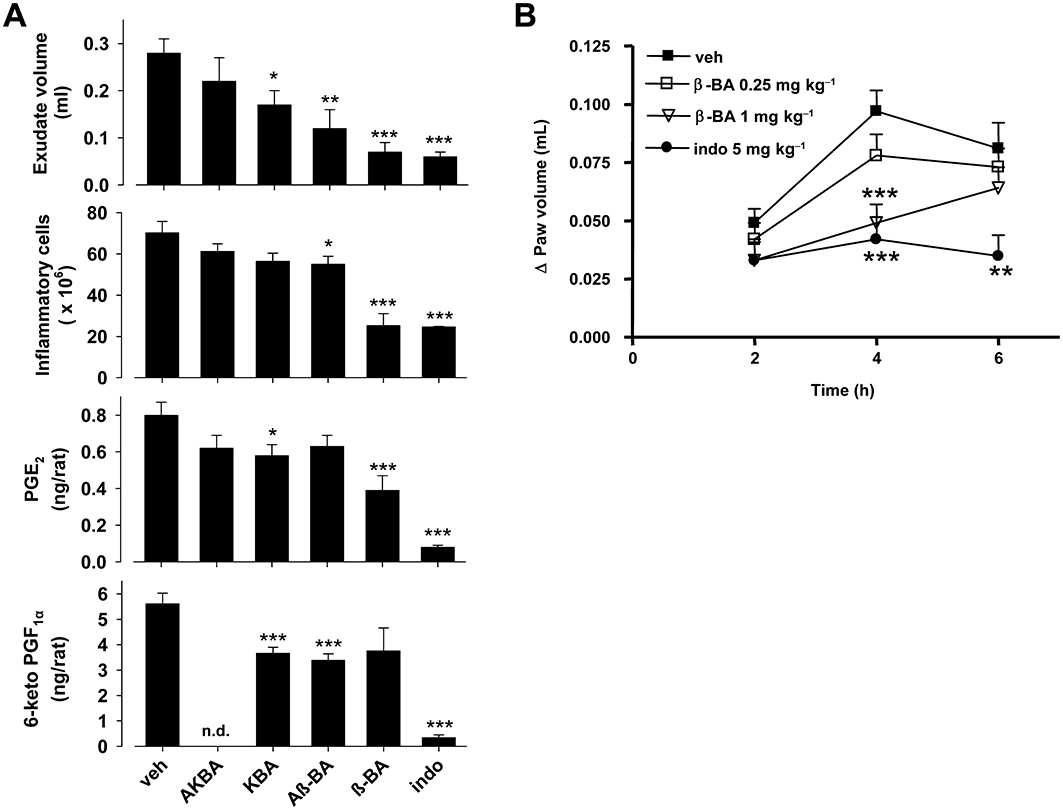
Effects of boswellic acids (BAs) in animal models in vivo. (A) Carrageenan-induced pleurisy in rats. Thirty minutes before intrapleural injection of carrageenan, rats (n = 10 for each experimental group) were treated i.p. with BAs (1 mg·kg−1 each), indomethacin (5 mg·kg−1) or vehicle (veh, DMSO 4%). Exudate volume, PGE2 and 6-keto PGF1α levels as well as inflammatory cell accumulation in pleural cavity were assessed 4 h after carrageenan injection. Data are expressed as mean ± SE, n = 10. *P < 0.05; **P < 0.01; ***P < 0.01 versus vehicle; n.d. = not determined. (B) Carrageenan-induced mouse paw oedema. Animals (n = 10 for each experimental group) were treated i.p. with 0.25 and 1 mg·kg−1β-BA, 5 mg·kg−1 indomethacin (indo) or veh (2% DMSO) 30 min before carrageenan subplantar injection. Data are given as mean + SE, *P < 0.05; **P < 0.01; ***P < 0.001 versus vehicle control. DMSO, dimethyl sulphoxide.
Next, β-BA was analysed for its anti-inflammatory efficacy in vivo after oral administration. As shown in Table 1, β-BA (1 mg·kg−1) caused potent inhibition of exudate formation and prevented infiltration of inflammatory cells, along with reduced levels of PGE2. The reference compound indomethacin (5 mg·kg−1), given p.o., completely prevented the formation of PGE2 associated with strong inhibition of exudate formation and cell infiltration (Table 1).
Table 1.
Effect of β-BA on carrageenan-induced pleurisy in rats
| Treatment | Exudate volume (mL) | Inflammatory cells × 106 | PGE2 (ng·rat−1) |
|---|---|---|---|
| Vehicle | 0.19 ± 0.020 | 47.1 ± 3.0 | 2.15 ± 0.20 |
| β-BA | 0.029 ± 0.016*** | 29.6 ± 2.8*** | 1.21 ± 0.14** |
| 1 mg·kg−1 | 85% | 37% | 44% |
| Indo | 0 | 17.0 ± 4.9** | n.d. |
| 5 mg·kg−1 | 100% | 64% | 100% |
P < 0.01;
P < 0.001 versus vehicle.
Thirty minutes before intrapleural injection of carrageenan, rats (n = 11 for each experimental group) were treated per os with 1 mg·kg−1β-BA, 5 mg·kg−1 indomethacin (indo) or vehicle (water containing 0.5% CMC, 10% Tween-20 and 1% sesame oil). Exudate volume and PGE2, as well as inflammatory cell accumulation in the pleural cavity were assessed 4 h after carrageenan injection. Data are expressed as mean ± SEM, n = 11.
β-BA, β-boswellic acid; n.d., not detectable, under the limit of detection of the assay (0.0625 ng·mL−1).
Finally, we explored the efficacy of β-BA in carrageenan-induced paw oedema, another rodent model of acute inflammation, to assess the pathophysiological role of mPGES1 in inflammation in vivo (Guay et al., 2004). The injection of carrageenan into the mouse paw produced a marked increase of paw volume, with a maximal effect after 4 h. Pretreatment (30 min) of mice with β-BA attenuated the inflammatory response at 4 h (Figure 7B). Thus, for mice treated (i.p.) with 0.25 and 1 mg·kg−1 of β-BA, the peak of the response to carrageenan at 4 h was reduced by 20 and 50% respectively. Indomethacin (5 mg·kg−1) caused 57% inhibition of the carrageenan response.
Discussion and conclusions
The modes of action and molecular targets of BAs and frankincense preparations are still incompletely understood. Most studies have focused on AKBA or KBA as the pharmacologically active principles and numerous targets (5-LOX, 12S-LOX, COX-1, HLE, IκB kinases and topoisomerases) have been proposed, but the interference with these targets in vivo has been largely neglected and thus, the pharmacological relevance is still unclear. Recently, we presented the identification of cathepsin G as a pharmacologically relevant target of all major BAs (Tausch et al., 2009). PGE2 is a key player in inflammation and pain, and mPGES1 is regarded as a potential target for the development of anti-inflammatory therapeutics (Samuelsson et al., 2007). Here, we present mPGES1 as a molecular target of BAs, and we provide evidence for a functional interaction of β-BA with mPGES1 in vivo which may contribute to its anti-inflammatory effectiveness.
The mPGES-1 bound to immobilized β-BA and KBA in a pull-down assay and SPR spectroscopy data confirmed a direct interaction. Pull-down assays using immobilized BAs were previously applied to demonstrate a direct interference of BAs with COX-1, 12S-LOX and cathepsin G (Poeckel et al., 2006b; Siemoneit et al., 2008; Tausch et al., 2009), supporting the suitability of this methodology for target identification. The SPR-based ligand-analyte studies revealed KD values of 5.2–23 µM for 3-O-oxaloyl-KBA, and these values fit the IC50 (5 µM) of 3-O-oxaloyl-KBA in the mPGES1 cell-free assay, suggesting specific binding to mPGES1 and hence a direct relation between enzyme binding and interference with mPGES1 activity.
AKBA, KBA and β-BA at low micromolar concentrations inhibited mPGES1-mediated PGE2 formation in cell-free assays, and also concentration dependently blocked PGE2 biosynthesis in intact A549 cells. Previous studies showed that BAs (up to 100 µM) hardly inhibited isolated COX-2 (Siemoneit et al., 2008) and all BAs (up to 30 µM) failed to reduce the formation of the COX-2-derived products 6-keto PGF1α and TXB2 in intact A549 cells and in human whole blood. Therefore, we conclude that impaired PGE2 biosynthesis is the result of the selective inhibition of the transformation of PGH2 to PGE2 by interference of BAs with mPGES1, rather than with COX-2 or other distal events (such as AA release). In particular β-BA, the major BA present in frankincense that reaches the highest plasma levels (up to 10.1 µM) among the BAs in treated humans (Buchele and Simmet, 2003; Tausch et al., 2009), suppressed PGE2 formation in human whole blood, again without significant reduction of 6-keto PGF1α or TXB2 levels. Interference with COX-1 was excluded as 12-HHT formation in whole blood was unaffected by β-BA, although BAs (in particular AKBA and KBA) may inhibit COX-1 in cell-free and cell-based models at higher concentrations (Siemoneit et al., 2008).
β-BA at a dose of 1 mg·kg−1 given i.p. or p.o. reduced the inflammatory reaction in two in vivo models of acute inflammation, carrageenan-induced mouse paw oedema and rat pleurisy, being about as effective as 5 mg·kg−1 indomethacin. During carrageenan-induced oedema formation, PGE2 levels are significantly elevated (Harada et al., 1982; Guay et al., 2004), and COX inhibitors prevent the inflammatory response (Gemmell et al., 1979). Results from studies using carrageenan-induced paw oedema and i.p. application of undefined mixtures of BAs concur with our data, although much higher doses (e.g. 125 mg·kg−1) were used in those studies (Singh et al., 2007). Whether or not BAs and related triterpenes present in frankincense extracts synergize or antagonize each other remains to be investigated. In the early phase of carrageenan-induced pleurisy, PGE2 plays a central role (Kawamura et al., 2000); and in fact, exudates from β-BA-treated rats showed markedly lower PGE2 levels. Thus, lowering PGE2 by inhibition of mPGES1 may contribute to the anti-inflammatory properties of β-BA. However, compared with indomethacin, β-BA was less potent in reducing PGE2 levels, but still efficiently suppressed exudate formation and infiltration of inflammatory cells. It is possible that other anti-inflammatory features of β-BA, such as inhibition of cathepsin G (Tausch et al., 2009), may contribute to the overall anti-inflammatory effects. The inhibition of cathepsin G may also explain the slight but still significant reduction of oedema formation and cell infiltration upon pretreatment with Aβ-BA, which was the least potent inhibitor of mPGES1 and failed to suppress PGE2 formation in whole blood. Similarly, interference with other pro-inflammatory components, such as cytokines and transcription factors (Syrovets et al., 2005; Kunnumakkara et al., 2009), may suppress COX-2 induction, explaining the reduced 6-keto PGF1α levels in the exudates of KBA- and Aβ-BA-treated rats.
Initially, the inhibition of 5-LOX activity and formation of leukotrienes (LTs) by BAs was proposed as an anti-inflammatory mechanism (Poeckel and Werz, 2006). Thus, AKBA and KBA blocked 5-LOX with IC50 values of 1.5–50 µM depending on the experimental settings (Safayhi et al., 1992; Siemoneit et al., 2009). However, in whole blood assays, AKBA and KBA failed to inhibit 5-LOX product synthesis, and the LTB4 plasma levels of human healthy volunteers treated with standard doses of frankincense were not affected (Siemoneit et al., 2009). Similarly, AKBA and KBA failed to suppress PGE2 formation in human whole blood and in rats, despite significant inhibition of mPGES1 in the cell-free assay. The failure of KBA and AKBA to suppress LT and PGE2 formation in vivo could be related to the marginal permeability of AKBA and moderate absorption of KBA (Kruger et al., 2009), resulting in poor bioavailability (Kruger et al., 2008) with fairly low plasma concentrations (0.3 and <0.1 µM) (Buchele and Simmet, 2003; Tausch et al., 2009). The marked loss of activity of AKBA in whole blood might be related to its strong plasma protein binding (Siemoneit et al., 2009). Together, the 11-keto moiety may hamper the cellular and biological availability of respective BAs in a physiological environment and thus compromise the overall anti-inflammatory effectiveness of AKBA in vivo. In contrast, β-BA (at 1 mg·kg−1) showed comparably high efficacy in the pleurisy model after p.o. or i.p. administration, suggesting clearly different and better bioavailability of this BA.
Our data favour a role of β-BA as the most relevant anti-inflammatory BA acting at least in part via inhibition of PGE2 formation. Pilot clinical studies indicated some therapeutic efficacy of frankincense, mainly in OA and RA (Ammon, 2006; Sengupta et al., 2008). Clinical studies using 5-LOX inhibitors (i.e. zileuton) and studies on 5-LOX- or 5-LOX-activating protein-deficient mice, exclude a prominent role of LTs in OA or RA (Werz and Steinhilber, 2006). However, PGE2 is a key mediator accounting for typical disease symptoms in OA and RA (Smith, 1989); and in patients suffering from RA or OA, a pivotal role for mPGES1 has been demonstrated (Westman et al., 2004; Li et al., 2005). Moreover, data from animal arthritis models support the relevance of mPGES1 to inflammatory joint diseases (Claveau et al., 2003; Trebino et al., 2003; Guay et al., 2004). Hence, the inhibition of PGE2 biosynthesis by β-BA may contribute to the beneficial effects of frankincense preparations observed in clinical studies and animal models of OA and RA (Ammon, 2006). Such speculations are favoured by the close correlation between steady-state plasma levels of β-BA (6.4–10.1 µM) in humans obtained after oral administration of frankincense preparations [containing 18.2% β-BA, 10.5% Aβ-BA, 6.1% KBA and 3.7% AKBA (Sterk et al., 2004)] in clinical trials (Buchele and Simmet, 2003; Tausch et al., 2009) and the effective concentrations of β-BA (≥3 µM) to suppress PGE2 synthesis in human whole blood.
The mPGES1 inhibitors might be alternatives to NSAIDs with reduced side effects (Koeberle and Werz, 2009). Less gastric and cardiovascular complications were evident in mPGES1-deficient mice, compared with COX-1- or COX-2-deficient mice or mice treated with NSAIDs (Cheng et al., 2006; Wang et al., 2006). In fact, the mPGES1 inhibitor MF63 relieved pyresis and inflammatory pain in animal models, without NSAID-like gastric toxicity (Xu et al., 2008). BAs showed gastric ulcer protective effects in different experimental models (Singh et al., 2008), and a 90-day placebo-controlled study supports evidence for the safety of Boswellia serrata extracts in OA patients (Sengupta et al., 2008).
In conclusion, we have shown that BAs directly and functionally interfered with mPGES1. The formation of PGE2 was selectively suppressed by β-BA in cell-based assays and in pleural exudates in vivo, and β-BA clearly exhibited anti-inflammatory effectiveness in the carrageenan-induced mouse paw oedema and rat pleurisy after i.p. or p.o. administration of fairly low doses. As the effective concentrations of β-BA, under physiologically relevant conditions, were in the range of β-BA levels in plasma of humans treated with frankincense, an interference with mPGES1 might represent a reasonable molecular mechanism contributing to some of the anti-inflammatory properties of frankincense extracts and rationalize its therapeutic use.
Acknowledgments
We thank Gertrud Kleefeld for expert technical assistance. We would like to acknowledge the financial support from Pharmasan GmbH (Freiburg, Germany), Medeon GmbH, Berlin, Aureliasan GmbH (Tuebingen, Germany) and the Deutsche Forschungsgemeinschaft.
Glossary
Abbreviations
- 5-LOX
5-lipoxygenase
- Aβ-BA
3-O-acetyl-β-boswellic acid
- AA
arachidonic acid
- AKBA
3-O-acetyl-11-keto-β-boswellic acid
- BA
boswellic acid
- COX
cyclooxygenase
- FCS
fetal calf serum
- GSH
glutathione
- 12-HHT
12(S)-hydroxy-5-cis-8,10-trans-heptadecatrienoic acid
- HLE
human leukocyte elastase
- IL-1β
interleukin-1β
- KBA
11-keto-β-boswellic acid
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- mPGES
microsomal prostaglandin E2 synthase
- NFκB
nuclear factor κB
- NSAID
non-steroidal anti-inflammatory drugs
- OA
osteoarthritis
- PBS
phosphate-buffered saline
- PG
prostaglandin
- RA
rheumatoid arthritis
- RU
resonance unit
- SPR
surface plasmon resonance
- TXB2
thromboxane B2
Conflicts of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammon HP. Boswellic acids in chronic inflammatory diseases. Planta Med. 2006;72:1100–1116. doi: 10.1055/s-2006-947227. [DOI] [PubMed] [Google Scholar]
- Asano K, Lilly CM, Drazen JM. Prostaglandin G/H synthase-2 is the constitutive and dominant isoform in cultured human lung epithelial cells. Am J Physiol. 1996;271:L126–L131. doi: 10.1152/ajplung.1996.271.1.L126. [DOI] [PubMed] [Google Scholar]
- Buchele B, Simmet T. Analysis of 12 different pentacyclic triterpenic acids from frankincense in human plasma by high-performance liquid chromatography and photodiode array detection. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;795:355–362. doi: 10.1016/s1570-0232(03)00555-5. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Wang M, Yu Y, Lawson J, Funk CD, Fitzgerald GA. Cyclooxygenases, microsomal prostaglandin E synthase-1, and cardiovascular function. J Clin Invest. 2006;116:1391–1399. doi: 10.1172/JCI27540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claveau D, Sirinyan M, Guay J, Gordon R, Chan CC, Bureau Y, et al. Microsomal prostaglandin E synthase-1 is a major terminal synthase that is selectively up-regulated during cyclooxygenase-2-dependent prostaglandin E2 production in the rat adjuvant-induced arthritis model. J Immunol. 2003;170:4738–4744. doi: 10.4049/jimmunol.170.9.4738. [DOI] [PubMed] [Google Scholar]
- Funk CD. Prostaglandins and leukotrienes: advances in eicosanoid biology. Science. 2001;294:1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- Gemmell DK, Cottney J, Lewis AJ. Comparative effects of drugs on four paw oedema models in the rat. Agents Actions. 1979;9:107–116. doi: 10.1007/BF02024141. [DOI] [PubMed] [Google Scholar]
- Giannetti AM, Koch BD, Browner MF. Surface plasmon resonance based assay for the detection and characterization of promiscuous inhibitors. J Med Chem. 2008;51:574–580. doi: 10.1021/jm700952v. [DOI] [PubMed] [Google Scholar]
- Guay J, Bateman K, Gordon R, Mancini J, Riendeau D. Carrageenan-induced paw edema in rat elicits a predominant prostaglandin E2 (PGE2) response in the central nervous system associated with the induction of microsomal PGE2 synthase-1. J Biol Chem. 2004;279:24866–24872. doi: 10.1074/jbc.M403106200. [DOI] [PubMed] [Google Scholar]
- Harada Y, Tanaka K, Uchida Y, Ueno A, Oh-Ishi S, Yamashita K, et al. Changes in the levels of prostaglandins and thromboxane and their roles in the accumulation of exudate in rat carrageenin-induced pleurisy – a profile analysis using gas chromatography-mass spectrometry. Prostaglandins. 1982;23:881–895. doi: 10.1016/0090-6980(82)90131-9. [DOI] [PubMed] [Google Scholar]
- Jakobsson PJ, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal, glutathione-dependent, inducible enzyme, constituting a potential novel drug target. Proc Natl Acad Sci USA. 1999;96:7220–7225. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauch J, Bergmann J. An efficient method for the large-scale preparation of 3-O-acetyl-11-oxo-beta-boswellic acid and other boswellic acids. Eur J Org Chem. 2003;24:4752–4756. [Google Scholar]
- Karlsson R, Falt A. Experimental design for kinetic analysis of protein-protein interactions with surface plasmon resonance biosensors. J Immunol Methods. 1997;200:121–133. doi: 10.1016/s0022-1759(96)00195-0. [DOI] [PubMed] [Google Scholar]
- Kawamura M, Hatanaka K, Saito M, Ogino M, Ono T, Ogino K, et al. Are the anti-inflammatory effects of dexamethasone responsible for inhibition of the induction of enzymes involved in prostanoid formation in rat carrageenin-induced pleurisy? Eur J Pharmacol. 2000;400:127–135. doi: 10.1016/s0014-2999(00)00377-0. [DOI] [PubMed] [Google Scholar]
- Koeberle A, Werz O. Inhibitors of the microsomal prostaglandin E(2) synthase-1 as alternative to non steroidal anti-inflammatory drugs (NSAIDs) – a critical review. Curr Med Chem. 2009;16:4274–4296. doi: 10.2174/092986709789578178. [DOI] [PubMed] [Google Scholar]
- Koeberle A, Siemoneit U, Buehring U, Northoff H, Laufer S, Albrecht W, et al. Licofelone suppresses prostaglandin E2 formation by interference with the inducible microsomal prostaglandin E2 synthase-1. J Pharmacol Exp Ther. 2008;326:975–982. doi: 10.1124/jpet.108.139444. [DOI] [PubMed] [Google Scholar]
- Koeberle A, Pollastro F, Northoff H, Werz O. Myrtucommulone, a natural acylphloroglucinol, inhibits microsomal prostaglandin E(2) synthase-1. Br J Pharmacol. 2009;156:952–961. doi: 10.1111/j.1476-5381.2009.00070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger P, Daneshfar R, Eckert GP, Klein J, Volmer DA, Bahr U, et al. Metabolism of boswellic acids in vitro and in vivo. Drug Metab Dispos. 2008;36:1135–1142. doi: 10.1124/dmd.107.018424. [DOI] [PubMed] [Google Scholar]
- Kruger P, Kanzer J, Hummel J, Fricker G, Schubert-Zsilavecz M, Abdel-Tawab M. Permeation of Boswellia extract in the Caco-2 model and possible interactions of its constituents KBA and AKBA with OATP1B3 and MRP2. Eur J Pharm Sci. 2009;36:275–284. doi: 10.1016/j.ejps.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Kunnumakkara AB, Nair AS, Sung B, Pandey MK, Aggarwal BB. Boswellic acid blocks signal transducers and activators of transcription 3 signaling, proliferation, and survival of multiple myeloma via the protein tyrosine phosphatase SHP-1. Mol Cancer Res. 2009;7:118–128. doi: 10.1158/1541-7786.MCR-08-0154. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Li X, Afif H, Cheng S, Martel-Pelletier J, Pelletier JP, Ranger P, et al. Expression and regulation of microsomal prostaglandin E synthase-1 in human osteoarthritic cartilage and chondrocytes. J Rheumatol. 2005;32:887–895. [PubMed] [Google Scholar]
- Murakami M, Nakatani Y, Tanioka T, Kudo I. Prostaglandin E synthase. Prostaglandins Other Lipid Mediat. 2002;68–69:383–399. doi: 10.1016/s0090-6980(02)00043-6. [DOI] [PubMed] [Google Scholar]
- Poeckel D, Werz O. Boswellic acids: biological actions and molecular targets. Curr Med Chem. 2006;13:3359–3369. doi: 10.2174/092986706779010333. [DOI] [PubMed] [Google Scholar]
- Poeckel D, Tausch L, George S, Jauch J, Werz O. 3-O-Acetyl-11-keto-boswellic acid decreases basal intracellular Ca2+ levels and inhibits agonist-induced Ca2+ mobilization and mitogen-activated protein kinase activation in human monocytic cells. J Pharmacol Exp Ther. 2006a;316:224–232. doi: 10.1124/jpet.105.089466. [DOI] [PubMed] [Google Scholar]
- Poeckel D, Tausch L, Kather N, Jauch J, Werz O. Boswellic acids stimulate arachidonic acid release and 12-lipoxygenase activity in human platelets independent of Ca2+ and differentially interact with platelet-type 12-lipoxygenase. Mol Pharmacol. 2006b;70:1071–1078. doi: 10.1124/mol.106.024836. [DOI] [PubMed] [Google Scholar]
- Roden LD, Myszka DG. Global analysis of a macromolecular interaction measured on BIAcore. Biochem Biophys Res Commun. 1996;225:1073–1077. doi: 10.1006/bbrc.1996.1297. [DOI] [PubMed] [Google Scholar]
- Safayhi H, Mack T, Sabieraj J, Anazodo MI, Subramanian LR, Ammon HP. Boswellic acids: novel, specific, nonredox inhibitors of 5-lipoxygenase. J Pharmacol Exp Ther. 1992;261:1143–1146. [PubMed] [Google Scholar]
- Safayhi H, Rall B, Sailer ER, Ammon HP. Inhibition by boswellic acids of human leukocyte elastase. J Pharmacol Exp Ther. 1997;281:460–463. [PubMed] [Google Scholar]
- Samuelsson B, Morgenstern R, Jakobsson PJ. Membrane prostaglandin E synthase-1: a novel therapeutic target. Pharmacol Rev. 2007;59:207–224. doi: 10.1124/pr.59.3.1. [DOI] [PubMed] [Google Scholar]
- Schwarz D, Klammt C, Koglin A, Lohr F, Schneider B, Dotsch V, et al. Preparative scale cell-free expression systems: new tools for the large scale preparation of integral membrane proteins for functional and structural studies. Methods. 2007;41:355–369. doi: 10.1016/j.ymeth.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Sengupta K, Alluri KV, Satish AR, Mishra S, Golakoti T, Sarma KV, et al. A double blind, randomized, placebo controlled study of the efficacy and safety of 5-Loxin for treatment of osteoarthritis of the knee. Arthritis Res Ther. 2008;10:R85. doi: 10.1186/ar2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemoneit U, Hofmann B, Kather N, Lamkemeyer T, Madlung J, Franke L, et al. Identification and functional analysis of cyclooxygenase-1 as a molecular target of boswellic acids. Biochem Pharmacol. 2008;75:503–513. doi: 10.1016/j.bcp.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Siemoneit U, Pergola C, Jazzar B, Northoff H, Skarke C, Jauch J, et al. On the interference of boswellic acids with 5-lipoxygenase: mechanistic studies in vitro and pharmacological relevance. Eur J Pharmacol. 2009;606:246–254. doi: 10.1016/j.ejphar.2009.01.044. [DOI] [PubMed] [Google Scholar]
- Singh S, Khajuria A, Taneja SC, Khajuria RK, Singh J, Qazi GN. Boswellic acids and glucosamine show synergistic effect in preclinical anti-inflammatory study in rats. Bioorg Med Chem Lett. 2007;17:3706–3711. doi: 10.1016/j.bmcl.2007.04.034. [DOI] [PubMed] [Google Scholar]
- Singh S, Khajuria A, Taneja SC, Khajuria RK, Singh J, Johri RK, et al. The gastric ulcer protective effect of boswellic acids, a leukotriene inhibitor from Boswellia serrata, in rats. Phytomedicine. 2008;15:408–415. doi: 10.1016/j.phymed.2008.02.017. [DOI] [PubMed] [Google Scholar]
- Smith WL. The eicosanoids and their biochemical mechanisms of action. Biochem J. 1989;259:315–324. doi: 10.1042/bj2590315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterk V, Buchele B, Simmet T. Effect of food intake on the bioavailability of boswellic acids from a herbal preparation in healthy volunteers. Planta Med. 2004;70:1155–1160. doi: 10.1055/s-2004-835844. [DOI] [PubMed] [Google Scholar]
- Syrovets T, Buchele B, Krauss C, Laumonnier Y, Simmet T. Acetyl-boswellic acids inhibit lipopolysaccharide-mediated TNF-alpha induction in monocytes by direct interaction with IkappaB kinases. J Immunol. 2005;174:498–506. doi: 10.4049/jimmunol.174.1.498. [DOI] [PubMed] [Google Scholar]
- Tausch L, Henkel A, Siemoneit U, Poeckel D, Kather N, Franke L, et al. Identification of human cathepsin G as a functional target of boswellic acids from the anti-inflammatory remedy frankincense. J Immunol. 2009;183:3433–3442. doi: 10.4049/jimmunol.0803574. [DOI] [PubMed] [Google Scholar]
- Thoren S, Jakobsson PJ. Coordinate up- and down-regulation of glutathione-dependent prostaglandin E synthase and cyclooxygenase-2 in A549 cells. Inhibition by NS-398 and leukotriene C4. Eur J Biochem. 2000;267:6428–6434. doi: 10.1046/j.1432-1327.2000.01735.x. [DOI] [PubMed] [Google Scholar]
- Trebino CE, Stock JL, Gibbons CP, Naiman BM, Wachtmann TS, Umland JP, et al. Impaired inflammatory and pain responses in mice lacking an inducible prostaglandin E synthase. Proc Natl Acad Sci USA. 2003;100:9044–9049. doi: 10.1073/pnas.1332766100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Zukas AM, Hui Y, Ricciotti E, Pure E, FitzGerald GA. Deletion of microsomal prostaglandin E synthase-1 augments prostacyclin and retards atherogenesis. Proc Natl Acad Sci USA. 2006;103:14507–14512. doi: 10.1073/pnas.0606586103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werz O, Steinhilber D. Therapeutic options for 5-lipoxygenase inhibitors. Pharmacol Ther. 2006;112:701–718. doi: 10.1016/j.pharmthera.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Westman M, Korotkova M, af Klint E, Stark A, Audoly LP, Klareskog L, et al. Expression of microsomal prostaglandin E synthase 1 in rheumatoid arthritis synovium. Arthritis Rheum. 2004;50:1774–1780. doi: 10.1002/art.20286. [DOI] [PubMed] [Google Scholar]
- Xu D, Rowland SE, Clark P, Giroux A, Cote B, Guiral S, et al. MF63 [2-(6-chloro-1H-phenanthro[9,10-d]imidazol-2-yl)-isophthalonitrile], a selective microsomal prostaglandin E synthase-1 inhibitor, relieves pyresis and pain in preclinical models of inflammation. J Pharmacol Exp Ther. 2008;326:754–763. doi: 10.1124/jpet.108.138776. [DOI] [PubMed] [Google Scholar]