

Abstract
BACKGROUND AND PURPOSE
An isothiourea derivative (2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl]isothiourea methane sulfonate (KB-R7943), a widely used inhibitor of the reverse Na+/Ca2+ exchanger (NCXrev), was instrumental in establishing the role of NCXrev in glutamate-induced Ca2+ deregulation in neurons. Here, the effects of KB-R7943 on N-methyl-D-aspartate (NMDA) receptors and mitochondrial complex I were tested.
EXPERIMENTAL APPROACH
Fluorescence microscopy, electrophysiological patch-clamp techniques and cellular respirometry with Seahorse XF24 analyzer were used with cultured hippocampal neurons; membrane potential imaging, respirometry and Ca2+ flux measurements were made in isolated rat brain mitochondria.
KEY RESULTS
KB-R7943 inhibited NCXrev with IC50= 5.7 ± 2.1 µM, blocked NMDAR-mediated ion currents, and inhibited NMDA-induced increase in cytosolic Ca2+ with IC50= 13.4 ± 3.6 µM but accelerated calcium deregulation and mitochondrial depolarization in glutamate-treated neurons. KB-R7943 depolarized mitochondria in a Ca2+-independent manner. Stimulation of NMDA receptors caused NAD(P)H oxidation that was coupled or uncoupled from ATP synthesis depending on the presence of Ca2+ in the bath solution. KB-R7943, or rotenone, increased NAD(P)H autofluorescence under resting conditions and suppressed NAD(P)H oxidation following glutamate application. KB-R7943 inhibited 2,4-dinitrophenol-stimulated respiration of cultured neurons with IC50= 11.4 ± 2.4 µM. With isolated brain mitochondria, KB-R7943 inhibited respiration, depolarized organelles and suppressed Ca2+ uptake when mitochondria oxidized complex I substrates but was ineffective when mitochondria were supplied with succinate, a complex II substrate.
CONCLUSIONS AND IMPLICATIONS
KB-R7943, in addition to NCXrev, blocked NMDA receptors in cultured hippocampal neurons and inhibited complex I in the mitochondrial respiratory chain. These findings are critical for the correct interpretation of experimental results obtained with KB-R7943 and a better understanding of its neuroprotective action.
Keywords: glutamate, excitotoxicity, calcium deregulation, cultured hippocampal neurons, Na+/Ca2+ exchanger, NMDA receptor, mitochondria, mitochondrial complex I
Introduction
In neurons exposed to glutamate, prolonged stimulation of glutamate receptors, particularly the N-methyl-D-aspartate (NMDA) subtype, results in massive Ca2+ influx into the cell, disruption of calcium homeostasis and eventual cell death (Choi, 1988; Manev et al. 1989). The plasmalemmal Na+/Ca2+ exchanger (NCX; channel and receptor nomenclature follows Alexander et al., 2009) operating in the forward mode plays an important role in lowering cytosolic Ca2+ concentration ([Ca2+]c) by exchanging one cytosolic Ca2+ for three external Na+ (Blaustein and Lederer, 1999). Predictably, inhibition of NCX forward mode by removing external Na+ leads to increased [Ca2+]c (Mattson et al., 1989). However, under conditions of prolonged exposure to glutamate or NMDA, when cytosolic Na+ is elevated and the plasma membrane is depolarized (Mayer and Westbrook, 1987), NCX can switch from forward to reverse mode bringing external Ca2+ into the cytosol (Kiedrowski et al., 1994; Hoyt et al. 1998). This may exacerbate disturbances of Ca2+ homeostasis caused by excessive stimulation of glutamate receptors. Therefore, inhibition of NCX operating in the reverse mode (NCXrev) represents an attractive target for pharmacological intervention aimed at protecting neurons against Ca2+ deregulation and glutamate/NMDA excitotoxicity.
An isothiourea derivative 2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl]isothiourea methanesulfonate (KB-R7943), was introduced in 1996 as a selective inhibitor of NCX isoform 1 (NCX1) operating in the reverse mode (Iwamoto et al., 1996). Later, it was demonstrated that KB-R7943 can also inhibit NCX isoforms 2 (NCX2) and 3 (NCX3) with higher affinity to NCX3 (see (Amran et al., 2003). Since its introduction, KB-R7943 remains the most widely used inhibitor of NCXrev. KB-R7943 antagonized elevations in [Ca2+]c and reduced neuronal death following ischemic episodes, mechanical brain trauma, oxygen/glucose deprivation and glutamate excitotoxicity (Schroder et al., 1999; Breder et al. 2000; Li et al., 2000; MacGregor et al. 2003; Luo et al., 2007). However, the exact mechanisms of neuroprotection evoked by KB-R7943 are not completely understood. This is because in addition to inhibiting NCXrev, KB-R7943 has several important effects on other targets. KB-R7943 inhibits L-type voltage-gated Ca2+ channels in dorsal column slices (Ouardouz et al., 2005) and store-operated Ca2+ influx in cultured neurons and astrocytes (Arakawa et al., 2000). In permeabilized HeLa cells, KB-R7943 was shown to inhibit mitochondrial Ca2+ uptake (Santo-Domingo et al., 2007). KB-R7943 also blocked transient receptor potential (TRP) channels expressed in HEK-293 cells (Kraft, 2007). In addition, some investigators reported that KB-R7943 blocks NMDA receptors (Sobolevsky and Khodorov, 1999), while other investigators did not find compelling evidence for that action (Hoyt et al., 1998; Czyz et al., 2002).
In a previous paper, we reported that KB-R7943-induced neuroprotection against glutamate excitotoxicity, at least in part, could be mediated by mild mitochondrial depolarization (Storozhevykh et al., 2010). Our findings are consistent with an early report indicating the protective role of mitochondrial depolarization against glutamate excitotoxicity (Stout et al., 1998). However, the mechanisms of KB-R7943-evoked mitochondrial depolarization remained unclear. In the present paper, we demonstrate for the first time that. in addition to inhibiting NCXrev, KB-R7943 inhibits complex I in the mitochondrial respiratory chain. This complex I inhibition with KB-R7943 suppresses mitochondrial respiration, sensitizes mitochondrial membrane potential (Δψ) to ion fluxes across the inner mitochondrial membrane, and limits Ca2+ uptake by mitochondria. In addition, we have found that KB-R7943 does block NMDA receptors. Thus, our data significantly broaden our understanding of the mechanisms of KB-R7943 action and will help the correct interpretation of results obtained with this widely used pharmacological agent.
Methods
Cell culturing
All animal care and experimental procedures complied with and were approved by the Institutional Animal Care and Use Committee (IACUC). Primary cultures of hippocampal neurons were prepared from postnatal day 1 rat pups as previously described (Dubinsky, 1993). For fluorescence measurements, neurons were plated on glass-bottomed Petri dishes without preplated glia as previously described (Dubinsky, 1993). For all platings, 35 µg·mL−1 uridine plus 15 µg·mL−1 5-fluoro-2′-deoxyuridine were added 24 h after plating to inhibit proliferation of non-neuronal cells. Neuronal cultures were maintained in a 5% CO2 atmosphere at 37°C in Earl's MEM supplemented with 10% NuSerum (BD Bioscience, Bedford, MA), 27 mM glucose and 26 mM NaHCO3 (Dubinsky et al., 1995).
Fluorescence imaging of cultured neurons
In these and other experiments, rat-cultured hippocampal neurons grown for 10–12 days in vitro (10–12 DIV) were used. For the imaging experiments, neurons were co-loaded at 37°C with 2.6 µM Fura-2FF-AM and 1.7 µM Rhodamine 123. Then, neurons were rinsed twice with a standard bath solution containing 139 mM NaCl, 3 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 10 mM NaHEPES, pH 7.4, 5 mM glucose and 65 mM sucrose. Fluorescence imaging was performed as described previously (Brustovetsky et al., 2009) with an inverted microscope Nikon Eclipse TE2000-S using Nikon objective Plan Fluor 20× 0.45 NA and back-thinned EM-CCD camera Hamamatsu C9100-12 (Hamamatsu Photonic Systems, Bridgewater, NJ, USA) controlled by Simple PCI software 6.1 (Compix Inc., Sewickley, PA, USA).
NAD(P)H fluorescence measurements in cultured hippocampal neurons
NAD(P)H autofluorescence was followed using the excitation light with maximum at 360 ± 20 nm and was delivered by a Lambda-LS system (Sutter Instruments, Novato, CA, USA). Fluorescence was recorded through a 400 nm dichroic mirror at 460 ± 25 nm using Nikon objective Super Fluor 40× 1.3 NA and back-thinned EM-CCD camera Hamamatsu C9100-12 (Hamamatsu Photonic Systems). The NAD(P)H fluorescence traces were constructed after subtracting the background and expressed as F/F0.
Isolation and purification of brain mitochondria
Brain mitochondria were isolated in mannitol-sucrose medium and purified on a discontinuous Percoll gradient as described previously (Brustovetsky et al., 2002). Mitochondrial protein was determined by the Bradford method (Bradford, 1976) using bovine serum albumin as a standard.
Measurements of mitochondrial respiration and Ca2+ uptake
The mitochondrial respiratory rates were measured as described previously (Li et al., 2008) at 37°C under continuous stirring in the closed 0.3 mL thermostated chamber using a Clark-type oxygen electrode in the standard incubation medium containing 125 mM KCl, 10 mM HEPES, pH 7.4, 0.5 mM MgCl2, 3 mM KH2PO4, 10 µM EGTA, supplemented with 3 mM pyruvate and 1 mM malate or with 3 mM succinate plus 3 mM glutamate. Here and in other experiments, glutamate was used to prevent oxaloacetate inhibition of succinate dehydrogenase (Lehninger et al., 1993). Mitochondrial Ca2+ uptake was measured as described previously (Storozhevykh et al., 2010) with a miniature Ca2+-selective electrode in a 0.3 mL chamber at 37°C under continuous stirring. A decrease in the external Ca2+ concentration indicated mitochondrial Ca2+ uptake. The incubation medium was supplemented with 0.1% bovine serum albumin (BSA, free from fatty acids), 0.1 mM ADP, and 1 µM oligomycin. In all experiments with isolated mitochondria, the concentration of mitochondrial protein in the chamber was 0.2 mg·mL−1. All experimental traces shown are representative of at least three separate experiments.
Experiments with individual mitochondria loaded with Rhodamine-123
Membrane potential of individual brain non-synaptic mitochondria was monitored following fluorescence of Rhodamine-123 loaded into mitochondria as described previously (Shalbuyeva et al., 2007). Briefly, isolated mitochondria were placed into the glass-bottomed 35 mm Petri dish and continuously perfused with a standard incubation medium supplemented with 0.2 µM Rhodamine 123. The standard incubation medium was supplemented with either 3 mM glutamate and 1 mM malate or with 3 mM succinate and 3 mM glutamate as indicated. A Nikon Eclipse TE2000-S inverted microscope equipped with objective Nikon CFI Plan Apo VC 100× 1.4 NA and CCD Hamamatsu C9100-12 camera (Hamamatsu, Photonic Systems) controlled by SimplePCI 6.1 software (Compix, Sewickley, PA, USA) were used to visualize individual mitochondria loaded with Rhodamine 123. In all experiments, mitochondria were perfused using a ValveBank 8 perfusion system (AutoMate Scientific, San Francisco, CA, USA).
Electrophysiological patch-clamp experiments
Whole-cell patch-clamp recordings were conducted at room temperature using a HEKA EPC-10 double amplifier. Data were acquired using the Pulse program (HEKA Electronic, Lambrecht/Pfalz, Germany). Fire-polished electrodes (0.9–1.3 MΩ) were fabricated from 1.7 mm capillary glass (VMR Scientific, West Chester, PA) using a Sutter P-97 puller (Sutter Instruments). When examining NMDA-induced currents, the composition of the electrode (‘intracellular’) solution was as follows: 140 mM CsF, 10 mM NaCl, 1.1 mM EGTA, and 10 mM HEPES, pH 7.3 adjusted with CsOH. The external solution was the same as in the fluorescence imaging experiments, except Mg2+ was omitted. The NCX-mediated ion currents were recorded using voltage ramp protocol as described previously (Convery and Hancox, 1999). The composition of the electrode solution used for recording voltage ramp currents mediated by NCX was as follows: 20 mM KCl, 100 mM K-aspartate, 20 mM tetraethylammonium-Cl, 10 mM HEPES, 0.01 mM K-EGTA, 4.5 mM MgCl2 and 4 mM Na-ATP, pH 7.3 adjusted with KOH (Smith et al., 2006). The external solution used for recording ramp currents was as follows: 129 mM NaCl, 10 mM CsCl (to block K+ channels), 3 mM KCl, 0.8 mM MgCl2, 1.8 mM CaCl2, 5 mM glucose, 10 mM Na-HEPES, pH 7.2, 65 mM sucrose, 0.01 mM nifedipine (to block voltage-gated Ca2+ channels), 0.02 mM ouabain and 0.001 mM TTX (to block Na+ channels). Coverslips containing hippocampal neurons were placed into an RC-26 Open Diamond Bath perfusion chamber (Warner Instruments, LLC, Hamden, CT, USA). The bath solution was removed using a Masterflex® C/L® Single Channel Variable Speed peristaltic pump (Cole-Parmer Instrument Company, Vernon Hills, IL, USA). An SF-77B Perfusion Fast-Step system (Warner Instruments) was used to deliver drugs focally onto isolated hippocampal neurons in whole-cell configuration. The drug delivery three-barrel tube was placed near the neuron, and its positioning was calibrated before each experiment. The position of the drug delivery tube was controlled manually by a stepper motor, which could change the positioning in about 20 milliseconds.
Cellular respirometry
Oxygen consumption rate (OCR) of cultured hippocampal neurons (10 DIV) was measured using Seahorse XF24 analyzer (Seahorse Bioscience, Billerica, MA, USA) following the manufacturer's instructions. Neurons were grown in the assay plates at 5 × 105 cells per well. Before measuring, the growth medium was replaced by the standard bath solution supplemented with 10 mM glucose and 15 mM pyruvate.
Data analysis
Data are shown as mean ± SEM of at least three independent experiments. Statistical analysis of the experimental results consisted of unpaired t-test or one-way ANOVA followed by Bonferroni's post hoc test (GraphPad Prism® 4.0, GraphPad Software Inc., San Diego, CA, USA). Every experiment was performed using at least three different preparations of isolated mitochondria or three separate neuronal platings.
Materials
Glutamate, glycine, EGTA, rotenone, gramicidin, and carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP) were purchased from Sigma (St. Louis, MO, USA). Bovine serum albumin (BSA), free from free fatty acids, was from MP Biomedicals (Irvine, CA, USA). Fura-2FF AM was bought from Teflabs (Austin, TX, USA) and Rhodamine-123 (FluoroPure™ grade) was purchased from Invitrogen (Carlsbad, CA, USA). KB-R7943 was from Tocris (Ellisville, MO, USA). KB-R7943 solutions were prepared freshly for each day's experiments.
Results
In our experiments, KB-R7943 (15µM) failed to protect cultured hippocampal neurons against delayed Ca2+ deregulation (Figure 1 and Supporting Figure S1). Moreover, KB-R7943 hastened delayed Ca2+ deregulation and mitochondrial depolarization induced by 25 µM glutamate (plus 10 µM glycine) and completely prevented a recovery of [Ca2+]c after glutamate removal (Figure 1B). Here and in other experiments, 100% of neurons responded to KB-R7943 by accelerating the delayed Ca2+ deregulation induced by 25 µM glutamate and by depolarizing mitochondria. Recently, we introduced a parameter: the time from the beginning of glutamate exposure to completion of the delayed Ca2+ deregulation (tDCD) to provide a statistical analysis of delayed Ca2+ deregulation (Brustovetsky et al., 2009; Li et al. 2009). The statistical analysis revealed that KB-R7943 significantly accelerated the onset of delayed Ca2+ deregulation in cultured neurons exposed to 25 µM glutamate (Figure 1): tDCD without KB-R7943 was 817 ± 27 s while tDCD with 15 µM KB-R7943 was 398 ± 38 s (mean ± SEM, P < 0.01, unpaired t-test, five independent experiments, 113 neurons total in the control with vehicle, 0.3% DMSO, and 108 neurons with KB-R7943 were analyzed). In contrast to KB-R7943, MK801 (10 µM), an inhibitor of the NMDA receptor, completely inhibited glutamate-induced calcium deregulation, whereas CNQX (20 µM), an inhibitor of AMPA/kainate receptor, was essentially without effect (Supporting Figure S2).
Figure 1.
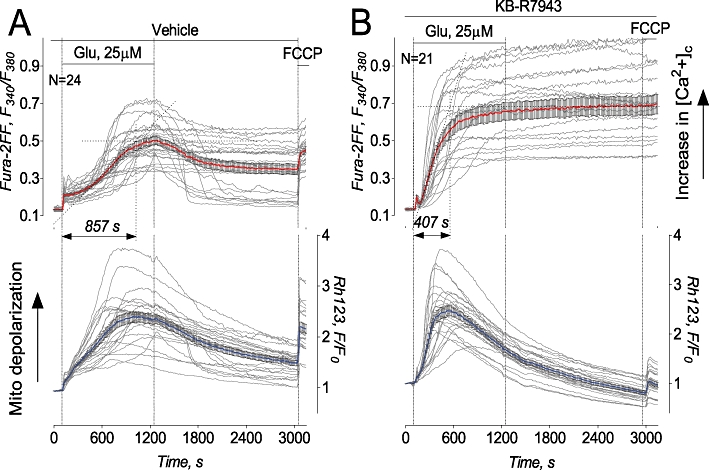
KB-R7943 accelerated delayed Ca2+ deregulation and mitochondrial depolarization in cultured hippocampal neurons exposed to glutamate. Here and in all other similar illustrations, thin, grey traces show signals from individual neurons while thick, red traces show averaged signals (mean ± SEM) for Fura-2FF ratio F340/F380 (upper panels) and thick, blue traces for Rhodamine 123 (Rh123) F/F0 (lower panels), respectively. Neurons were derived from postnatal day one (PN1) Sprague-Dawley rat pups and were 10–12 days in vitro on the day of the experiment. In A and B, neurons were treated with 25 µM glutamate (Glu). In both cases, glutamate was applied in combination with 10 µM glycine. In A, 0.3% DMSO was applied to neurons as a vehicle. In B, 15 µM KB-R7943 was applied to neurons as indicated. Here and in other similar experiments, 1 µM FCCP was applied at the end of experiments to completely depolarize mitochondria in the Ca2+-free bath solution. Here and in other similar illustrations, N is the number of neurons examined in each individual representative experiment.
Despite failing to prevent delayed Ca2+ deregulation in our experiments, KB-R7943 (15 µM) inhibited an increase in [Ca2+]c induced by gramicidin, an ionophore for monovalent cations which does not transport Ca2+ on its own (Figure 2A,C). Gramicidin collapses the Na+ gradient across the plasma membrane and depolarizes cells, resulting in a reversal of NCX (Czyz and Kiedrowski, 2002). The exact cause of variations in the delay of gramicidin-induced calcium deregulation evident in individual neurons treated with KB-R7943 (Figure 2C) is unknown, but it might be due to different expressions of various proteins involved in calcium signalling and, correspondingly, due to different resistances of individual cells to Ca2+ overload. Replacement of Na+ for K+ in the bath solution prevented the gramicidin-induced increase in [Ca2+]c indicating that this increase depended on Na+ and could not be induced solely by plasma membrane depolarization (Figure 2B). Using the calcium imaging protocol, we determined an IC50value of 5.7 ± 2.1 µM (mean ± SEM, N= 3 independent experiments) for KB-R7943-induced inhibition of NCXrev (Figure 2D,E).
Figure 2.
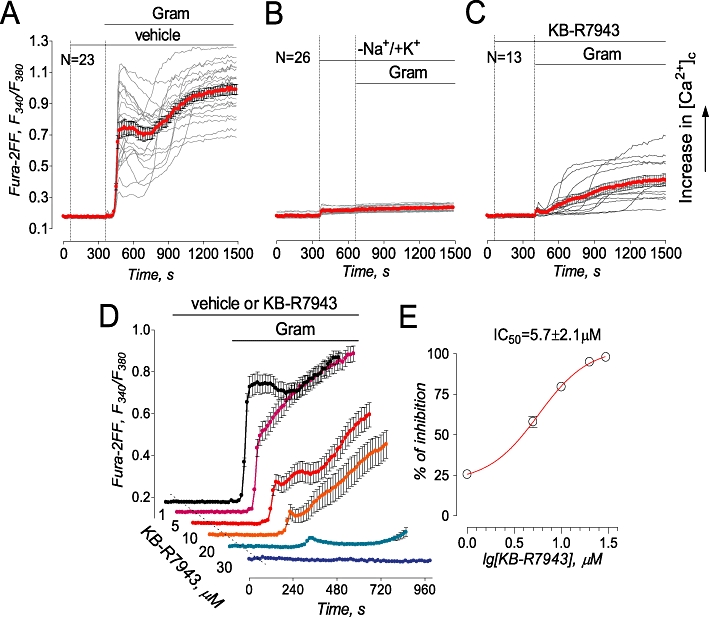
KB-R7943 inhibited gramicidin-induced increase in cytosolic Ca2+ concentration ([Ca2+]c) in cultured hippocampal neurons (A–C). In A, 0.3% DMSO was applied to neurons as a vehicle. In A–C, 5 µM gramicidin (Gram) and 15 µM KB-R7943 were applied as indicated. In B, where indicated, 139 mM NaCl in the bath solution was replaced by 139 mM KCl. In A–C, bath solution was supplemented with 5 µM nifedipine, a blocker of voltage-gated Ca2+ channels, and 1 mM ouabain, an inhibitor of the Na+/K+-ATPase. In A–C, each experiment was performed in triplicate with the total number of examined neurons 51 (A), 56 (B), and 41 (C). In D, traces are averages ± SEM from individual experiments (N= 18–24 neurons per experiment) performed in triplicate. Where indicated, vehicle (veh, 0.3% DMSO, black trace) or various concentrations of KB-R7943 (1–30 µM) were applied and present in the bath solution until the end of the experiment. Gramicidin (Gram, 5 µM) was applied as indicated. The activity of NCXrev was evaluated by measuring the area under the curve for 90 s following the onset of [Ca2+]c elevation, the dose-dependence graph was plotted and IC50 was calculated using GraphPad Prism® 4.0 (GraphPad Software Inc., San Diego, CA, USA).
In addition to calcium imaging, we used the electrophysiological patch-clamp technique to evaluate the effect of KB-R7943 on NCXrev activity in cultured hippocampal neurons. Figure 3 shows measurements of whole-cell outward ion currents obtained in response to repeated application of the voltage ramp protocol shown in Figure 3A. KB-R7943 (15 µM) inhibited ion currents produced in response to the voltage ramp protocol (Figure 3B,C). Previously, it has been shown that this type of ion current is mediated by NCX in forward mode at negative voltages (i.e. −120 mV) and by NCX in reverse mode (NCXrev) at positive voltages (i.e. +80 mV) (Convery and Hancox, 1999; Watanabe and Kimura, 2000). For comparison, we evaluated Ni2+-sensitivitiy of the ramp currents (Figure 3D), which has been previously attributed to NCX activity (Smith et al., 2006; Reppel et al. 2007). Memantine (10 µM), an NMDA receptor inhibitor (Chen et al., 1992), did not affect the ramp currents (not shown). Overall, these results suggested that KB-R7943 inhibited NCXrev in cultured hippocampal neurons.
Figure 3.
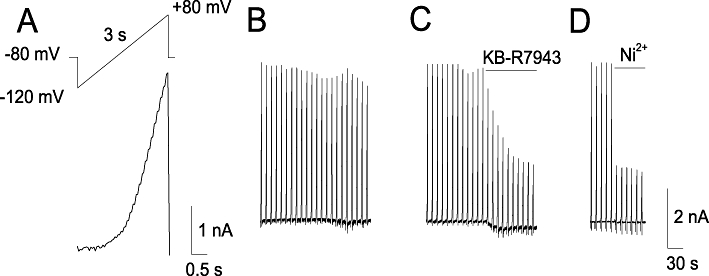
NCXrev currents obtained with cultured rat hippocampal neurons. In A, the ascending voltage ramp protocol used in these experiments and associated currents obtained with and without KB-R7943 (B, C) and Ni2+ (D). Where indicated, KB-R7943, 15 µM, or Ni2+, 5 mM were applied. For further details, see Materials and Methods.
In addition to NCXrev, KB-R7943 dose-dependently and reversibly blocked ion currents elicited by NMDA (Figure 4A,B). As a positive control, MK801, an inhibitor of NMDA receptors (Clifford et al., 1989), completely blocked NMDA-induced ion current (Figure 4C). In addition, for the first time, we demonstrated that KB-R7943 dose-dependently inhibited NMDA-induced increases in [Ca2+]c with IC50= 13.4 ± 3.6 µM (mean ± SEM, N= 3 independent experiments) (Figure 5) confirming the inhibition of NMDA receptors observed in electrophysiological experiments (Figure 4A,B). Thus, our experiments showed that KB-R7943 inhibited NMDA receptors in addition to inhibiting NCXrev.
Figure 4.
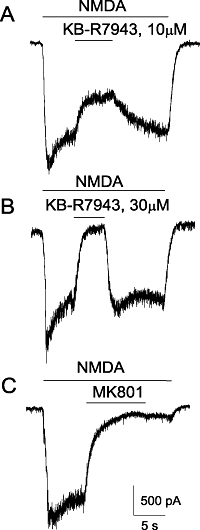
Electrophysiological patch-clamp recordings of NMDA-induced whole-cell currents obtained with rat-cultured hippocampal neurons. The effects of 10 or 30 µM KB-R7943 (A, B), and 20 µM MK801 (C). In all experiments, 30 µM NMDA (plus 10 µM glycine) were applied as indicated. Holding voltage was −100 mV. For further details, see Materials and Methods.
Figure 5.
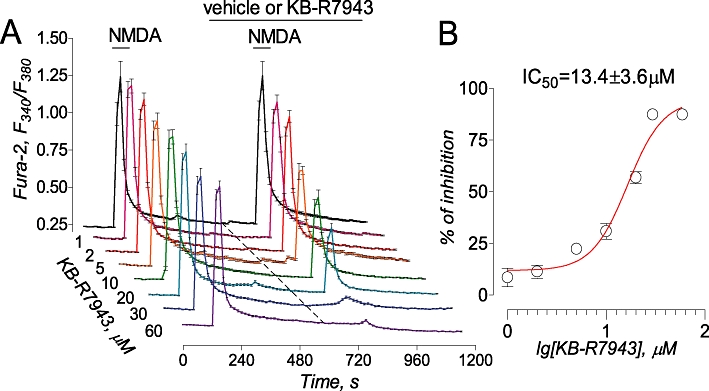
KB-R7943 inhibited NMDA-induced increases in cytosolic Ca2+. Traces are averages ± SEM from individual experiments (N= 19–26 neurons per experiment) performed in triplicate. Where indicated, vehicle (veh, 0.3% DMSO, black trace) or various concentrations of KB-R7943 (2–60 µM) were applied and present in the bath solution until the end of the experiment. NMDA (30 µM, plus 10 µM glycine) was applied twice for 30 s as indicated. The activity of NMDA receptors was evaluated by measuring amplitude of the increases in Fura-2 F340/F380 ratio triggered by the second application of NMDA. The concentration-response graph was plotted, and IC50 was calculated using GraphPad Prism® 4.0 (GraphPad Software Inc., San Diego, CA, USA).
In our previous paper, we demonstrated that KB-R7943 depolarized mitochondria (Storozhevykh et al., 2010). Mitochondrial depolarization inhibits Ca2+ uptake by these organelles (Bernardi, 1999) and strongly contributes to collapse of calcium homeostasis in cultured neurons (Pivovarova et al., 2004). Therefore, it is possible that mitochondrial depolarization produced by KB-R7943 could interfere with a protective effect of this drug on [Ca2+]c. The mechanism of KB-R7943-induced mitochondrial depolarization demonstrated in our previous paper (Storozhevykh et al., 2010) remained unclear. In the present study, we hypothesized that KB-R7943 depolarizes mitochondria by inhibiting electron flow in the mitochondrial respiratory chain. To test this hypothesis, we examined autofluorescence of NAD(P)H in cultured hippocampal neurons exposed to glutamate with or without KB-R7943. In the presence of external Ca2+ (1.8 mM), glutamate produced a gradual decrease in NAD(P)H fluorescence, reflecting NAD(P)H oxidation and conversion into NAD(P)+, which does not fluoresce (Chance and Williams, 1956) (Figure 6A). This NAD(P)H oxidation could be due to accelerated electron flow in the respiratory chain associated with the increased Δψ dissipation in response to augmented ATP synthesis.
Figure 6.
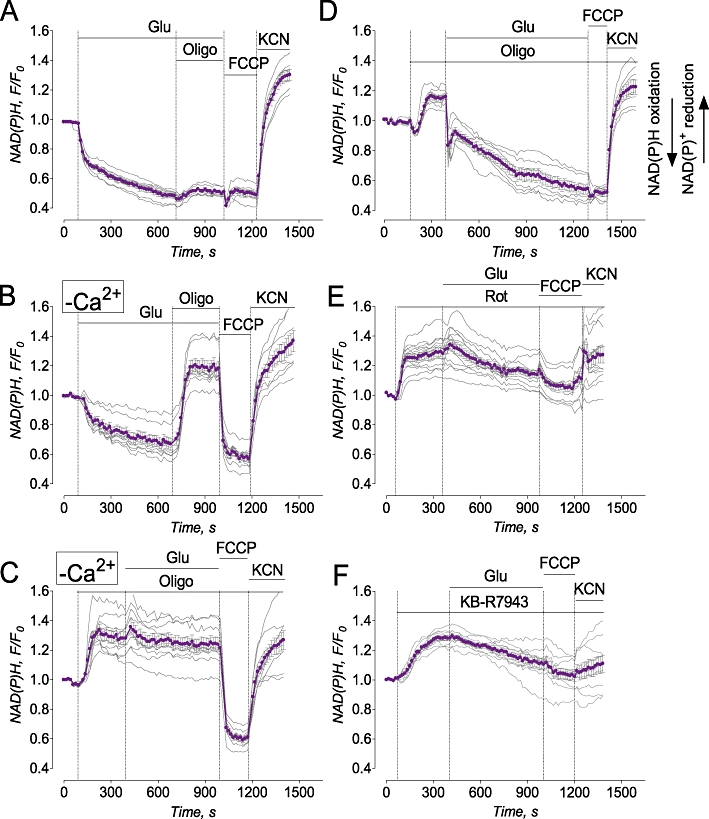
Glutamate evoked NAD(P)H oxidation which was either coupled (in the absence of external Ca2+) or uncoupled (with external Ca2+) from ATP synthesis. KB-R7943 increased NAD(P)H fluorescence under resting conditions and suppressed glutamate-induced NAD(P)H oxidation similar to rotenone. In A and D–F, bath solution was with 1.8 mM Ca2+; in B and C, bath solution was nominally Ca2+-free. Where indicated, 25 µM glutamate (Glu, plus 10 µM glycine), 1 µM oligomycin (Oligo), 1 µM rotenone, 1 µM FCCP, or 10 mM KCN were applied to neurons. In F, 15 µM KB-R7943 was applied as indicated. Thin, grey traces show signals from individual neurons from the same dish while thick, purple traces show averaged signals (mean ± SEM) for NAD(P)H F/F0. In A-F, each experiment was performed five times. The total number of examined neurons was 38 (A), 37 (B), 40 (C), 44 (D), 38 (E), and 45 (F).
Alternatively, NAD(P)H oxidation could be due to accelerated electron flow resulting from the uncoupling of respiration and ATP synthesis in mitochondria. Oligomycin (1 µM), an inhibitor of the mitochondrial ATP synthase, failed to restore NAD(P)H levels, suggesting that the decrease in NAD(P)H was not due to augmentation of ATP synthesis (Figure 6A). FCCP, a protonophore that depolarized mitochondria and, hence, accelerated electron flow in the respiratory chain, failed to influence NAD(P)H, suggesting that the NAD(P)H pool already had been maximally oxidized (Figure 6A). On the other hand, KCN, an inhibitor of cytochrome oxidase, stopped electron flow in the entire respiratory chain, leading to recovery of NAD(P)H (Figure 6A). Omitting Ca2+ from the bath solution did not prevent oxidation of NAD(P)H induced by glutamate (Figure 6B). However, in this case, oligomycin produced a strong increase in NAD(P)H, indicating that oxidation and ATP synthesis were tightly coupled and mitochondria were healthy. FCCP strongly decreased NAD(P)H, whereas KCN added after FCCP restored NAD(P)H. In the absence of external Ca2+, oligomycin added prior to glutamate increased NAD(P)H and completely prevented NAD(P)H oxidation following application of glutamate (Figure 6C). In the bath solution with 1.8 mM Ca2+, oligomycin added prior to glutamate also increased NAD(P)H, indicating tight coupling of oxidative phosphorylation under resting conditions (Figure 6D). Interestingly, glutamate added after oligomycin still produced oxidation of NAD(P)H, suggesting uncoupling between mitochondrial oxidation and ATP synthesis. Moreover, in the presence of oligomycin, glutamate caused faster NAD(P)H oxidation. In the presence of oligomycin, ATP supply for plasma membrane Ca2+-ATPase could be depleted, leading to less Ca2+ extruded from the cytosol. In addition, by inhibiting ATP synthesis oligomycin hyperpolarizes mitochondria. Mitochondrial hyperpolarization accelerates Ca2+ uptake because mitochondrial membrane potential is a driving force for Ca2+ uptake (Bernardi, 1999). Correspondingly, elevated cytosolic Ca2+ could be taken up by mitochondria faster, resulting in more rapid depolarization, more robust activation of electron flow in the respiratory chain, and faster NAD(P)H oxidation.
At the end of the experiment, FCCP failed to further decrease NAD(P)H, while KCN produced robust NAD(P)H recovery (Figure 6D). Rotenone (1 µM), an inhibitor of complex I, prevented NAD(P)H oxidation by the NAD(P)H dehydrogenase (complex I). This was manifested in the increased NAD(P)H fluorescence (Figure 6E) resembling the effect of oligomycin (Figure 5D). However, in the presence of rotenone, the effects of glutamate, FCCP, and KCN were greatly diminished because of inhibited complex I and strongly suppressed electron flow in the respiratory chain (Figure 6E). Similar to rotenone and oligomycin, KB-R7943 (15 µM) increased NAD(P)H fluorescence (Fig, 6F). In contrast to oligomycin and similar to rotenone, the increase in NAD(P)H induced by KB-R7943 was almost insensitive to glutamate, FCCP, and KCN. This strongly suggested that KB-R7943, like rotenone, inhibited complex I in the respiratory chain.
Consistent with complex I inhibition, KB-R7943 depolarized neuronal mitochondria which otherwise maintained a stable membrane potential (Figure 7A,B). Depolarization occurred gradually with some lag. In the experiments with cultured neurons, Rhodamine 123 was used in the quenching mode. Mitochondrial depolarization caused a release of dye from mitochondria accompanied by an increase in Rhodamine 123 fluorescence because the Rhodamine 123 was ‘unquenched’. However, the decrease in Rhodamine 123 fluorescence following the initial fluorescence increase (Figure 7B) most probably was not due to mitochondrial re-polarization but rather due to dye leakage out of the cells. Following mitochondrial depolarization, [Ca2+]c was gradually increased probably due to impaired Ca2+ extrusion and sequestration mechanisms. A slight increase in proton permeability of the inner mitochondrial membrane produced with very low concentrations of FCCP (2.5 nM) did not depolarize mitochondria on its own but significantly accelerated depolarization in the presence of KB-R7943 (Figure 7C,D). Importantly, KB-R7943-induced mitochondrial depolarization did not depend on Ca2+ and occurred without external Ca2+ as well (Supporting Figure S3). Overall, these data suggested that KB-R7943 depolarized mitochondria by inhibiting the respiratory chain and that this inhibition might be responsible for mitochondrial depolarization.
Figure 7.
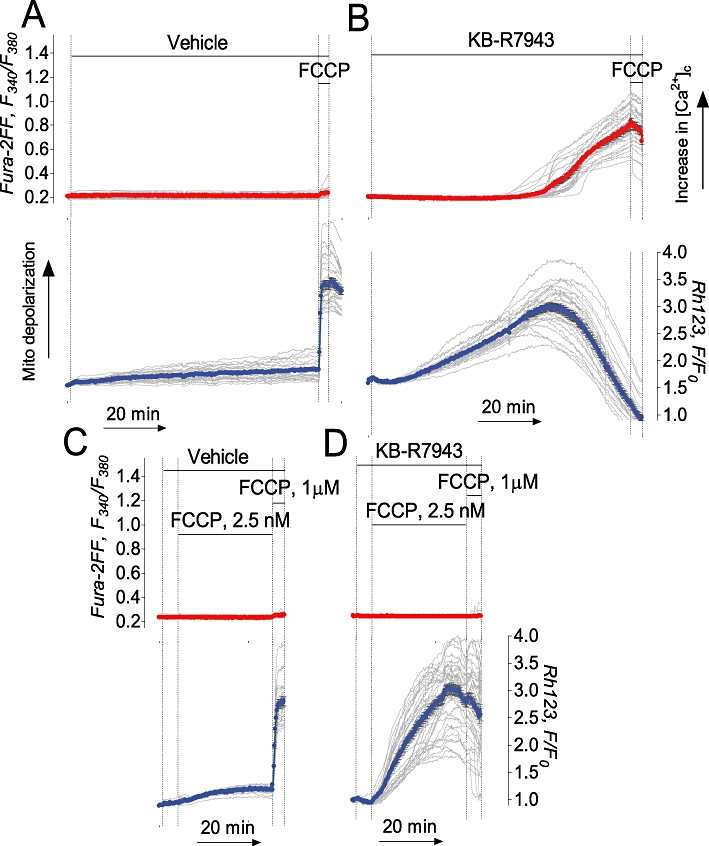
KB-R7943 depolarized mitochondria in cultured hippocampal neurons. Ultra-low concentration of FCCP accelerated mitochondrial depolarization evoked by KB-R7943. In A and C, 0.3% DMSO was applied to neurons as a vehicle. In B and D, 15 µM KB-R7943 was applied as indicated. In C and D, 2.5 nM FCCP was applied as indicated. In A–D, each experiment was performed three times. The total number of examined neurons was 81 (A), 79 (B), 84 (C), and 91 (D).
An increase in NAD(P)H (Figure 6F) accompanied by mitochondrial depolarization (Figure 7B,D) suggested an inhibition of electron transport in the respiratory chain and suppression of respiration. We evaluated the effect of KB-R7943 on neuronal respiration using Seahorse XF24 analyzer (Seahorse Bioscience). The Seahorse technology allows precise measurements of oxygen consumption by as few as 2–6 × 105 cells. In our experiments, oligomycin inhibited respiration coupled with ATP synthesis while 2,4-dinitrophenol (2,4-DNP), a protonophore, uncoupled oxidation and phosphorylation in mitochondria. This resulted in maximal oxygen consumption rate (OCR), reflecting the activity of the respiratory chain. KB-R7943 dose-dependently inhibited both basal and 2,4-DNP-stimulated respiration (Figure 8) with IC50= 11.4 ± 2.4 µM for 2,4-DNP-stimulated respiration (mean ± SEM, N= 3 independent experiments). Thus, the inhibition of 2,4-DNP-stimulated respiration confirmed inhibition of the electron transport in the respiratory chain by KB-R7943.
Figure 8.
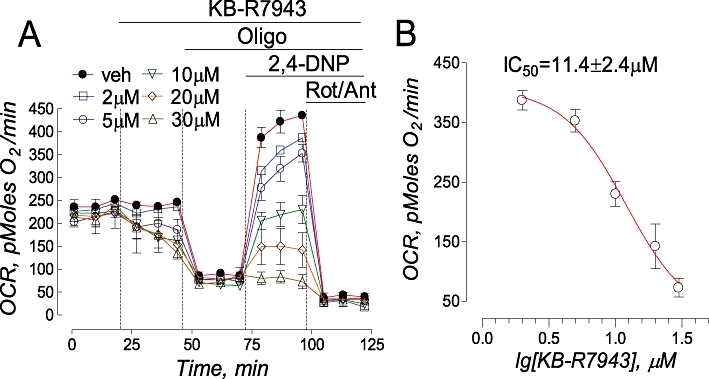
KB-R7943-induced inhibition of cellular respiration. In A, oxygen consumption rate (OCR) was measured in triplicate with a Seahorse XF24 flux analyzer. KB-R7943 (2–30 µM), oligomycin (Oligo, 1 µM), 2,4-dinitrophenol (2,4-DNP, 50 µM), and a combination of rotenone (Rot, 1 µM) and antimycin A (Ant, 1 µM) were applied to neurons as indicated. 0.3% DMSO was used as a vehicle (veh). At the beginning of the experiment, basal respiration was measured prior to addition of KB-R7943. After KB-R7943 was applied, it was present in the bath solution until the end of the experiment. In B, the concentration-response graph was plotted and IC50 was calculated using GraphPad Prism® 4.0 (GraphPad Software Inc., San Diego, CA, USA).
To substantiate our conclusions about inhibition of complex I with KB-R7943, we performed experiments with isolated brain mitochondria. First, we evaluated the effect of KB-R7943 on mitochondrial membrane potential followed with Rhodamine 123 in individual mitochondria attached to a cover slip (Shalbuyeva et al., 2007). In these experiments, Rhodamine 123 was used in the non-quenching mode in which a decrease in Rhodamine 123 fluorescence reflected mitochondrial depolarization. Figure 9A,B shows representative images demonstrating fluorescence of Rhodamine 123-loaded isolated mitochondria with high membrane potential (A) and depolarized with 1 µM FCCP (B). In these experiments, KB-R7943 dose-dependently depolarized mitochondria supplied with malate and glutamate, oxidative substrates for complex I (Figure 9C–E), but did not depolarize mitochondria supplied with succinate plus glutamate (Figure 9F,G). Here and in other experiments, glutamate was used to prevent oxaloacetate inhibition of succinate dehydrogenase (Lehninger et al., 1993). This strongly suggests specific inhibition of complex I. In addition, KB-R7943 inhibited mitochondrial respiration when mitochondria oxidized malate plus glutamate (Figure 10A) but failed to influence mitochondrial respiration when mitochondria oxidized succinate in the presence of glutamate (Figure 10B). Interestingly, respiration of isolated brain mitochondria appeared to be less sensitive to KB-R7943 than mitochondrial respiration in situ in live cells (Figure 8). The reason for that is not clear. Figure 10C and D summarize the respirometry data and show statistical analyses of mitochondrial respiratory rates. Finally, KB-R7943 inhibited mitochondrial Ca2+ uptake when mitochondria were incubated with malate plus glutamate (Figure 11A–C) but failed to inhibit Ca2+ uptake when mitochondria were incubated with succinate plus glutamate (Figure 11D,E). Moreover, after inhibition with KB-R7943, the Ca2+ uptake could be restored by addition of succinate (Figure 11C). Thus, experiments with isolated mitochondria confirmed that KB-R7943 inhibited complex I and demonstrated possible ramifications of this inhibition: a decrease in respiration, mitochondrial depolarization and suppression of mitochondrial Ca2+ uptake.
Figure 9.
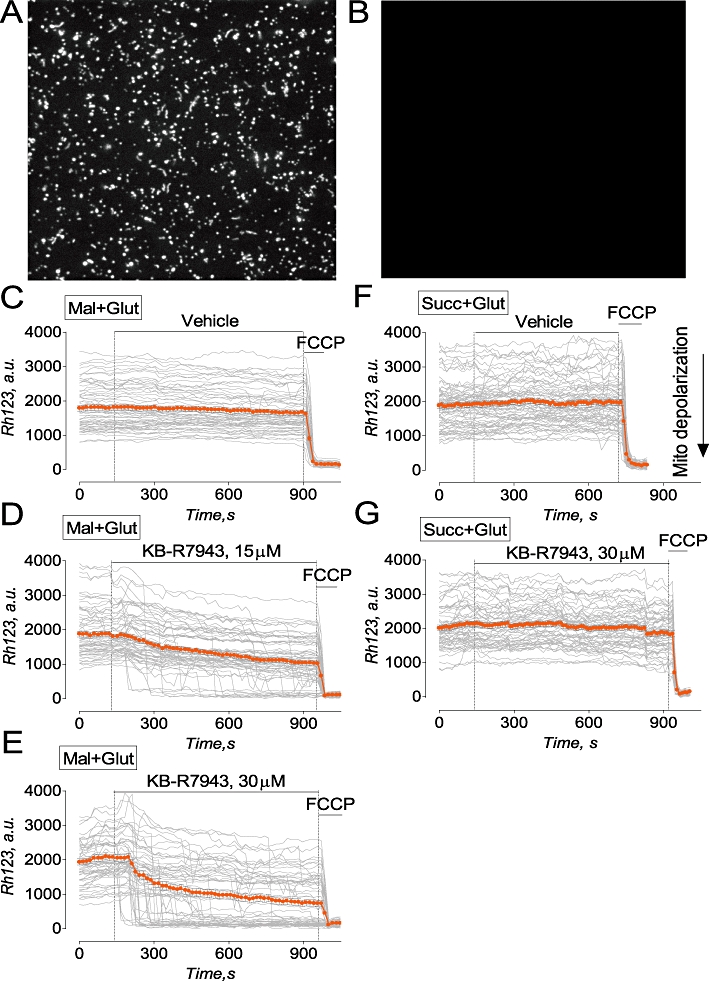
KB-R7943 depolarized isolated brain mitochondria supplied with glutamate and malate, a mixture of complex I substrates, but failed to depolarize mitochondria fuelled with succinate, a complex II substrate. In A and B, representative images of isolated mitochondria prior to (a) and after depolarization with 1 µM FCCP (B). Mitochondria were attached to cover slip and loaded with 200 nM Rhodamine-123 (Rh123) during continuous perfusion (Shalbuyeva et al., 2007). In C–E, mitochondria were perfused with the standard mitochondrial incubation medium supplemented with 1 mM malate plus 3 mM glutamate (Mal+Glut). Here and in other experiments with succinate, glutamate was used to prevent oxaloacetate inhibition of succinate dehydrogenase (Oestreicher et al., 1969; Lehninger et al. 1993). In F and G, mitochondria were perfused with the standard mitochondrial incubation medium supplemented with 3 mM succinate plus 3 mM glutamate (Succ+Glut). In C and F, 0.3% DMSO was applied as a vehicle. In D, E and G, KB-R7943 was applied as indicated. Rh123 fluorescence is expressed in arbitrary units (a.u.). Thin, grey traces demonstrate signals from individual mitochondria while thick orange traces demonstrate averaged signals (mean ± SEM) for Rh123 fluorescence expressed in arbitrary units (a.u.). In C-G, each experiment was performed three times. The total number of examined mitochondria was 201 (C), 227 (B), 198 (D), 186 (E), 214 (F) and 231 (G).
Figure 10.
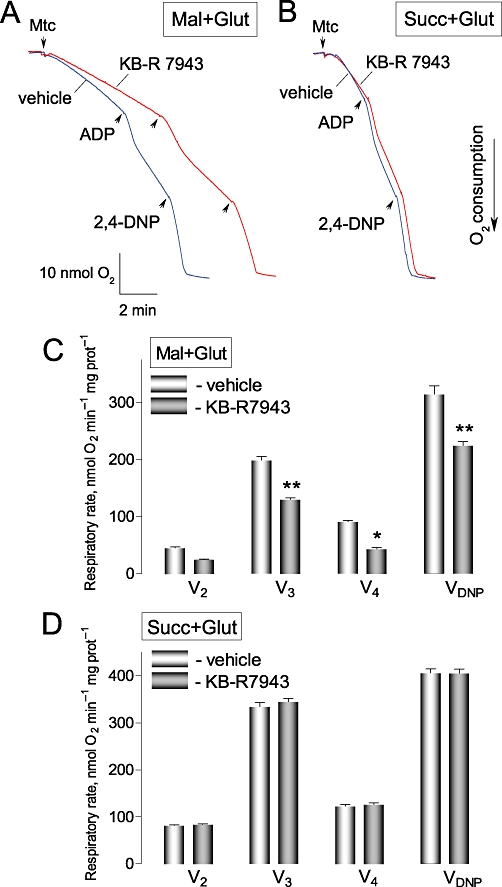
KB-R7943 inhibited respiration of isolated brain mitochondria oxidizing malate plus glutamate, but failed to affect mitochondrial respiration supported by succinate. In A and B, representative respiratory traces obtained with and without 30 µM KB-R7943 are shown. The traces are overlapped for comparison. In A, the standard incubation medium was supplemented with 1 mM malate and 3 mM glutamate (Mal+Glut); in B, with 3 mM succinate plus 3 mM glutamate (Succ+Glut). ADP (100 µM) and 2,4-dinitrophenol (2,4-DNP, 80 µM) were applied as indicated. 0.3% DMSO was applied as a vehicle. Mtc, mitochondria. V2, V3, V4, and VDNP are respiratory rates prior to ADP addition, with added ADP, after ADP depletion, and with 2,4-DNP, respectively. Blue traces show experiments with vehicle; red traces – with KB-R7943. In C, data are mean ± SEM, N= 4, *P < 0.01, **P < 0.001 comparing respiratory rates without and with KB-R7943.
Figure 11.
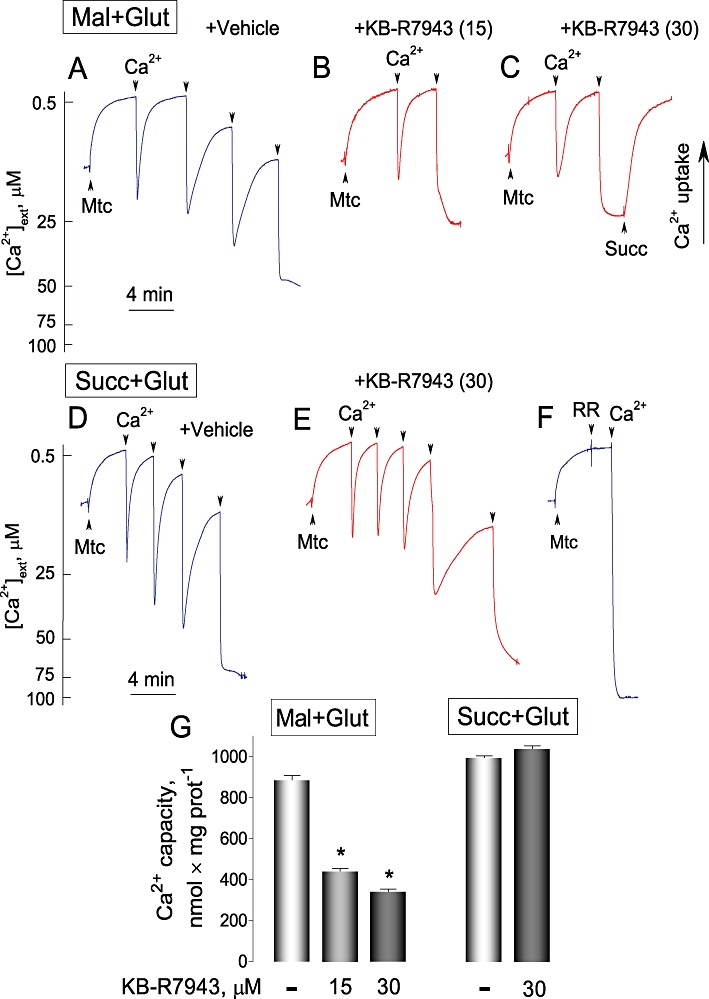
KB-R7943 hindered Ca2+ uptake by isolated brain mitochondria oxidizing glutamate and malate, but failed to inhibit Ca2+ uptake by mitochondria oxidizing succinate. In A–F, 100 µM Ca2+ was added to mitochondria as indicated. In A–C, the standard incubation medium was supplemented with 1 mM malate plus 3 mM glutamate (Mal+Glut); in D–F, with 3 mM succinate plus 3 mM glutamate (Succ+Glut). In all cases, the medium was supplemented with 0.1% BSA (free from free fatty acids), 100 µM ADP, and 1 µM oligomycin. In A and D, 0.3% DMSO was applied as a vehicle. In B, C and E, KB-R7943 was added to the incubation medium prior to mitochondria. Numbers in the parentheses indicate KB-R7943 concentration in µM. In C, succinate (Succ, 3 mM) restored Ca2+ uptake by mitochondria supplied with glutamate plus malate and incubated with 30 µM KB-R7943. In F, 1 µM ruthenium red (RR) completely blocked Ca2+ uptake by mitochondria. Blue traces show experiments with vehicle; red traces – with KB-R7943. In G, data are mean ± SEM, N= 4, *P < 0.001 comparing Ca2+ capacity without and with KB-R7943.
Discussion
Sustained elevation in [Ca2+]c represents a serious danger for neurons due to possible activation of various degradative enzymes such as Ca2+-dependent proteases (e.g. calpains) (Bano et al., 2005; Brustovetsky et al. 2010) and phospholipases (Farooqui et al., 2006). It has been proposed that the reversal of NCX could significantly contribute to Ca2+ influx into neurons and thus promote Ca2+ deregulation under conditions of prolonged glutamate exposure and/or oxygen/glucose deprivation (Kiedrowski et al., 1994; Hoyt et al. 1998). Therefore, inhibitors of NCXrev have attracted attention as research tools as well as potentially valuable therapeutic agents. Despite new additions to the panel of NCXrev inhibitors (SEA0400, YM-244769, SN-6) (Matsuda et al., 2001; Iwamoto & Kita, 2006; Watanabe et al., 2006), KB-R7943 remains the most often used pharmacological agent used to attenuate NCXrev activity in different experimental settings (Namekata et al., 2006; Araujo et al., 2007; Chen et al., 2007; Dietz et al. 2007; Kiedrowski, 2007; Wu et al. 2008). Similar to many other pharmacological agents, KB-R7943 has some side effects that might affect the interpretation of experimental results and KB-R7943 applicability. For example, in addition to inhibiting NCXrev, KB-R7943 was reported to inhibit L-type of voltage gated Ca2+ channels (Ouardouz et al., 2005), TRP channels (Kraft, 2007), NMDA receptors (Sobolevsky and Khodorov, 1999) and mitochondrial Ca2+ uptake (Santo-Domingo et al., 2007). In the present paper, we present the first evidence that KB-R7943 inhibits mitochondrial complex I and that this inhibition might influence neuronal response to excitotoxic glutamate. In addition, by using electrophysiological patch-clamp technique and calcium imaging, we unequivocally demonstrated that KB-R7943 attenuated NMDA-induced increases in [Ca2+]c and blocked ion current via NMDA receptors. Our observations indicate that KB-R7943 indeed inhibits NMDA receptors, and thus, our data support conclusions made in an early electrophysiological study (Sobolevsky and Khodorov, 1999).
In the cell, up to 80% of NAD(P)H is located in mitochondria (Duchen and Biscoe, 1992). The increase in NAD(P)H concentration takes place when NAD(P)H oxidation is slowed by suppressing the electron flow through the respiratory chain. FCCP depolarizes mitochondria, accelerating electron flow in the respiratory chain and leading to NAD(P)H oxidation. NAD(P)+ does not fluoresce (Chance and Williams, 1956) and therefore a decrease in fluorescence was observed following addition of FCCP. On the other hand, KCN, an inhibitor of cytochrome oxidase (complex IV), stops electron flow in the entire respiratory chain, leading to recovery of NAD(P)H. Glutamate could stimulate NAD(P)H oxidation by different mechanisms. Glutamate causes massive Na+ influx into neurons, increasing Na+/K+-ATPase activity and ADP production. In turn, augmented ADP production stimulates oxidative phosphorylation. Oligomycin inhibits ATP synthase, so NAD(P)H increase induced by oligomycin reflects oxidation coupled with ATP synthesis. Alternatively, a massive influx of Ca2+ induced by glutamate could cause uncoupling of oxidative phosphorylation due to activation of the permeability transition pore (Bernardi et al., 2006) or Ca2+ cycling across the inner mitochondrial membrane (Carafoli, 1979). The failure of oligomycin to recover NAD(P)H following glutamate application in the Ca2+-containing bath solution supports this scenario. In contrast, robust oligomycin-induced recovery of NAD(P)H in Ca2+-free bath solution indicates tight coupling of oxidative phosphorylation and suggests a key role of Ca2+ in the uncoupling of oxidative phosphorylation in neurons exposed to glutamate.
An increase in NAD(P)H fluorescence induced by KB-R7943 in our experiments indicated a reduction of NAD(P)+ to NAD(P)H. The similarity between the effects of KB-R7943 and rotenone, an inhibitor of complex I, suggested that KB-R7943 could inhibit complex I as well. However, oligomycin, an inhibitor of ATP synthase, also increased NAD(P)H under resting conditions. This raised the question whether KB-R7943 inhibited complex I, ATP synthase, or both. The answer to this question comes from the observation that KB-R7943, similar to rotenone and in contrast to oligomycin, greatly suppressed the effects of glutamate, FCCP, and KCN. This strongly suggested that KB-R7943 inhibited complex I in neuronal mitochondria. Of note, the experiments with isolated brain mitochondria fuelled with succinate, in which KB-R7943 failed to affect State 3 respiration (in the presence of ADP), ruled out inhibition of the ATP synthase.
Despite KB-R7943-induced inhibition of complex I, mitochondrial depolarization did not occur immediately, presumably due to a very low proton permeability of the inner mitochondrial membrane in live neurons. A slight increase in proton permeability induced by a very low concentration of the protonophore FCCP, ineffective on its own for in terms of the membrane potential, produced rapid depolarization in the presence of KB-R7943. This suggested that the ability of mitochondria to compensate for an increased H+ influx back into the matrix by increasing H+ extrusion via proton pumps of the respiratory chain was diminished by KB-R7943. Importantly, the effect of KB-R7943 on mitochondrial membrane potential was Ca2+-independent and could be observed in the Ca2+-free medium as well.
The increase in NAD(P)H autofluorescence produced with KB-R7943 suggested inhibition of complex I. However, the same effect on NAD(P)H fluorescence could produce inhibition of complex III or IV. Isolated brain mitochondria were instrumental in distinguishing between these possibilities. With isolated mitochondria, we could deliberately choose oxidative substrates, which are oxidized either by complex I (malate plus glutamate or malate plus pyruvate) or by complex II (succinate). It appeared that KB-R7943 selectively inhibited mitochondrial respiration, produced mitochondrial depolarization and suppressed mitochondrial Ca2+ uptake when mitochondria oxidized complex I substrates but not succinate, a complex II substrate. This indicated that in our experiments, KB-R7943 specifically inhibited complex I but did not inhibit other components of the respiratory chain (e.g. complexes II, III or IV) or mitochondrial Ca2+ uniporter. In fact, a recovery of Ca2+ uptake with succinate suggested that KB-R7943 might inhibit the Ca2+ uptake by limiting the amount of energy available for Ca2+ transport rather than directly inhibiting the Ca2+ uniporter (Santo-Domingo et al., 2007). However, we cannot completely exclude the latter possibility. Thus, the experiments with isolated mitochondria corroborated our conclusions about the mechanism of KB-R7943 action on mitochondrial membrane potential and Ca2+ handling in cultured hippocampal neurons.
What consequences might complex I inhibition with KB-R7943 have for the whole cell? As we demonstrated in the present paper, complex I inhibition decreases mitochondrial capacity to generate membrane potential thus limiting the ability of mitochondria to contribute to clearance of elevated cytosolic Ca2+. This explains why KB-R7943 accelerated the onset of delayed Ca2+ deregulation in the experiments with neurons exposed to 25 µM glutamate. On the other hand, in the experiments with 100 µM glutamate, the effect of KB-R7943 on delayed Ca2+ deregulation was less pronounced probably because its onset was very rapid. Under these conditions, mitochondria apparently became injured and lost their ability to accumulate Ca2+ very quickly even in the absence of KB-R7943. Interestingly, the indirect inhibition of mitochondrial Ca2+ uptake due to mild mitochondrial depolarization with KB-R7943 might protect mitochondria from irreversible damage imposed by excessive Ca2+ accumulation and this could be protective for the whole neuron. Previously, a transient depolarization of neuronal mitochondria with FCCP leading to inhibition of mitochondrial Ca2+ uptake was found to be neuroprotective in the experiments with excitotoxic glutamate applied to cultured neurons (Stout et al., 1998). In our previous study, KB-R7943 protected the ability of cultured neurons to recover low [Ca2+]c after glutamate removal (Storozhevykh et al., 2010) suggesting that KB-R7943 prevented mitochondrial injury and preserved neuronal mitochondria for Ca2+ accumulation in the post-glutamate period. This supports the idea that prevention of mitochondrial Ca2+ overload could be neuroprotective in the experimental model of glutamate excitotoxicity (Stout et al., 1998).
In summary, here for the first time, we presented strong evidence suggesting that KB-R7943 inhibits complex I in the mitochondrial respiratory chain. This could contribute to mitochondrial depolarization, limit Ca2+ uptake by the organelles, and thus preserve mitochondria from the Ca2+-induced damage in neurons exposed to excitotoxic glutamate. In turn, preventing mitochondrial damage could facilitate recovery of Ca2+ homeostasis and promote neuronal survival after excitotoxic challenge with glutamate. Our data also indicate that, in addition to mitochondrial complex I and NCXrev, KB-R7943 inhibits NMDA receptors. Therefore, the whole range of KB-R7943 effects has to be taken into consideration when interpreting the results obtained with this widely used pharmacological agent.
Acknowledgments
This study was supported by the NIH/NINDS R01 NS 050131, by grant A70-0-079212 from Indiana State Department of Health – Indiana Spinal Cord and Brain Injury Research Fund and by grant from Ralph W. and Grace M. Showalter foundation to NB. We are very grateful to Dr Robert Harris (Richard L. Roudebush VA Medical Center, Indianapolis) for providing access to Seahorse XF24 flux analyzer and Dr Jay Dunn (Seahorse Bioscience) for training and invaluable advices.
Glossary
Abbreviations
- Δψ
mitochondrial membrane potential
- [Ca2+]c
cytosolic Ca2+ concentration
- CNQX
6-cyano-7-nitroquinoxaline-2,3-dione
- DMSO
dimethyl sulfoxide
- FCCP
carbonylcyanide-p-trifluoromethoxyphenylhydrazone
- KB-R7943
(2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl]isothiourea methane sulfonate
- MK801
(5R,10S)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate
- NAD(P)+
oxidized nicotinamide adenine dinucleotide phosphate
- NAD(P)H
reduced nicotinamide adenine dinucleotide phosphate
- NCX
plasmalemmal Na+/Ca2+ exchanger
- NCXrev
reverse Na+/Ca2+ exchanger
- NMDA
N-methyl-D-aspartate
- TRP channels
transient receptor potential channels
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 KB-R7943 failed to protect cultured hippocampal neurons against delayed calcium deregulation and mitochondrial depolarization. The thin, grey traces show signals from individual neurons, while thick, red traces show averaged signals (mean ± SEM) for Fura-2FF ratio F340/F380 (upper panels) and thick, blue traces for Rhodamine 123 (Rh123) F/F0 (lower panels), respectively. Neurons were derived from postnatal day one (PN1) Sprague-Dawley rat pups and were 10–12 days in vitro on the day of the experiment. In A and B, neurons were treated with 100 μM glutamate (Glu). In both cases, glutamate was applied in combination with 10 μM glycine. In A, 0.3% DMSO was applied to neurons as a vehicle. In B, 15 μM KB-R7943 was applied to neurons as indicated. Here and in other similar experiments, 1 μM FCCP was applied at the end of experiments to completely depolarize mitochondria in the Ca2+-free bath solution.
Figure S2 MK801, an inhibitor of NMDA receptor, inhibited glutamate- and NMDA-induced calcium deregulation while CNQX, an inhibitor of AMPA/kainate receptor, was without effect. Where indicated, 30 μM glutamate (plus 10 μM glycine), 30 μM NMDA (plus 10 μM glycine), 10 μM MK801, or 20 μM CNQX were added.
Figure S3 In cultured hippocampal neurons, KB-R7943 depolarized mitochondria in a Ca2+-independent manner. In A and B, KB-R7943 was applied as indicated. In B, Ca2+ was omitted from the bath solution. FCCP, 1 μM.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl 2):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amran MS, Homma N, Hashimoto K. Pharmacology of KB-R7943: a Na+-Ca2+ exchange inhibitor. Cardiovasc Drug Rev. 2003;21:255–276. doi: 10.1111/j.1527-3466.2003.tb00121.x. [DOI] [PubMed] [Google Scholar]
- Arakawa N, Sakaue M, Yokoyama I, Hashimoto H, Koyama Y, Baba A, et al. KB-R7943 inhibits store-operated Ca(2+) entry in cultured neurons and astrocytes. Biochem Biophys Res Commun. 2000;279:354–357. doi: 10.1006/bbrc.2000.3968. [DOI] [PubMed] [Google Scholar]
- Araujo IM, Carreira BP, Pereira T, Santos PF, Soulet D, Inacio A, et al. Changes in calcium dynamics following the reversal of the sodium-calcium exchanger have a key role in AMPA receptor-mediated neurodegeneration via calpain activation in hippocampal neurons. Cell Death Differ. 2007;14:1635–1646. doi: 10.1038/sj.cdd.4402171. [DOI] [PubMed] [Google Scholar]
- Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, et al. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005;120:275–285. doi: 10.1016/j.cell.2004.11.049. [DOI] [PubMed] [Google Scholar]
- Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Krauskopf A, Basso E, Petronilli V, Blalchy-Dyson E, Di Lisa F, et al. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273:2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Breder J, Sabelhaus CF, Opitz T, Reymann KG, Schroder UH. Inhibition of different pathways influencing Na(+) homeostasis protects organotypic hippocampal slice cultures from hypoxic/hypoglycemic injury. Neuropharmacology. 2000;39:1779–1787. doi: 10.1016/s0028-3908(00)00027-7. [DOI] [PubMed] [Google Scholar]
- Brustovetsky N, Brustovetsky T, Jemmerson R, Dubinsky JM. Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J Neurochem. 2002;80:207–218. doi: 10.1046/j.0022-3042.2001.00671.x. [DOI] [PubMed] [Google Scholar]
- Brustovetsky T, Li V, Brustovetsky N. Stimulation of glutamate receptors in cultured hippocampal neurons causes Ca2+-dependent mitochondrial contraction. Cell Calcium. 2009;46:18–29. doi: 10.1016/j.ceca.2009.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky T, Bolshakov A, Brustovetsky N. Calpain activation and Na(+)/Ca(2+) exchanger degradation occur downstream of calcium deregulation in hippocampal neurons exposed to excitotoxic glutamate. J Neurosci Res. 2010;88:1317–1328. doi: 10.1002/jnr.22295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carafoli E. The calcium cycle of mitochondria. FEBS Lett. 1979;104:1–5. doi: 10.1016/0014-5793(79)81073-x. [DOI] [PubMed] [Google Scholar]
- Chance B, Williams GR. The respiratory chain and oxidative phosphorylation. Adv Enzymol Relat Subj Biochem. 1956;17:65–134. doi: 10.1002/9780470122624.ch2. [DOI] [PubMed] [Google Scholar]
- Chen H, Kintner DB, Jones M, Matsuda T, Baba A, Kiedrowski L, et al. AMPA-mediated excitotoxicity in oligodendrocytes: role for Na(+)-K(+)-Cl(-) co-transport and reversal of Na(+)/Ca(2+) exchanger. J Neurochem. 2007;102:1783–1795. doi: 10.1111/j.1471-4159.2007.04638.x. [DOI] [PubMed] [Google Scholar]
- Chen HS, Pellegrini JW, Aggarwal SK, Lei SZ, Warach S, Jensen FE, et al. Open-channel block of N-methyl-D-aspartate (NMDA) responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. J Neurosci. 1992;12:4427–4436. doi: 10.1523/JNEUROSCI.12-11-04427.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Clifford DB, Zorumski CF, Olney JW. Ketamine and MK-801 prevent degeneration of thalamic neurons induced by focal cortical seizures. Exp Neurol. 1989;105:272–279. doi: 10.1016/0014-4886(89)90130-1. [DOI] [PubMed] [Google Scholar]
- Convery MK, Hancox JC. Comparison of Na+-Ca2+ exchange current elicited from isolated rabbit ventricular myocytes by voltage ramp and step protocols. Pflugers Arch. 1999;437:944–954. doi: 10.1007/s004240050866. [DOI] [PubMed] [Google Scholar]
- Czyz A, Kiedrowski L. In depolarized and glucose-deprived neurons, Na+ influx reverses plasmalemmal K+-dependent and K+-independent Na+/Ca2+ exchangers and contributes to NMDA excitotoxicity. J Neurochem. 2002;83:1321–1328. doi: 10.1046/j.1471-4159.2002.01227.x. [DOI] [PubMed] [Google Scholar]
- Czyz A, Baranauskas G, Kiedrowski L. Instrumental role of Na+ in NMDA excitotoxicity in glucose-deprived and depolarized cerebellar granule cells. J Neurochem. 2002;81:379–389. doi: 10.1046/j.1471-4159.2002.00851.x. [DOI] [PubMed] [Google Scholar]
- Dietz RM, Kiedrowski L, Shuttleworth CW. Contribution of Na(+)/Ca(2+) exchange to excessive Ca(2+) loading in dendrites and somata of CA1 neurons in acute slice. Hippocampus. 2007;17:1049–1059. doi: 10.1002/hipo.20336. [DOI] [PubMed] [Google Scholar]
- Dubinsky JM. Intracellular calcium levels during the period of delayed excitotoxicity. J Neuroscience. 1993;13:623–631. doi: 10.1523/JNEUROSCI.13-02-00623.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubinsky JM, Kristal BS, Elizondo-Fournier M. An obligate role for oxygen in the early stages of glutamate-induced, delayed neuronal death. J Neurosci. 1995;15:7071–7078. doi: 10.1523/JNEUROSCI.15-11-07071.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Biscoe TJ. Mitochondrial function in type I cells isolated from rabbit arterial chemoreceptors. J Physiol. 1992;450:13–31. doi: 10.1113/jphysiol.1992.sp019114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqui AA, Ong WY, Horrocks LA. Inhibitors of brain phospholipase A2 activity: their neuropharmacological effects and therapeutic importance for the treatment of neurologic disorders. Pharmacol Rev. 2006;58:591–620. doi: 10.1124/pr.58.3.7. [DOI] [PubMed] [Google Scholar]
- Hoyt KR, Arden SR, Aizenman E, Reynolds IJ. Reverse Na+/Ca2+ exchange contributes to glutamate-induced intracellular Ca2+ concentration increases in cultured rat forebrain neurons. Mol Pharmacol. 1998;53:742–749. [PubMed] [Google Scholar]
- Iwamoto T, Kita S. YM-244769, a novel Na+/Ca2+ exchange inhibitor that preferentially inhibits NCX3, efficiently protects against hypoxia/reoxygenation-induced SH-SY5Y neuronal cell damage. Mol Pharmacol. 2006;70:2075–2083. doi: 10.1124/mol.106.028464. [DOI] [PubMed] [Google Scholar]
- Iwamoto T, Watano T, Shigekawa M. A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem. 1996;271:22391–22397. doi: 10.1074/jbc.271.37.22391. [DOI] [PubMed] [Google Scholar]
- Kiedrowski L. NCX and NCKX operation in ischemic neurons. Ann N Y Acad Sci. 2007;1099:383–395. doi: 10.1196/annals.1387.035. [DOI] [PubMed] [Google Scholar]
- Kiedrowski L, Brooker G, Costa E, Wroblewski JT. Glutamate impairs neuronal calcium extrusion while reducing sodium gradient. Neuron. 1994;12:295–300. doi: 10.1016/0896-6273(94)90272-0. [DOI] [PubMed] [Google Scholar]
- Kraft R. The Na+/Ca2+ exchange inhibitor KB-R7943 potently blocks TRPC channels. Biochem Biophys Res Commun. 2007;361:230–236. doi: 10.1016/j.bbrc.2007.07.019. [DOI] [PubMed] [Google Scholar]
- Lehninger AL, Nelson DL, Cox MM. Principles of Biochemistry. New York: Worth Publishers; 1993. [Google Scholar]
- Li S, Jiang Q, Stys PK. Important role of reverse Na(+)-Ca(2+) exchange in spinal cord white matter injury at physiological temperature. J Neurophysiol. 2000;84:1116–1119. doi: 10.1152/jn.2000.84.2.1116. [DOI] [PubMed] [Google Scholar]
- Li T, Brustovetsky T, Antonsson B, Brustovetsky N. Oligomeric BAX induces mitochondrial permeability transition and complete cytochrome c release without oxidative stress. Biochim Biophys Acta. 2008;1777:1409–1421. doi: 10.1016/j.bbabio.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li V, Brustovetsky T, Brustovetsky N. Role of cyclophilin d-dependent mitochondrial permeability transition in glutamate-induced calcium deregulation and excitotoxic neuronal death. Exp Neurol. 2009;218:171–182. doi: 10.1016/j.expneurol.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Wang Y, Chen X, Chen H, Kintner DB, Shull GE, et al. Increased tolerance to ischemic neuronal damage by knockdown of Na+-Ca2+ exchanger isoform 1. Ann N Y Acad Sci. 2007;1099:292–305. doi: 10.1196/annals.1387.016. [DOI] [PubMed] [Google Scholar]
- MacGregor DG, Avshalumov MV, Rice ME. Brain edema induced by in vitro ischemia: causal factors and neuroprotection. J Neurochem. 2003;85:1402–1411. doi: 10.1046/j.1471-4159.2003.01772.x. [DOI] [PubMed] [Google Scholar]
- Manev H, Favaron M, Guidotti A, Costa E. Delayed increase of Ca2+ influx elicited by glutamate: role in neuronal death. Mol Pharmacol. 1989;36:106–112. [PubMed] [Google Scholar]
- Matsuda T, Arakawa N, Takuma K, Kishida Y, Kawasaki Y, Sakaue M, et al. SEA0400, a novel and selective inhibitor of the Na+-Ca2+ exchanger, attenuates reperfusion injury in the in vitro and in vivo cerebral ischemic models. J Pharmacol Exp Ther. 2001;298:249–256. [PubMed] [Google Scholar]
- Mattson MP, Guthrie PB, Kater SB. A role for Na+-dependent Ca2+ extrusion in protection against neuronal excitotoxicity. FASEB J. 1989;3:2519–2526. doi: 10.1096/fasebj.3.13.2572500. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL. Permeation and block of N-methyl-D-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. J Physiol. 1987;394:501–527. doi: 10.1113/jphysiol.1987.sp016883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekata I, Kawanishi T, Iida-Tanaka N, Tanaka H, Shigenobu K. Quantitative fluorescence measurement of cardiac Na+/Ca2+ exchanger inhibition by kinetic analysis in stably transfected HEK293 cells. J Pharmacol Sci. 2006;101:356–360. doi: 10.1254/jphs.scj06002x. [DOI] [PubMed] [Google Scholar]
- Oestreicher AB, Bergh SG, Slater EC. The inhibition by 2,4-dinitrophenol of the removal of oxaloacetate formed by the oxidation of succinate by rat-liver and -heart mitochondria. Biochim Biophys Acta. 1969;180:45–55. doi: 10.1016/0005-2728(69)90192-3. [DOI] [PubMed] [Google Scholar]
- Ouardouz M, Zamponi GW, Barr W, Kiedrowski L, Stys PK. Protection of ischemic rat spinal cord white matter: dual action of KB-R7943 on Na+/Ca2+ exchange and L-type Ca2+ channels. Neuropharmacology. 2005;48:566–575. doi: 10.1016/j.neuropharm.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Pivovarova NB, Nguyen HV, Winters CA, Brantner CA, Smith CL, Andrews SB. Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. J Neurosci. 2004;24:5611–5622. doi: 10.1523/JNEUROSCI.0531-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reppel M, Sasse P, Malan D, Nguemo F, Reuter H, Bloch W, et al. Functional expression of the Na+/Ca2+ exchanger in the embryonic mouse heart. J Mol Cell Cardiol. 2007;42:121–132. doi: 10.1016/j.yjmcc.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Santo-Domingo J, Vay L, Hernandez-SanMiguel E, Lobaton CD, Moreno A, Montero M, et al. The plasma membrane Na+/Ca2+ exchange inhibitor KB-R7943 is also a potent inhibitor of the mitochondrial Ca2+ uniporter. Br J Pharmacol. 2007;151:647–654. doi: 10.1038/sj.bjp.0707260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder UH, Breder J, Sabelhaus CF, Reymann KG. The novel Na+/Ca2+ exchange inhibitor KB-R7943 protects CA1 neurons in rat hippocampal slices against hypoxic/hypoglycemic injury. Neuropharmacology. 1999;38:319–321. doi: 10.1016/s0028-3908(98)00198-1. [DOI] [PubMed] [Google Scholar]
- Shalbuyeva N, Brustovetsky T, Brustovetsky N. Lithium desensitizes brain mitochondria to calcium, antagonizes permeability transition, and diminishes cytochrome C release. J Biol Chem. 2007;282:18057–18068. doi: 10.1074/jbc.M702134200. [DOI] [PubMed] [Google Scholar]
- Smith GL, Elliott EE, Kettlewell S, Currie S, Quinn FR. Na(+)/Ca(2+) exchanger expression and function in a rabbit model of myocardial infarction. J Cardiovasc Electrophysiol. 2006;17(Suppl 1):S57–S63. doi: 10.1111/j.1540-8167.2006.00384.x. [DOI] [PubMed] [Google Scholar]
- Sobolevsky AI, Khodorov BI. Blockade of NMDA channels in acutely isolated rat hippocampal neurons by the Na+/Ca2+ exchange inhibitor KB-R7943. Neuropharmacology. 1999;38:1235–1242. doi: 10.1016/s0028-3908(99)00040-4. [DOI] [PubMed] [Google Scholar]
- Storozhevykh TP, Senilova YE, Brustovetsky T, Pinelis VG, Brustovetsky N. Neuroprotective effect of KB-R7943 against glutamate excitotoxicity is related to mild mitochondrial depolarization. Neurochem Res. 2010;35:323–335. doi: 10.1007/s11064-009-0058-x. [DOI] [PubMed] [Google Scholar]
- Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Kimura J. Inhibitory effect of amiodarone on Na(+)/Ca(2+) exchange current in guinea-pig cardiac myocytes. Br J Pharmacol. 2000;131:80–84. doi: 10.1038/sj.bjp.0703527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Koide Y, Kimura J. Topics on the Na+/Ca2+ exchanger: pharmacological characterization of Na+/Ca2+ exchanger inhibitors. J Pharmacol Sci. 2006;102:7–16. doi: 10.1254/jphs.fmj06002x2. [DOI] [PubMed] [Google Scholar]
- Wu MP, Kao LS, Liao HT, Pan CY. Reverse mode Na+/Ca2+ exchangers trigger the release of Ca2+ from intracellular Ca2+ stores in cultured rat embryonic cortical neurons. Brain Res. 2008;1201:41–51. doi: 10.1016/j.brainres.2008.01.042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.