

The 26 S proteasome contains a 20 S catalytic core, which has a cylindrical shape and on its own is capable of degrading small peptides (1). However, eukaryotic proteins that are designated for degradation are frequently marked by polyubiquitination, which apparently increases the bulk of the protein to make it difficult to enter the small catalytic chamber. To degrade polyubiquitinated proteins, the 19 S regulatory particle is needed, and these 19 S particles cap the two ends of the 20 S catalytic core to produce the 26 S proteasome. In order to understand how the 26 S proteasome efficiently and selectively degrades proteins, it is critical to ascertain how a 26 S proteasome is properly assembled.
A 19 S regulatory particle can be further divided into a lid and a base subcomplex, each of which contains approximately eight subunits. Intriguingly, more than half of the lid subunits contain a proteasome-COP9-initiation factor (PCI)4 domain, thus named because it is frequently found in these three protein complexes, which, besides the proteasome, include the COP9/signalosome and eIF3 translation initiation complex (2, 3). The common presence of PCI domains in these protein complexes supports a model in which the PCI domain can play a role in assembling subunits within or even between these complexes (4).
The PCI domains remain structurally poorly defined. So far, the only relevant crystal structure of the PCI domain is that of human eIF3k (5), since this protein is a more distant member in the PCI family (6). In the structural study of Wei et al. (5), the authors have concluded that the PCI domain in eIF3k actually contains two subdomains, and this two-subdomain architecture is probably also true for more typical PCI domains. The eIF3k PCI N-terminal subdomain was termed HEAT analogous motif (HAM) because it resembles the HEAT helical repeats found in numerous proteins whose main roles appear to be binding and assembling other proteins. In a previous bioinformatic analysis of PCI architecture (6), we predicted that the N-terminal subdomain of typical PCI domains most likely resembles the tetratricopeptide repeat (TPR), which consists of pairs of α-helices that are oriented in an antiparallel manner. Compared with the HEAT repeats, the functionally similar TPR has a slightly different arrangement of helices.
The C-terminal subdomain in the eIF3k PCI was determined to be a winged helix (5), which is also likely to be conserved in the other PCI domains. Together with the TPR/HEAT repeat subdomain, these two subdomains create two large surfaces, one of which is concave and one of which is convex, resulting in an earlike shape. The presence of these two large surfaces may allow a PCI protein to provide ample binding areas for many proteins. However, no detailed structure-function analyses have yet been performed to investigate whether the presence of these two subdomains is physiologically relevant.
Our laboratory has characterized a PCI protein called Yin6 in the fission yeast Schizosaccharomyces pombe (7). Yin6 is the S. pombe ortholog of the mammalian Int6 protein that has been implicated in breast tumorigenesis (8-10), although its molecular functions are poorly defined. Int6 is also known as eIF3e, because it was found to co-purify with the eIF3 complex (11). We and others have shown that Yin6 is not essential for global translation initiation (12). By contrast, we have found that a major evolutionarily conserved function of Yin6 is to regulate the 26 S proteasome by preferentially binding and controlling the localization and/or assembly of a PCI proteasome lid subunit, Rpn5 (13, 14). In wild type S. pombe cells, the proteasome is concentrated in the inner layer of the nuclear membrane. By contrast, in yin6 null (yin6Δ) cells as well as rpn5Δ cells, the proteasome appears more diffuse, as measured by microscopy, and lacks many subunits, as revealed by affinity pull-down.
To better define the role of Yin6, in this study we carried out a high copy suppressor screen seeking S. pombe genes that when overexpressed rescued the cold-dependent growth defect of yin6Δ cells. In support of the model in which Yin6 regulates the proteasome, we report herein the isolation of another PCI proteasome subunit, Rpn7, from the screen. Since together with Yin6 and Rpn5, we now have three well characterized PCI proteins, we performed a structure-function analysis to identify amino acid resides in the PCI domain that may be important for the function of many PCI proteins. We have thus identified a Leu residue that is critical for the functions of these three proteins. Bioinformatics analysis showed that this Leu appears to reside in a region that is in close proximity to both the TPR-like and the WH subdomains. The hydrophobic interactions provided by the identified Leu may influence how each of these two subdomains folds, how they are arranged relative to one another, or both. Collectively, these data suggest that maintaining proper conformation of these two subdomains is indeed important for the function of many PCI proteins.
EXPERIMENTAL PROCEDURES
Strains and Growth Conditions
The parental wild-type S. pombe strain used in this paper is SP870 (h90, ade6-M210, leu1-32, ura4-D18). The yin6Δ, rpn5Δ, and pus1/rpn10-PA strains were as described (13, 14). Cells were grown in either yeast extract medium (YEAU) or synthetic minimal medium (MM) with appropriate supplements. To test for canavanine sensitivities, stock solutions of canavanine sulfate (50 mg/ml in water) were prepared and added to media after autoclaving. We carried out all the experiments with cells pregrown to early logarithmic phase (2–5 × 106 cells/ml). For growth experiments on plates, cells were serially diluted 1:5.
HeLa cells were cultured at 37 °C in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and penicillin/streptomycin, and the CO2 was maintained at 5%. For transfection, HeLa cells were grown to 40–50% confluence in 24-well glass bottom culture plates (MatTek), 100 ng of plasmid DNA was used, and the transfection reagent was Fugene 6 (Roche Applied Science).
High Copy Suppressor Screen
The YIN6K strain (yin6::kanR) (7) was transformed with a cDNA library whose cDNA clones are carried by the pREP3 vector that contains a thiamine-repressible nmt1 promoter (15). Transformed cells were incubated at 20 °C for 6 weeks, and plasmid DNA was isolated from colonies that emerged. These cDNA clones were retested with a fresh batch of YIN6K cells, and those that failed to rescue the growth defects were discarded. The final cDNA clones were sequenced using two primers: 5′-CAATCTCATTCTCACTTTCTGAC-3′ and 5′-TTGAATGGGCTTCCATAGTTTG-3′. The former anneals in the nmt1 promoter, whereas the latter anneals to the nmt1 terminator. The obtained sequences were submitted to the BLAST server of the S. pombe sequencing project at the Sanger Institute (16). In addition to yin6, which was isolated six times, two identical rpn7 clones (SPBC582.07c) were isolated. These two clones do not have the coding sequence for the first 8 amino acids. The 12th amino acid in the Rpn7 protein is a methionine; therefore, we assumed that protein expression from these two plasmids starts from this second methionine. All of the Rpn7 vectors described in this study contain the full-length rpn7 gene, however. This screen has also isolated other genes, but they will be described elsewhere.
Plasmid Constructions
The construction of pREP1YIN6, pGADYIN6, pLBDMOE1, pGADRPN9, and pLBDRPN5 was as described (7, 13, 14). A BamHI rpn7 cDNA fragment was amplified by PCR from the cosmid SPBC582 (16) and subcloned into pVJL11 (17), pREP1, pREP41, and pREP41GFP to create pLBDRPN7, pREP1RPN7, pREP41RPN7, and pREPGFPRPN7. All PCR products in this study have been verified by sequencing to make sure that there is no mutation altering the coding sequence. In a separate project, we5 have constructed an expression vector that was derived from pNR210gck (18) and uses the cdc42 promoter to express fusion proteins that are N-terminally tagged by a monomeric form of RFP called mCherry (19). We named it mcRFP in this study. To tag Rpn7 with mcRFP, the rpn7 cDNA was modified by PCR to contain SalI and SphI sites for cloning, and the resulting vector is named pMCRPN7. All site-directed mutagenesis was performed by the QuikChange kit (Stratagene). pAL1YIN6g contains the genomic version of the wild type yin6 gene in a high copy Leu+ vector, pAL1 (7). The coding sequence of wild type yin6 in this vector was replaced with the mutated ones, and the final vector expressing Yin6L332D was named pYIN6LD. The wild type human int6 in pHAHINT6 (7) and the wild type yin6 in pHAYIN6 (7) were similarly mutated to create pHAHINT6LD and pHAYIN6LD. Wild type or mutant human int6 were subcloned into the YFP-N1 vector (BD Biosciences) to create pYFPHINT6 and pYFPHINT6LD. The mutant rpn5 and rpn7 gene products were also subcloned into pVJL11, pREP41, and pREP41GFP to create pLBDRPN5LD, pLBDRPN7LD, and pREPGFPRPN7LD. Site-directed mutagenesis was performed to mutate rpn9 in pGADRPN9 to create pGADRPN9FD, pGADRPN9MD, and pGADRPN9FDMD.
Strain Constructions
rpn7Δ/+ heterozygous diploid cells were created by homologous recombination of an rpn7::ura4 fragment amplified from the KS-ura4 plasmid as described (20), and this was also the method that we first chose to tag Rpn7 with GFP at the C terminus by the pFA6a-GFP(S65T)-kanMX6 plasmid. This approach of tagging does not deliberately create an amino acid spacer between Rpn7 and GFP, and the resulting strain displayed a temperature-sensitive growth defect. We thus named this strain RPN7TS. To properly perform gene tagging using PCR and homologous recombination, we used a different plasmid, pYM27, as described by another group (21). This approach creates a poly(AG) spacer between the gene of interest and GFP. Proper tagging was confirmed by Western blots (supplemental Fig. S1). The resulting strain was named RPN7GFP. The same method was used to tag Rpn7 with GFP in yin6Δ, rpn5Δ, and pus1/rpn10-PA cells to create strain Y6AR7GFP, R5AR7GFP, and R10PAR7GFP. We used a similar method to tag Rpn11 and α4 with GFP, and the resulting strains were named RPN11GFP and A4GFP, respectively, and Rpn5, Rpn7, and Rpn9 were tagged in SP870 by HA6, GFP, and Myc9 to create strain R5HAR7GFPR9MYC.
The pYIN6LD vector was linearized and allowed to integrate to the chromosome of yin6Δ cells (strain YIN6K) (7) to create strain YIN6LD, such that the only copy of yin6 in this strain is the mutant version. In parallel, the same mutant yin6 was integrated into a wild type strain to make sure that it did not induce a yin6Δ-like phenotype (data not shown). Other yin6 mutant genes were examined similarly. S. pombe contains two copies of rpn5 genes, rpn5a and rpn5b, that encode an identical protein (14). The expression of these two loci is tightly regulated, such that when one of them is deleted, the expression of the second locus will increase to maintain the same level of total Rpn5 protein. Site-directed mutagenesis was carried out to similarly mutate the rpn5 coding sequence in pBSRPN5-HAKanR (14) to create pBSR5LD-HAKanR. This plasmid was digested by XhoI and SmaI to release a fragment containing rpn5, and this fragment was used to transform a rpn5bΔ strain (RPN5bA) (14). After homologous recombination, its rpn5a locus was replaced by rpn5L316L-HAKanR. We named the resulting strain RPN5LD.
Fluorescence Microscopy
The general procedures for 4′,6-diamidino-2-phenylindole staining of S. pombe cells were as described (22). Samples were examined via an UPlanFI ×100/1.30 oil objective on an Olympus BX61 microscope, and the images were captured by a Q-Imaging Retiga camera. Deconvolution of images (Nearest Neighbor) was performed using Slidebook software (Intelligent Imaging Innovations) on a stack of eight images taken at 0.5-μm intervals. For the protein colocalization study, images in each focal plane were examined to confirm colocalization. Openlab software (Improvision) was used to quantify the GFP intensity.
To visualize YFP-tagged Int6 in HeLa cells, 48 h after transfection, YFP signals were captured using the Olympus IX70 microscope via a ×60/1.4 oil objective. To visualize the nuclei of live cells, Hoechst 33342 was added 10 min prior to image acquisition. YFP intensity was quantified by Slidebook software. Images were deconvolved (constrained iterative) using Slidebook on a stack of 12 images collected at 0.5-μm intervals.
26 S Proteasome Pull-down
Approximately 30 OD units (~3 × 108) of cells were lysed in ice-cold 26 S-binding buffer (25 mm Tris, 50 mm NaCl, 10 mm MgCl2, 1 mm dithiothreitol, 5 mm ATP, 0.1% Triton X-100, 20% glycerol, pH 7.0–7.4). Total crude lysates were centrifuged twice at 20,800 × g for 20 min. Supernatants with the same amount of protein were incubated with either 50 μl of IgG-Sepharose (Invitrogen) or Protein A-Sepharose control (Sigma) overnight at 4 °C. After eight washes in the 26 S-binding buffer, beads were incubated with the tobacco etch virus protease (Invitrogen) twice for 1 h at 30 °C to release the 26 S proteasome. Eluted proteins were analyzed by SDS-PAGE and Western blots. Antibodies against Mts4/Rpn1, Pus1/Rpn10, and Mts3/Rpn12 were as described (13) and used at a 1:1000 dilution. Antibodies against proteasome 20 S core α subunits (MCP231; 1:1000), GFP (1:1000), HA (1:1000), and Myc (1:1000) were from BIOMOL, Fitzgerald, Covance, and Abcam, respectively. The anti-Cdc8 antibody was a gift from M. Balasubramanian (University of Singapore) (23). The existing antibody against the α core does not recognize the α4 subunit, so we generated a polyclonal rabbit antibody using CIVKEIQDEKEAEAARKKGR as a peptide antigen. The confirmation of this antibody (1:2000) is shown in supplemental Fig. S2. To quantify protein levels, fluorescently conjugated secondary antibodies were used, and the signals were measured by the Odyssey infrared imaging system (Li-COR Biosciences).
Proteasome Subunit Co-immunoprecipitation
Cells (strain R5HAR7GFPR9MYC) were grown to 5 × 106 cells/ml in YEAU at 30 °C. Lysates were prepared in PEM buffer (100 mm PIPES, 1mm EGTA, 1 mm MgSO4, pH 6.9) plus 0.5% IGEPAL CA-630 (Sigma) and cleared by centrifuging twice at 20,800 × g for 15 min. Supernatants containing 5 mg of total proteins were mixed with the antibody (12CA5 or 9E10) at 4 °C for 1 h. Protein A-agarose beads (Sigma) were then added and incubated at 4 °C overnight. To perform salt extraction (24), protein A beads containing immunoprecipitated sample were mixed with the same lysis buffer containing 0, 0.5, or 1 m NaCl and incubated at 30 °C for 1 h with gentle rocking. The samples were analyzed by Western blotting, and protein levels were quantified by the Odyssey system (see “26 S Proteasome Pull-down”).
Glycerol Gradient Centrifugation
The cell lysate was prepared following the same procedure as that in the proteasome pull-down experiment. Glycerol gradient centrifugation was performed as described (25), except that 300-μl fractions were collected. Samples were analyzed by SDS-PAGE and Western blots.
Yeast Two-hybrid Assay
The yeast two-hybrid assay was performed as described (17). Briefly, the reporter strain was L40, which carries the reporter gene cassettes lexA-HIS3 and lexA-lacZ. The β-galactosidase activity was assayed by a color filter assay using 5-bromo-4-chloro-3-indolyl-β-d-galactoside (X-gal) as substrate, whereas the HIS3 expression was assayed by plating cells in medium lacking histidine. To quantify β-galactosidase activity, a colorimetric assay using ortho-nitrophenyl-β-galactoside as substrate was performed as described (17).
Detection of Polyubiquitinated Proteins
To detect ubiquitinated proteins, cells were broken by glass beads in phosphate-buffered saline (pH 7.4). Total crude extracts containing equal amounts of proteins were analyzed by immunoblotting with an ubiquitin antibody (1:100; Sigma).
RESULTS
Isolation of Rpn7 as a High Copy Suppressor That Rescues yin6Δ Phenotypes
The cDNA encoding a putative proteasome subunit in the lid of the 19 S cap particle was isolated twice in a genetic screen in which we sought S. pombe cDNAs that when overexpressed rescued the cold-dependent growth defects of yin6Δ cells (Fig. 1A). We named it Rpn7 based on its sequence homology to other Rpn7 proteins.
FIGURE 1. Rpn7 is a high copy suppressor that rescues yin6Δ phenotypes.
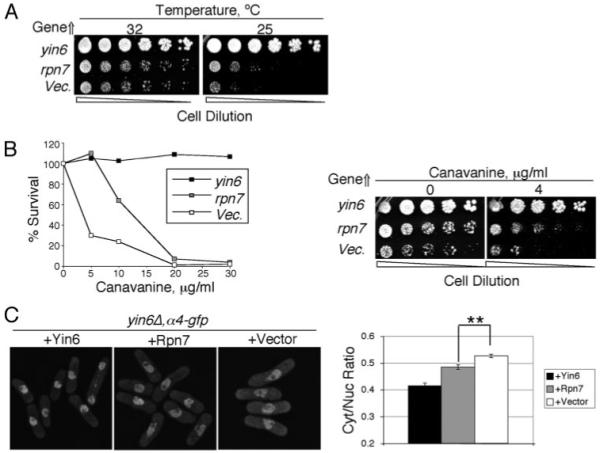
A, yin6Δ cells (strain YIN6K) were transformed with either a vector control (pREP1) or the same vector carrying the yin6 or rpn7 (pREP1YIN6 or pREP1RPN7). Serially diluted cells were spotted on MM plates and incubated at the indicated temperatures. B, left, the same transformed yin6Δ cells as in A were pregrown in MM at 32 °C, and equal numbers were spread on MM plates with the indicated amount of canavanine and incubated at 32 °C for 10 days. The number of colonies that emerged without canavanine was taken as 100% survival. Right, the same transformed yin6Δ cells as in A were serially diluted and spotted on MM plates with the indicated concentrations of canavanine and incubated at 32 °C. C, the α4 subunit was tagged in a yin6Δ strain by GFP via homologous recombination, and Rpn7 and Yin6 were overexpressed in the resulting strain. On the right, the ratio of GFP signal in the cytoplasm versus the nucleus is quantified from the microscopy data. **, p< 0.01 (n = 60 cells).
Canavanine is an arginine analog; cells defective in the proteasome, such as yin6Δ cells, are hypersensitive to this drug. We found that overexpressing Rpn7 also rescued canavanine hypersensitivity of yin6Δ cells (Fig. 1B). Proteasome subunits in normal cells concentrate in the nuclear membrane, but in yin6Δ cells they are more diffused throughout the cell (Fig. 1C) (13). We overexpressed rpn7 in yin6Δ cells and measured the ratio of the catalytic α4 subunit in the cytoplasm versus that in the nucleus. As shown in Fig. 1C, in rpn7-overexpressed cells, substantially more α4 subunit could now be found in the nucleus, suggesting that rpn7 overexpression can directly rescue proteasome mislocalization/disassembly in yin6Δ cells.
Rpn7 Is Essential for Proteasome Functioning
To determine whether rpn7 encodes an essential proteasome subunit, we deleted and replaced it with the ura4 selectable marker via homologous recombination in a diploid cell, and the deletion (rpn7::ura4) was confirmed by PCR. Tetrad analysis of its meiotic products showed that only two of the four spores in a given tetrad formed colonies (Fig. 2A), and they were all auxotrophic for uracil (Ura−) (data not shown), indicating that a single essential gene had been replaced by ura4. Finally, we transformed the heterozygous rpn7Δ/+ diploid cells with a plasmid expressing the rpn7 cDNA. By random spore analysis, we were able to easily identify cells that were uracil-prototrophic (Ura+) (data not shown), indicating that the expression of rpn7 cDNA rescued the lethality of rpn7Δ cells, as expected. These data collectively indicate that rpn7 is a gene essential for viability.
FIGURE 2. Rpn7 is an essential protein required for proteasome functioning.
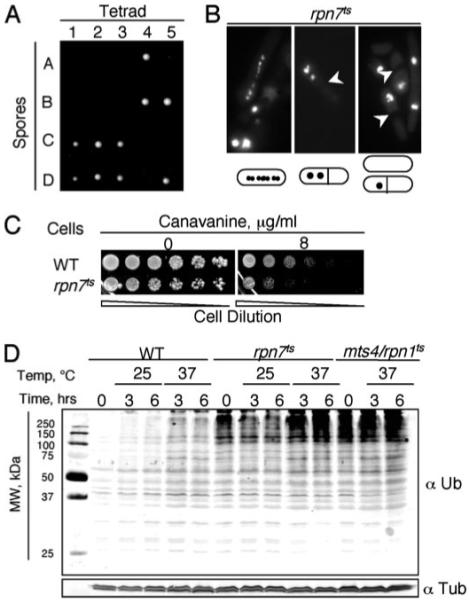
A, rpn7Δ/+ diploid cells were sporulated, the resulting tetrads were dissected, and their spores were incubated at 30 °C. A total of 16 tetrads were dissected, and five are shown. Only two of the four spores in a given tetrad formed colonies, and they were all Ura−. rpn7Δ spores could only divide a few times after germination and then stopped. B, rpn7ts cells were grown in YEAU at 30 °C, an intermediate temperature at which the cells showed mitotic defects, although they did manage to grow, and stained with 4′,6-diamidino-2-phenylindole. Cells with fragmented chromosomes (left) or unevenly distributed chromosomes (middle and right) were readily observed for rpn7ts cells. The arrowheads mark the septa. C, rpn7ts and WT cells were serially diluted and spotted on MM plates with the indicated concentration of canavanine and grown at 25 °C. D, cells (strains SP870, RPN7TS, and MTS4-288) were pregrown at room temperature, and at t = 0, they were shifted to either 25 or 37 °C. The levels of polyubiquitinated proteins over time were analyzed by Western blotting using an anti-ubiquitin (Ub) antibody. Tubulins were examined as the loading control. In our laboratory, accumulation of polyubiquitinated proteins can be readily detected in the rpn1ts mutant even when it is grown at room temperature.
During the course of an unsuccessful gene-tagging experiment, we found that Rpn7 that was C-terminally tagged with GFP without a proper amino acid spacer induced growth defects that were temperature-sensitive (ts), and this ts phenotype could be rescued by overexpressing rpn7 (data not shown). These data indicate that the ts growth defect is caused by loss of Rpn7 function. We thus used this strain, called RPN7TS, to more efficiently analyze Rpn7 functions and its relationship to the proteasome, and the rpn7 allele in this strain is called rpn7ts. In Fig. 2B, we show that at even semipermissive temperature, abnormal mitotic RPN7TS cells accumulated. In particular, cells that had apparently undergone abnormal chromosome segregation were frequently observed, which is consistent with the fact that sister-chromatid separation during anaphase is a key event regulated by the proteasome. To determine whether such a phenotype can be similarly detected in rpn7Δ cells, we sporulated diploid cells heterozygous for rpn7::ura4 in medium lacking uracil, so that only rpn7Δ (rpn7::ura4) cells could grow. We found that all germinated cells divided just a few times and displayed the same mitotic abnormalities as seen in RPN7TS cells (data not shown). We conclude that the rpn7ts allele recapitulates the deficiencies in rpn7Δ cells. Cells defective in the proteasome are expected to show hypersensitivity to canavanine and to accumulate polyubiquitinated proteins, and indeed, both phenotypes were readily detected in RPN7TS cells (Fig. 2, C and D, respectively). These data demonstrated that Rpn7 is critical for proper proteasome functions.
Rpn7 Associates with the Proteasome
Having concluded that Rpn7 is critical for proteasome functions, we went on to determine whether Rpn7 binds the proteasome. We used a different tagging strategy to introduce a spacer between Rpn7 and GFP (see “Experimental Procedures”). In the resulting strain, Rpn7-GFP is expressed from its authentic chromosomal locus by its own promoter. This strain showed no detectable growth defects in a wide range of temperatures and was not hypersensitive to canavanine (data not shown), indicating that unlike the Rpn7-GFP in the RPN7TS strain, this Rpn7-GFP is fully functional. In Fig. 3A, we show that Rpn7-GFP clearly concentrated at the nuclear envelope. To determine whether Rpn7 colocalized with other proteasome subunits by deconvolution microscopy, we tagged Rpn7 with a monomeric RFP (mcRFP) and tagged two proteasome subunits (Rpn11 and α4) with GFP and found that indeed they colocalize (Fig. 3A; data not shown). By contrast, Rpn7-GFP in the RPN7TS strain mislocalized in the nucleoplasm (supplemental Fig. S2). Furthermore, like other proteasome subunits (14), Rpn7-GFP is mislocalized in rpn5Δ and yin6Δ cells (Fig. 3B). These data together strongly suggest that, like other proteasome subunits, Rpn7 is concentrated at the nuclear membrane, and its mislocalization weakens the proteasome.
FIGURE 3. Rpn7 associates with the proteasome and preferentially binds Rpn9.
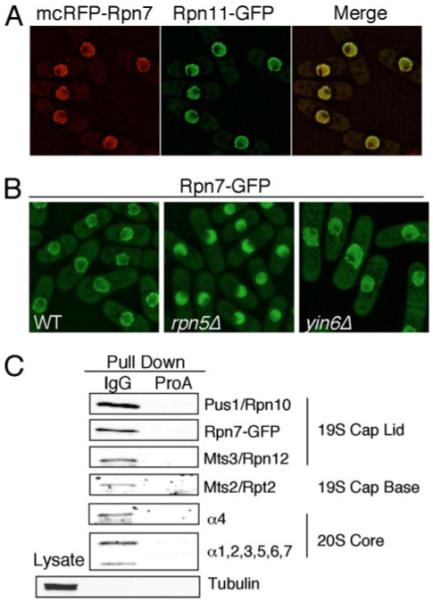
A, monomeric RFP (mcRFP)-Rpn7 was expressed from pMCRPN7 in strains (RPN11GFP and A4GFP) in which Rpn11 or α4 (not shown) was tagged with GFP. The green and red images were deconvolved and merged, and the co-localization was further confirmed by examining each optical section manually. B, endogenous Rpn7 in various strains was tagged by GFP, and the cells were grown in MM at 30 °C and photographed. The strains used in this experiment were RPN7GFP, Y6AR7GFP, and R5AR7GFP. C, cell lysate from strain R10PAR7GFP, whose Rpn10/Pus1 proteasome subunit was tagged with protein A, were pulled down with the IgG bead or the control protein A bead. The pulled down proteins were analyzed by Western blots using antibodies specific for proteins as indicated on the right.
To biochemically examine whether Rpn7 is indeed part of the entire proteasome, we performed a proteasome affinity pull-down and detected the presence of Rpn7 with other proteasome subunits (Fig. 3C). We conclude from these data that Rpn7 is a proteasome subunit and essential for viability and normal mitosis.
Preferential Binding between Rpn7, Rpn5, and Rpn9
We first employed the yeast two-hybrid assay to ascertain to which available proteasome subunit Rpn7 binds. Our data showed that Rpn7 bound Rpn9 (Fig. 4A), but not Rpn5, Rpn10/Pus1, Rpn1/Mts4, Rpt1, Rpt2/Mts2, Rpt5, or Rpt6.
FIGURE 4. Protein-protein interaction between Rpn5, Rpn7, and Rpn9.
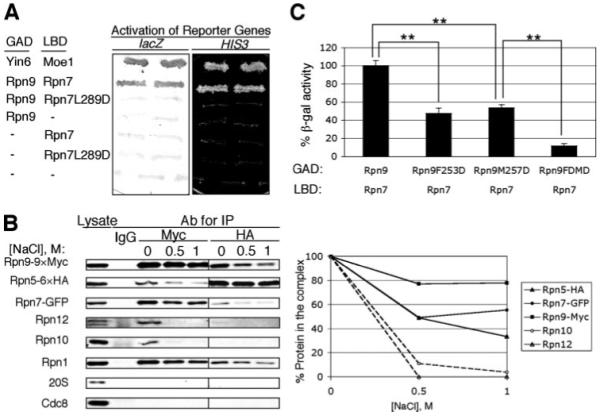
A, proteins fused with the LexA DNA binding domain (LBD) and the Gal4 activation domain (GAD) for the yeast two-hybrid assay are as indicated, and the activation of both HIS3 and lacZ reporters is shown. The plasmids used to express fusion Rpn7 and Rpn9 proteins are pGADRPN9 (13), pLBDRPN7, and pLBDRPN7LD. pVJL11 and pGADgh were used as vector controls, whereas pGADYIN6 and pLBDMOE1 were used as positive controls (7). B, lysates were prepared from cells expressing chromosomally tagged Rpn5-HA6, Rpn7-GFP, and Rpn9-Myc9 (strain R5HAR7-GFPR9MYC). Immunoprecipitation was performed using either anti-Myc or HA antibodies; purified mouse IgG was the antibody control. Immunoprecipitated protein samples were then washed with the indicated concentrations of NaCl. The proteins remaining on the beads were revealed and quantified after Western blots. C, Rpn7 and various forms of Rpn9 were tested for binding using the yeast two-hybrid system. The activity of the β-galactosidase reporter was measured by a colorimetric method. **, p < 0.01 (n = 4 colonies). The plasmids used to express fusion Rpn7 and Rpn9 proteins are pLBDRPN7, pGADRPN9 (13), pGADRPN9MD, pGADRPN9FD, and pGADRPN9FDMD. pVJL11 and pACTII were used as vector controls and showed no activation of the reporter.
In our previous study (14), we found that Rpn5, like Rpn7, also preferentially bound Rpn9. One interpretation of these data is that Rpn5, Rpn7, and Rpn9, three PCI components in the lid, may preferentially bind one another in S. pombe. To investigate this, we epitope-tagged rpn5, rpn7, and rpn9 chromosomally such that they were expressed from their authentic promoters. We then immunoprecipitated under conditions (e.g. lack of ATP) that do not favor the formation of a stable proteasome. Our data in Fig. 4B showed that when Rpn9 (Myctagged) was immunoprecipitated, Rpn5 and Rpn7 were readily detected in the same protein complex; α subunits in the proteasome catalytic core did not stably associate with these three proteins, although at least two components from the lid (Rpn12 and Rpn10) and one from the base (Rpn1) did associate with them.
We then washed the immunoprecipitated protein complexes with salt in increasing concentrations to disrupt the binding between them. Our data showed that although Rpn10 and Rpn12 were efficiently washed away, Rpn5, Rpn7, and Rpn9 stayed in the complex (Fig. 4B), suggesting that under conditions that disrupt the association between S. pombe proteasome lid subunits, Rpn5, Rpn7, and Rpn9 can still bind one another with high affinity. This conclusion was largely confirmed by immunoprecipitating the HA-tagged Rpn5 (Fig. 4B), during which hardly any Rpn12 and no Rpn10 were detectable in the co-precipitated sample. We note that the base subunit Rpn1 also bound this complex strongly, although we did not detect pairwise bindings between them in the two-hybrid assay. We will revisit this issue later under “Discussion.”
Identification of an Essential Leucine Residue in the PCI Domain of Yin6
Rpn7, as well as Yin6 and many proteasome subunits in the lid, contains the PCI domain; however, the exact function of the PCI domain remains elusive. The truncated Int6 identified in the mouse mammary tumors lacks the entire PCI domain (8), and we have found that these truncated human Int6 proteins are not functional in S. pombe (7, 13). These results suggest that the PCI domain is critical for the function of Int6. Since the MMTV insertion in int6 removes nearly one-third of the protein, which may broadly and severely affect the function of any protein, we conducted a structure-function analysis to identify single essential amino acid residues in the PCI domain and then analyzed the function of the resulting proteins.
We first focused on amino acid residues that are highly conserved among many PCI proteins, as illustrated in our original protein sequence analysis from which the PCI domain was defined (3). We then cross-referenced this with protein alignment analyses of nearly all known Int6 proteins available to us at the time to identify those residues that are also conserved in these Int6 proteins (Fig. 5A, arrows). Site-directed mutagenesis was performed to alter them one by one. We then replaced the wild type yin6 with the mutated ones and analyzed the phenotypes of the resulting cells. We have thus concluded that replacing the Leu at position 332 with aspartate (L332D) is particularly detrimental to the function of Yin6, since the cell carrying Yin6L332D displayed the same phenotype as yin6Δ cells (cold-dependent growth defect, abnormally fat and bent cell morphology, and hypersensitivity to canavanine) (Fig. 6A; data not shown). In proteasome pull-down experiments, Yin6L332D did not efficiently bind the proteasome (Fig. 6A). These results are consistent with the idea that the Yin6L332D mutant loses its ability to bind and regulate the proteasome.
FIGURE 5. Sequence alignment of the PCI region among int6 genes and among genes encoding PCI proteins.
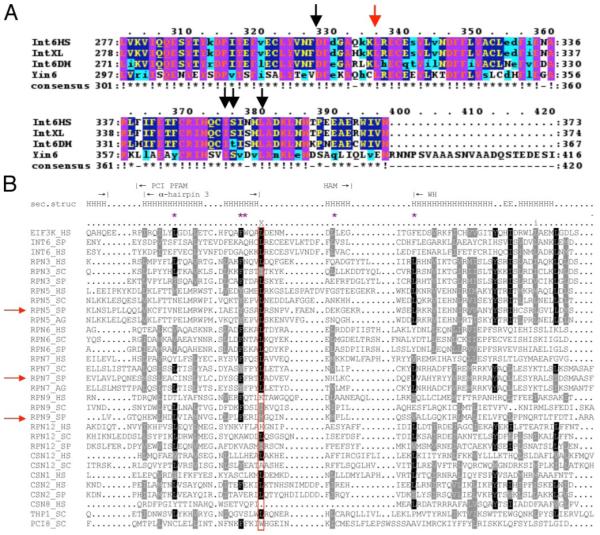
A, sequence alignment was performed using the ClustalW program (31) to seek evolutionarily conserved amino acid residues in the PCI domains of many Int6 proteins. The arrows indicate residues that have been chosen as targets for mutagenesis, and the red arrow marks the described leucine residue that is critical for Yin6 function. B, sequence alignment of a number of genes encoding PCI proteins across different species was performed as described (6). The red rectangle encloses the residues corresponding to the identified conserved leucine. The purple asterisks mark those hydrophobic residues that may interact with Leu121 as revealed in Fig. 8B. S. pombe Rpn5 and Rpn7 are marked by red arrows. The boundaries of the α3 helix (Fig. 8A), the end of the HAM subdomain, and the beginning of the WH subdomain are marked with vertical lines. Residues that are part of a helix are marked H.
FIGURE 6. Identification of an essential leucine residue in three PCI domains.
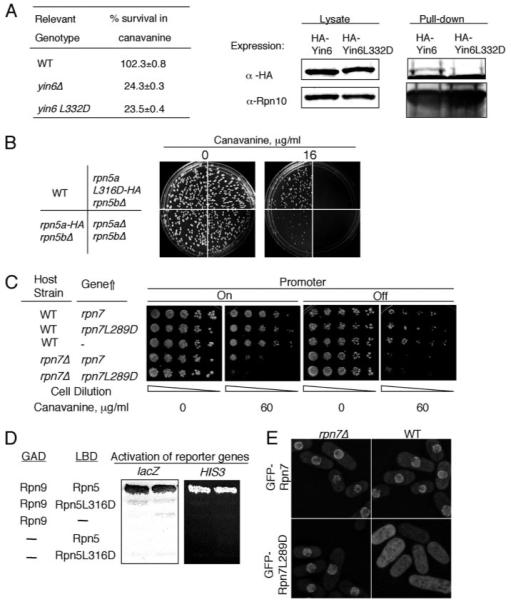
A, left, various strains were pregrown in YEAU at 30 °C to log phase. Equal numbers of cells were spread on MM plates without or with 12 μg/ml canavanine sulfate and incubated at 30 °C for 14 days. For each strain, the number of colonies that emerged on plates without canavanine was taken as 100% survival. The specific strains used were SP870 (WT), YIN6K (yin6Δ), and YIN6LD (yin6L332D). Right, cells whose Rpn10 is tagged with protein A were transformed with pHA-YIN6 or pHA-YIN6LD, and lysates were prepared and pulled down with either IgG or control protein A beads. The pulled-down proteins were analyzed by Western blots using antibodies specific for either HA or Rpn10. B, there are two copies of rpn5 genes, rpn5a and rpn5b. Equal numbers of cells from strains with the indicated genotype were spread on MM plates containing the indicated concentration of canavanine sulfate. The plates were incubated at 30 °C. The specific strains used for this study were as follows (see “Experimental Procedures” and Ref. 14): SP870 (WT), RPN5LD (rpn5aL316D-HA rpn5bΔ), RPN5bARPN5aHA (rpn5a-HA rpn5bΔ), and RPN5UA (rpn5aΔ rpn5bΔ). C, WT or rpn7Δ cells were transformed with the vector control (–, pREP41) or the same vector expressing the indicated genes (pREP41Rpn7 and pREP41RPN7LD). The transformed cells were then serially diluted and spotted on MM plates containing the indicated concentration of canavanine and incubated at 30 °C for 5 days. The expression of the gene was turned off by adding thiamine (20 μM). D, hybrid proteins fused with the LexA DNA binding domain (LBD) and the Gal4 activation domain (GAD) are as indicated, and the activation of both HIS3 and lacZ is shown. The plasmids used for expressing these fusion genes are pGADRPN9, pLBDRPN5, and pLBDRPN5LD. E, WT or rpn7Δ cells were transformed with linearized pREPGF-PRPN7 or pREPGFPRPN7LD to allow integration of these plasmids to the chromosome. These cells were grown in YEAU at 30 °C to log phase before being photographed. The images were captured under identical conditions to reveal the different intensities of GFP signals between samples.
An analogous mutation in human Int6 (Int6L312D) also inactivated Int6, since it could not rescue yin6Δ cells (Fig. 7A). In human cells, Int6 can be found in both the nucleus and cytoplasm (26-28). In live HeLa cells, we expressed YFP-tagged wild type and mutant Int6 proteins and then measured the ratio of YFP signal in the cytoplasm versus that in the nucleus. Our data show that although wild type Int6 is mostly cytoplasmic, Int6L312D frequently accumulates in the nucleus (Fig. 7, B–D). These data are consistent with the idea that the function of this leucine in the PCI domain is evolutionarily conserved among Int6 proteins.
FIGURE 7. The function of the identified Leu is conserved.
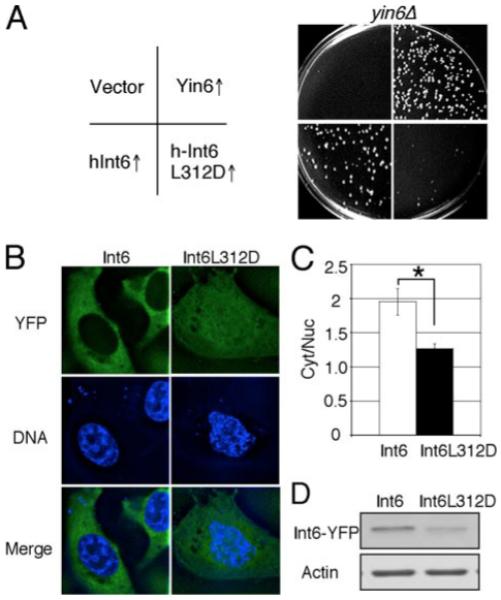
A, the same number of yin6Δ cells (strain YIN6K) transformed with a vector control (pSLF173) or the same vector carrying the indicated genes (pHAYIN6, pHAHINT6, and pHAHINT6LD) were spread on MM plates and incubated at 20 °C for 6 days. The growth of wild-type cells was unaffected by wild-type or mutated yin6 expression (data not shown). B, HeLa cells were transiently transfected with vectors expressing the indicated proteins that were YFP-tagged. The DNA was stained by Hoechst 33342 to mark the nucleus. Images were deconvolved. YFP and DNA signals in the same focal plane are shown and merged. C, the ratios of cytoplasmic versus nuclear YFP signal intensity in the cell were calculated from six cells (*, p < 0.05). D, ectopically expressed Int6 proteins were examined by Western blots, and β-actin was examined as loading control. Int6L312D was expressed at a level lower than that of wild type Int6, thus ruling out the possibility that its mislocalization is simply due to gross overexpression. The plasmids used to express wild-type or mutant int6 in HeLa cells were pYFPHINT6 and pYFPHINT6LD, respectively.
The Same Leucine Is Important for Many Proteasome PCI Subunits
Since the first publication of the PCI sequence alignment, more PCI protein sequences have become available, and we have thus performed another alignment to identify Leu residues in other PCI proteins that might be structurally (and thus also functionally) analogous to Leu332 in Yin6 (Fig. 5B). Besides Yin6, we have characterized two more PCI proteins, Rpn5 and Rpn7, and the alignment data suggest that Leu316 in Rpn5 and Leu289 in Rpn7 are good candidates.
As shown in Fig. 6B, like rpn5Δ cells, the rpn5L316D mutant cells are hypersensitive to canavanine, and similarly Rpn7L289D was not as efficient as wild type Rpn7 in rescuing canavanine hypersensitivity of rpn7Δ cells (Fig. 6C) or that of yin6Δ cells (data not shown).
We have shown in this and a previous study (14) that both Rpn5 and Rpn7 bind Rpn9. However, neither Rpn5L316D (Fig. 6D) nor Rpn7L289D bound Rpn9 (Fig. 4A). Rpn9 is slightly divergent from most PCI proteins in that the S. pombe Rpn9 contains a Met instead of Leu in the position of interest (Fig. 5B). Despite this difference, we found that the binding to Rpn7 was substantially reduced (Fig. 4C) when this Met was changed to Asp (Rpn9M257D). Furthermore, we speculate (see “Discussion”) that the residue in this position can interact with perhaps five hydrophobic residues, such as the phenylalanine at position 253, which are also highly conserved among PCI proteins (Fig. 5B, purple asterisk). Indeed, Rpn9F253D also showed much reduced binding to Rpn7; more importantly, Rpn9-F253D-M257D showed no detectable binding to Rpn7 at all (Fig. 4C).
To further ascertain whether these PCI mutations weaken the ability of these proteasome subunits to properly localize and assemble with the rest of the proteasome subunits in live cells, we analyzed the localization patterns of GFP-Rpn7L289D and GFP-Rpn7 in both wild type and rpn7Δ cells. Our data showed that although Rpn7 localized efficiently to the nuclear envelope in both wild type and rpn7Δ cells (Fig. 6E), Rpn7L289D-GFP mislocalized throughout the cell in wild type cells and slightly mislocalized in the nucleoplasm of rpn7Δ cells. Rpn5L316D was similarly tested and yielded the same results (data not shown). These localization data suggest that the tested PCI mutant proteasome subunits are not nearly as efficient as their normal counterparts in localizing and assembling at the nuclear membrane into the proteasome; thus, the presence of the wild type versions easily competed out the assembly and localization of the mutant versions. Collectively, we conclude from these results that the identified Leu residue in the PCI domain plays a critical role in mediating the binding between PCI proteins in the proteasome, thus affecting how the proteasome is assembled and localized in the cell.
DISCUSSION
To better define the role of the PCI domain in proteasome functioning, we have isolated a new PCI-containing proteasome subunit, Rpn7, from S. pombe. Our data firmly support the idea that Rpn7 is a proteasome subunit because it associates with the proteasome and because its inactivation appears to block the removal of polyubiquitinated proteins as well as abnormal proteins that contain canavanine instead of arginine. Guided by protein sequence alignments, we performed structure-function analyses to identify in the PCI domain a Leu residue that is critical for the functions of at least three different PCI proteins; i.e. Yin6L332D, Rpn5L316D, and Rpn7L289D cannot fully replace their wild type counterparts, and this observation correlates with the finding that they do not bind efficiently to the proteasome. These data support the hypothesis that the identified Leu residue is critical for many PCI domains to function properly.
Besides Asp, we have mutated this Leu residue to an alanine in Yin6, and the resulting mutant protein showed very little loss of function (supplemental Fig. S3). These data are consistent with the possibility that this Leu is in contact with other residues, residing either elsewhere in the PCI domain or in its binding partners, via hydrophobic interactions, which can be disrupted by the presence of an Asp residue. Based on the study of Rpn9, we further speculate that this Leu may be substituted by other amino acids with hydrophobic side chains, such as Met.
The Leu (or Met) identified from this study corresponds to Leu121 in human eIF3k, whose crystal structure has been reported. We have created a figure based on the reported structure of eIF3k to better illustrate and summarize our findings (Fig. 8). Leu121 resides at the end of the last pair of antiparallel α-helices in the TPR-like subdomain (α3 helix; red in Fig. 8A). Within a 5-Å radius of this Leu, five hydrophobic amino acid residues (Leu105, Phe117, Trp118, Leu128, and Phe134; Fig. 8B, purple) are so positioned that their side chains may interact with this Leu. Although it is evident that most of these hydrophobic amino acids reside within the TPR-like subdomain, Phe134 is in the WH subdomain. It is thus possible that the identified Leu can modulate the proper folding and/or spatial arrangement either within the TPR-like subdomain or between the TPR and the WH subdomains by interacting with these hydrophobic side chains. The importance of such hydrophobic interaction is further supported by the observation that three of the five described potential hydrophobic partners (e.g. Leu105, Phe117, and Phe134) are highly conserved among many PCI proteins (Fig. 5B), and we have validated the importance of Phe117 in Rpn9 (by altering the corresponding Phe253) (Fig. 4C).
FIGURE 8. Three-dimensional structure of the PCI domain in human eIF3k.
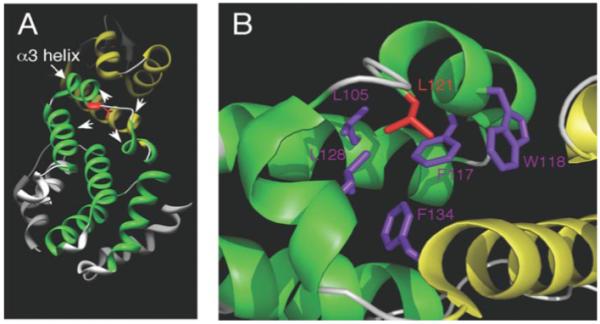
A, the N-terminal TPR/HAM subdomain is colored green, whereas the C-terminal WH subdomain is colored yellow. The described leucine residue corresponds to Leu121 (red) in eIF3k and is located at the end of the α3 helix in the N-terminal TPR/HAM subdomain. Three helices from the TPR-like subdomain and one helix from the WH subdomain (arrowheads) contain hydrophobic amino acid residues that may interact with Leu121 (see B). B, within a 5-Å radius from this leucine, at least 5 hydrophobic amino acids colored in purple (Leu105, Phe117, Trp118, Leu128, and Phe134) can be readily identified that may interact with this leucine. We note that although most of these neighboring hydrophobic residues reside in the TPR/HAM domain, Phe134 is in the WH domain. These images were created by Chimera (32) and PyMOL software.
Isono et al. (29) have identified a number of ts Rpn7 mutants in budding yeast. Each of these Rpn7 ts mutants contains more than one amino acid substitution, at least one of which in each mutant protein is in the PCI domain. Interestingly, none of these mutations can singularly inactivate budding yeast Rpn7; in contrast, mutating just the Leu identified in this study substantially weakens the function of S. pombe Rpn7. One interpretation of this is that by potentially modulating the conformation and folding of both the TPR-like and the WH subdomains, a substitution of this Leu to Asp may more globally and profoundly influence the structure of the PCI domain, thus leading to a greater loss of activity. We cannot exclude the possibility that this Leu may be part of a binding pocket, with which critical PCI binding partners interact.
Since each of the two subdomains in the PCI domain contains ample surface for protein-protein interactions, it is reasonable to assume that the main role of the PCI domain is to assemble a protein complex. We have previously observed that the S. pombe lid PCI protein Rpn5 is grossly mislocalized only in the lid mutants, whereas in the base mutants, Rpn5 can at least still concentrate in the nucleus (14). A recent study of budding yeast proteasome has similarly shown that mutations affecting the lid, but not the base, lead to a grossly mislocalized lid (30). These results are consistent with the possibility that at least some components in the lid preferentially bind one another and that they do so in a highly cooperative manner.
In this and other studies of ours (14), by the yeast two-hybrid assay, we found that among proteasome subunits available for the two-hybrid assay, Rpn9 preferentially binds both Rpn5 and Rpn7. By contrast, Rpn7 does not bind Rpn5. We performed glycerol gradient centrifugation experiments and found that all of the detectable Rpn7 co-fractionated with other proteasome subunits (supplemental Fig. S4). This observation does not support the hypothesis that a separate Rpn5-9-7 ternary complex can be readily detectable biochemically. However, our data in Fig. 4B reveal that Rpn5, Rpn9, and Rpn7 do bind one another with affinity higher than that with other proteasome subunits. In passing, we speculate that our two-hybrid data are also consistent with the possibility that Rpn9 may bridge the binding between Rpn5 and Rpn7 to result in a Rpn5-9-7 configuration. We note that this may explain why, when Rpn9 is first immunoprecipitated, high levels of both Rpn5 and Rpn7 can be co-immunoprecipitated. By contrast, although Rpn5 can efficiently bring down Rpn9, it is less efficient in bringing down Rpn7. Finally, our results surprisingly show that a base subunit, Rpn1, also tightly associates with Rpn5-7-9, although it does not bind any of them in the two-hybrid assay. One interpretation of this is that Rpn1 may associate with Rpn5-7-9 via other proteins in the same complex.
Supplementary Material
Acknowledgments
We thank Chin-tung (Tommy) Chen for the expert help in the initial genetic screen and Gary Chamness for critically reading the manuscript. We also thank Nick Rhind, Chris Norbury, Roger Tsien, and Colin Gordon for kindly providing materials that were critical for this study. We thank Drs. Wah Chiu and Matt Baker from the National Center for Macromolecular Imaging at Baylor College of Medicine for computational assistance. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by National Institutes of Health Grant P41 RR-01081).
Footnotes
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
- PCI
- proteasome-COP9-initiation factor
- WH
- winged helix
- eIF3
- eukaryotic initiation factor-3
- TPR
- tetratricopeptide repeat
- HAM
- HEAT analogous motif
- HEAT
- Huntington elongation factor 3, a subunit of protein phosphatase 2A
- YEAU
- yeast extract medium
- MM
- minimal medium
- RFP
- red fluorescent protein
- YFP
- yellow fluorescent protein
- GFP
- green fluorescent protein
- PIPES
- 1,4-piperazinediethanesulfonic acid
- ts
- temperature-sensitive
X. Fu and E. Chang, unpublished results.
REFERENCES
- 1.Voges D, Zwickl P, Baumeister W. Annu. Rev. Biochem. 1999;68:1015–1068. doi: 10.1146/annurev.biochem.68.1.1015. [DOI] [PubMed] [Google Scholar]
- 2.Aravind L, Ponting CP. Protein Sci. 1998;7:1250–1254. doi: 10.1002/pro.5560070521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hofmann K, Bucher P. Trends Biochem. Sci. 1998;23:204–205. doi: 10.1016/s0968-0004(98)01217-1. [DOI] [PubMed] [Google Scholar]
- 4.Chang EC, Schwechheimer C. EMBO Rep. 2004;5:1041–1045. doi: 10.1038/sj.embor.7400275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei Z, Zhang P, Zhou Z, Cheng Z, Wan M, Gong W. J. Biol. Chem. 2004;279:34983–34990. doi: 10.1074/jbc.M405158200. [DOI] [PubMed] [Google Scholar]
- 6.Scheel H, Hofmann K. BMC Bioinformatics. 2005;6:71. doi: 10.1186/1471-2105-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yen HS, Chang EC. Proc. Natl. Acad. Sci. U. S. A. 2000;97:14370–14375. doi: 10.1073/pnas.97.26.14370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marchetti A, Buttitta F, Miyazaki S, Gallahan D, Smith G, Callahan R. J. Virol. 1995;69:1932–1938. doi: 10.1128/jvi.69.3.1932-1938.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yen HC, Chang EC. Cell Cycle. 2003;2:81–83. [PubMed] [Google Scholar]
- 10.Sap J, Chang EC. AfCS-Nature Molecule Pages. 2005 10.1038/mp.a003493.01. [Google Scholar]
- 11.Asano K, Merrick WC, Hershey JW. J. Biol. Chem. 1997;272:23477–23480. doi: 10.1074/jbc.272.38.23477. [DOI] [PubMed] [Google Scholar]
- 12.Bandyopadhyay A, Lakshmanan V, Matsumoto T, Chang EC, Maitra U. J. Biol. Chem. 2002;277:2360–2367. doi: 10.1074/jbc.M107790200. [DOI] [PubMed] [Google Scholar]
- 13.Yen HC, Gordon C, Chang EC. Cell. 2003;112:207–217. doi: 10.1016/s0092-8674(03)00043-6. [DOI] [PubMed] [Google Scholar]
- 14.Yen HC, Espiritu C, Chang EC. J. Biol. Chem. 2003;278:30669–30676. doi: 10.1074/jbc.M302093200. [DOI] [PubMed] [Google Scholar]
- 15.Norbury C, Moreno S. Methods Enzymol. 1997;283:44–59. doi: 10.1016/s0076-6879(97)83006-6. [DOI] [PubMed] [Google Scholar]
- 16.Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, Stewart A, Sgouros J, Peat N, Hayles J, Baker S, Basham D, Bowman S, Brooks K, Brown D, Brown S, Chillingworth T, Churcher C, Collins M, Connor R, Cronin A, Davis P, Feltwell T, Fraser A, Gentles S, Goble A, Hamlin N, Harris D, Hidalgo J, Hodgson G, Holroyd S, Hornsby T, Howarth S, Huckle EJ, Hunt S, Jagels K, James K, Jones L, Jones M, Leather S, McDonald S, McLean J, Mooney P, Moule S, Mungall K, Murphy L, Niblett D, Odell C, Oliver K, O’Neil S, Pearson D, Quail MA, Rabbinowitsch E, Rutherford K, Rutter S, Saunders D, Seeger K, Sharp S, Skelton J, Simmonds M, Squares R, Squares S, Stevens K, Taylor K, Taylor RG, Tivey A, Walsh S, Warren T, Whitehead S, Woodward J, Volckaert G, Aert R, Robben J, Grymonprez B, Weltjens I, Vanstreels E, Rieger M, Schafer M, Muller-Auer S, Gabel C, Fuchs M, Fritzc C, Holzer E, Moestl D, Hilbert H, Borzym K, Langer I, Beck A, Lehrach H, Reinhardt R, Pohl TM, Eger P, Zimmermann W, Wedler H, Wambutt R, Purnelle B, Goffeau A, Cadieu E, Dreano S, Gloux S, Lelaure V, Mottier S, Galibert F, Aves SJ, Xiang Z, Hunt C, Moore K, Hurst SM, Lucas M, Rochet M, Gaillardin C, Tallada VA, Garzon A, Thode G, Daga RR, Cruzado L, Jimenez J, Sanchez M, del Rey F, Benito J, Dominguez A, Revuelta JL, Moreno S, Armstrong J, Forsburg SL, Cerrutti L, Lowe T, McCombie WR, Paulsen I, Potashkin J, Shpakovski GV, Ussery D, Barrell BG, Nurse P. Nature. 2002;415:871–880. doi: 10.1038/nature724. [DOI] [PubMed] [Google Scholar]
- 17.Chang EC, Barr M, Wang Y, Jung V, Xu H, Wigler HM. Cell. 1994;79:131–141. doi: 10.1016/0092-8674(94)90406-5. [DOI] [PubMed] [Google Scholar]
- 18.Sivakumar S, Porter-Goff M, Patel PK, Benoit K, Rhind N. Methods. 2004;33:213–219. doi: 10.1016/j.ymeth.2003.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shu X, Shaner NC, Yarbrough CA, Tsien RY, Remington SJ. Biochemistry. 2006;45:9639–9647. doi: 10.1021/bi060773l. [DOI] [PubMed] [Google Scholar]
- 20.Bähler J, Wu JQ, Longtine MS, Shah NG, McKenzie A, 3rd, Steever AB, Wach A, Philippsen P, Pringle JR. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 21.Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, Knop M. Yeast. 2004;21:947–962. doi: 10.1002/yea.1142. [DOI] [PubMed] [Google Scholar]
- 22.Alfa C, Fantes P, Hyams J, McLeod M, Warbrick E. Experiments with Fission Yeast. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1993. [Google Scholar]
- 23.Balasubramanian MK, Helfman DM, Hemmingsen SM. Nature. 1992;360:84–87. doi: 10.1038/360084a0. [DOI] [PubMed] [Google Scholar]
- 24.Leggett DS, Glickman MH, Finley D. In: Ubiquitin-Proteasome Protocols. Patterson C, Cyr DM, editors. Humana Press; Totowa, NJ: 2005. pp. 57–70. [Google Scholar]
- 25.Stone M, Hartmann-Petersen R, Seeger M, Bech-Otschir D, Wallace M, Gordon C. J. Mol. Biol. 2004;344:697–706. doi: 10.1016/j.jmb.2004.09.057. [DOI] [PubMed] [Google Scholar]
- 26.Watkins SJ, Norbury CJ. Cell Proliferation. 2004;37:149–160. doi: 10.1111/j.1365-2184.2004.00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rasmussen SB, Kordon E, Callahan R, Smith GH. Oncogene. 2001;20:5291–5301. doi: 10.1038/sj.onc.1204624. [DOI] [PubMed] [Google Scholar]
- 28.Guo J, Sen G. J. Virol. 2000;74:1892–1899. doi: 10.1128/jvi.74.4.1892-1899.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Isono E, Saeki Y, Yokosawa H, Toh-e A. J. Biol. Chem. 2004;279:27168–27176. doi: 10.1074/jbc.M314231200. [DOI] [PubMed] [Google Scholar]
- 30.Isono E, Nishihara K, Saeki Y, Yashiroda H, Kamata N, Ge L, Ueda T, Kikuchi Y, Tanaka K, Nakano A, Toh EA. Mol. Biol. Cell. 2007;18:569–580. doi: 10.1091/mbc.E06-07-0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson JD, Higgins DG, Gibson TJ. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.