

Abstract
Epithelial-mesenchymal transition (EMT) is a key event in the generation of invasive tumor cells. A hallmark of EMT is the repression of E-cadherin expression, which is regulated by various signal transduction pathways including extracellular signal regulated-kinase (ERK) and Wnt. These pathways are highly interconnected via multiple coupled feedback loops (CFLs). As the function of such coupled feedback regulations are difficult to analyze experimentally, we used a systems biology approach where computational models were designed to predict biological effects that result from the complex interplay of coupled feedback loops. Using EGF and Wnt as input and E-cadherin transcriptional regulation as output we established an ordinary differential equation (ODE) model of the ERK and Wnt signaling network containing six feedback links and used extensive computer simulations to analyze the effects of these feedback links in isolation and different combinations. The results show that the feedbacks can generate a rich dynamic behavior leading to various dose response patterns and have a decisive role in determining network responses to EGF and Wnt. In particular, we made two important findings. First, that coupled positive feedback loops (CPFLs) composed of phosphorylation of Raf kinase inhibitor RKIP by ERK and transcriptional repression of RKIP by Snail have an essential role in causing a switch-like behavior of E-cadherin expression. Second, that RKIP expression inhibits EMT progression by preventing E-cadherin suppression. Taken together, our findings provide us with a system-level understanding on how RKIP can regulate EMT progression and may explain why RKIP is down-regulated in so many metastatic cancer cells.
Keywords: Systems biology, ERK signaling pathway, Wnt signaling pathway, Epithelial-Mesenchymal Transition, feedback loop, RKIP, dynamics, mathematical modeling
INTRODUCTION
Epithelial-mesenchymal transition (EMT) plays an important role in embryonic development and the occurrence of EMT during tumor progression allows benign (i.e., noninvasive and nonmetastatic) tumor cells to acquire the capacity to infiltrate surrounding tissue and to ultimately metastasize to distinct sites (1-3). During this transition, epithelial cells undergo a morphological change that is appropriate for migration in the extracellular matrix and settlement in an area involved in organ formation or metastasis. EMT is characterized by loss of cell adhesion, repression of E-cadherin, and increased cell mobility. Many oncogenic pathways activated by growth factors such as epidermal growth factor (EGF), tumor growth factor (TGF), and hepatocyte growth factor (HGF), Src, Ras, integrin, Wnt/β-catenin, and Notch can induce EMT (4). In particular, ERK and Wnt/β-catenin signaling pathways have been shown to activate the transcriptional repressors Snail and Slug that suppress E-cadherin expression, leading to the development of EMT (4-6).
Polypeptides including EGF and Wnt secreted in many malignant tumor cells have been reported to be involved in EMT and the corresponding signal transduction pathways form a highly interconnected network through multiple coupled feedback loops (CFLs). Table 1 summarizes all possible positive and negative feedback loops suggested from previous experiments (7-24), which comprise CFLs between the EGF and Wnt signaling pathways. The compilation of feedback loops and topologies of the networks considered here are based mainly on studies done in colorectal cancer, and to a smaller extent in breast and prostate cancer cells. These observations indicate a highly complex regulation through a variety of feedback loop motifs. They further suggest that feedback loops play key roles in the regulation of E-cadherin and EMT. As little is known about the functional roles of the feedback loops that interconnect the ERK and Wnt signaling pathways, our focus was to identify the functional roles of such CFLs. However, it is difficult to experimentally analyze the effects of such CFLs or in fact even combinations of simple feedback regulations and to envisage how the behaviors of such a complex nonlinear systems can be understood without utilizing a formal in silico approach. In this paper, we used computational modeling to predict the behavior of the system and its responses to perturbations of the coupled feedback loops. For this purpose, we integrated experimental data related to ERK and Wnt signaling pathways and constructed a map of the protein interactions and regulations. We note, however, that the constructed network might not be conserved in all cancer cells because of their inherent heterogeneity as well as tumor and cell type specific differences. This implies that there could be different network topologies depending on tumors, tumor cell types and heterogeneity between tumor cells that arise from stochastic processes or interactions with different neighboring cells. To address this problem, we considered different possible network topologies, and analyzed their respective dynamical characteristics. In particular, we first developed a system-level signaling network model that contains protein interactions and regulations reported in the ERK and Wnt signaling pathways by integrating available experimental results and employing an established basic mathematical model of each pathway. As our model was developed mainly on data from colorectal cancer cell lines it is most likely applicable to this type of cancer. However, it is generic in the sense that it also will apply to other cancers with a similar network topology and protein expression profiles such as breast and prostate cancers (25). Then, we compared different network topologies by introducing individual or combinations of feedbacks through extensive computational simulations. We found that the coupled positive feedback loops (CPFLs), formed by ERK phosphorylation of RKIP and transcriptional repression of RKIP expression by Snail, cooperatively cause a switch-like behavior of E-cadherin expression. Furthermore, we revealed that the RKIP expression inhibits the EMT progression by counteracting the suppression of E-cadherin.
Table 1.
All feedback loops between the EGF & Wnt signaling pathways and key effects of these feedback loops suggested from the simulation study. PFL and NFL stand for positive feedback loop and negative feedback loop, respectively. The arrow denotes activation and the blunted arrow denotes inhibition.
| Type | Possible feedback loops | Related tumors | Experimental observation |
Key effects of the feedback loops suggested from this study |
|---|---|---|---|---|
| PFL | β-catenin/TCF complex → Ras → ERK 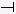 GSK3β ; β- GSK3β ; β-catenin/TCF complex |
Colon and liver cancer |
(7-14) | · It increases the sensitivity of Snail and E- cadherin expression to EGF stimulation |
| PFL | ERK GSK3β PKCδ → ERK |
Colon and prostate cancer |
(10, 15) | · It induces the switch-like behavior of E-cadherin expression in response to EGF stimulation. · It increases a large state change of E-cadherin in response to EGF stimulation |
| PFL | ERK RKIP MEK → ERK |
Colon cancer | (16-18) | · It induces the switch-like behavior of E-cadherin expression. · It decreases the state transition interval in the state change of E-cadherin |
| PFL | ERK → Snail RKIP MEK → ERK |
Prostate cancer | (19) | · If one of these feedback loops is blocked, the switch-like behavior disappears. |
| PFL | ERK GSK3β Snail RKIP MEK → ERK |
Prostate cancer | (20) | |
| NFL | ERK Ras → ERK |
Skin cancer | (21) | · This feedback loop, together with the PFL mediated by RKIP, increases EC50 of E-cadherin expression in response to EGF. |
| NFL | β-catenin/TCF complex→ Axin β-catenin/TCF complex |
Colon and liver cancer |
(22-23) | · It does not have any significant effect on the cellular responses considered here. |
MATERIALS AND METHODS
Mathematical modeling and model reduction
The ordinary differential equations for the mathematical model of ERK and Wnt signaling pathways including their transcriptional regulations are presented in Supplementary Data A where the reduction procedure of the developed mathematical model is also described. The numerical integration was performed using Matlab R2009a software. The kinetic parameters were obtained or modified from the previous model (18, 26) and those not available from the literature were estimated through iterative simulations such that they are qualitatively well in accord with the experimental evidences. The parameter estimates used in the numerical simulation are summarized in Table S2 of Supplementary Data A.
Quantification of the results and identification of dominant feedback combinations using K-map analysis
To quantify the simulation results and to characterize the dose-response curves, we introduced the following three output indexes (Fig. 1): STI, SD and EC50. STI denotes the stimulus range corresponding to the state transition between 80% (RR80) and 20% (RR20) of the E-cadherin response range (RR), respectively. SD indicates the range of E-cadherin expression level between RR20 and RR80. EC50 denotes the range of stimulus over which the expression of E-cadherin decreases by 50%. Using these three output indexes, we quantified the dose-response curves and summarized them in a table describing the relationships between 32 feedback combinations and output indexes (Fig. 1, step 1, and see also Tables S1-S3 of Supplementary Data D).
Figure 1. Identification of feedback combinations that have a significant effect on E-cadherin expression.

To identify the feedback combinations that have a significant effect on E-cadherin expression, we introduced three output indexes, SD, STI and EC50, and quantified the dose-response curves of 32 network topologies for oncogenic stimulation. These are summarized in ‘the feedback combination-output indexes table’ (step 1). The real-valued outputs of each output index are discretized. Then a truth table was constructed from the previous feedback combination table based on the logical representation of each value (step 2). By employing the K-map analysis method, we determined the simplest form of each Boolean function over the 32 feedback combinations and output indexes (step 3&4). Each term of the Boolean function denotes a particular feedback combination that has a significant effect on each output index. In the output indexes of a dose-response curve, RR20, RR50, and RR80 denote 20%, 50%, and 80% of the response range, respectively, where x-axis represents the normalized stimulation (‘0’ denotes low and ‘1’ denotes high) and y-axis represents the expression level of E-cadherin.
In order to discretize the continuous values of output indexes, we identified the feedback combinations that have a statistically significant effect on output indexes. It should be noted that the 32 values of each output index are observed to follow almost the normal distribution. We let ZSD, ZEC50 and ZSTI be the normalized probability variables of XSD, XEC50 and XSTI, respectively, and selected those with Z ≥ Z0 for ZSD and ZEC50 where the statistical significance p(Z ≥ Z0) = 0.1 and Z ≤ −Z0 for ZSTI where p(Z ≥ −Z0) = 0.1, and then assigned them with the logical value of ‘1’; the non-significant values were assigned with the logical value of ‘0’ (Fig. 1, step 2). Then, we constructed a truth table where each possible feedback combination is represented by a binary string of length five. Each bit of the string represents an individual feedback link (1 if the corresponding link is connected and 0 if not). The three output indexes are also represented by binary strings of length three (Fig. 1, Truth table). By scanning out each line of the truth table, we could enumerate all possible feedback combinations that have a significant effect on each output index. We then constructed a Boolean function by summing up those with the Boolean operator (+). However, as the resulting function can take a complex form, we then further apply K-map analysis in order to obtain a simplified form. This enables us to determine the simplest form of the logic function that represents the relationship between the feedback combinations and the corresponding output index (Fig.1, step 3&4). For instance, the Boolean function representing the relationship between the feedback combinations and SD was expressed as 7 feedback combinations as follows: , , , , , , , (Fig. 1, bottom). By applying the K-map analysis method, this Boolean function can be simplified as three feedback combinations: , , . See Supplementary Data C for further details.
RESULTS
Mathematical model
A diagrammatic molecular interaction map
In order to develop a mathematical model, we assembled the interactions between ERK and Wnt signaling pathways including their transcriptional regulations in the form of a diagrammatic interaction map (Fig. 2). This map organizes the protein interactions and functional regulations and provides a basis for further development of a mathematical model. The map consists of three modules: the ERK and Wnt signaling pathway modules and the gene transcriptional regulation module (see Supplementary Data A for details). The interaction map shows multiple CFLs that are formed by protein interactions and regulations (Fig. 3A). We have investigated the role of these feedback loops in the EMT progression through the regulation of E-cadherin.
Figure 2. The interaction map of the ERK and Wnt signaling pathways.

The interaction map consists of the ERK and Wnt signaling modules and the transcriptional regulation module. Here, xi denotes the state variable representing the concentration of a signaling molecule and vi (referred to as a ‘process’) denotes the protein interaction or regulation. Details on each process of biochemical reaction can be found in Table S1 of Supplementary Data A.
Figure 3. Multiple feedback loops connect the ERK and Wnt signaling pathways.

(A) The ERK and Wnt signaling pathways are highly interconnected with each other through multiple feedback loops. Red lines with a bar-end denote negative feedback links, while blue lines with an arrow-end denote positive feedback links. The feedback link F1 represents Ras stabilization by the β-catenin/TCF complex, F2 represents the inhibition of MEK by RKIP, F3 represents the inhibition of SOS/Grb2 complex by ERK, F4 represents the inhibition of GSK3β by ERK, F5 represents the inhibition of PKCδ by GSK3β, and F6 represents the expression of Axin by β-catenin/TCF complex. (B) Principal component analysis (PCA) of the network topologies that are determined by different feedback combinations. The 32 network topologies were analyzed using PCA with respect to the five outputs: the activation of ERK, the expression of Snail, Slug and E-cadherin, and the formation of the β-catenin/TCF complex, and then the results were projected onto two dimensions for which 96% of output variances were accounted. Label of each feedback combination is shown in the right of this panel.
Simulation results
Cellular responses to oncogenic stimulation in the ERK and Wnt signaling pathways are greatly affected by the different feedback combinations
We have reconstructed a complex network model of the ERK and Wnt signal transduction pathways by integrating protein interactions and regulations from the literature. To investigate how diverse cellular responses are produced from this signaling network, we considered various network topologies obtained from the combination of all possible protein interactions and regulations that are likely to play essential roles in the ERK and Wnt signaling pathways by forming coupled feedbacks. Such interactions and regulations include Ras stabilization by the β-catenin/TCF complex (F1), inhibition of the physical interaction between Raf-1 and MEK by RKIP (F2), inhibition of the SOS/Grb2 complex by active ERK (F3), inhibition of GSK3β by active ERK (F4), inhibition of PKCδ by GSK3β (F5) and expression of Axin by the β-catenin/TCF complex (F6) (Fig. 3A). By considering the protein level of RKIP F2 implicitly encompasses the transcriptional regulation of RKIP by Snail. As each of these interactions and regulations (F1-F6) is a key element within a feedback loop, we call it a feedback link. It should be noted that the feedback link denotes protein interaction or regulation but not the feedback loop itself. By considering all possible combinations of such feedback links (shortly, ‘feedback combinations’), we have 64 (26=64 possible combinations) network topologies in total (Note that feedback links can have two possible states: ‘connected’ and ‘non-connected’). We have simulated the cellular responses of each network topology to normalized oncogenic stimuli of EGF and Wnt. The activation of ERK, the expression of Snail, Slug and E-cadherin, and the formation of the β-catenin/TCF complex were considered as outputs. As in the initial stage of simulation we found that the feedback link F6 does not have a significant effect on the change of those responses (data not shown), we excluded it from further analysis. Thus, we have total 32 network topologies (25=32 possible combinations (Fig. 3B)). To investigate how the outputs of the signaling networks vary depending on a specific network topology, the 32 network topologies were analyzed using principal component analysis (PCA). Here, PCA was carried out with respect to five outputs: the activation of ERK, the expression of Snail, Slug and E-cadherin, and the formation of the β-catenin/TCF complex, and then the results were projected onto two dimensions for which 96% of the output variances were accounted (Fig. 3B). The 32 network topologies can be clustered into four groups: Group A that has a dominant effect on both the first principal component (PC1) and the second principal component (PC2) both in a positive way, Group B on PC1 in a positive way, Group D on PC1 in a negative way, and Group C on PC2 in a negative way. Each group has a distinct characteristic of feedback link combinations: networks in Group A have both the feedback links F1 and F4 but not F3, networks in Group B contain F1 but not F3 and F4, networks in Group C have F2 and F3 but not F1 and F4, and networks in Group D have F4. Although the degree of group separation may depend on the parameter values used in the numerical simulation, these results suggest that different feedback topologies in the network can cause different cellular responses to the same oncogenic stimulation and that some particular feedback combinations have a profound effect on the output of signaling networks. This result also could potentially explain how biological specificity can be encoded by combinatorial linkage of a limited array of components.
The coupled positive feedback loops mediated by RKIP cause a switch-like behavior of E-cadherin expression
E-cadherin plays crucial and direct roles in EMT at developmental stages as well as during the progression of malignant tumor cells, i.e., loss of E-cadherin expression is an important mark of EMT (1-3). To systematically identify feedback combinations that have a significant effect on E-cadherin expression, we simulated the dose-response characteristics of E-cadherin expression for 32 network topologies as three-dimensional surfaces with respect to two oncogenic stimuli, i.e. EGF and Wnt. We found that E-cadherin expression is remarkably affected by different feedback combinations (Fig. S1A&B of Supplementary Data C). To further investigate, we considered the dose-response curves of E-cadherin expression to EGF (and Wnt) stimulation for a fixed level of Wnt (and EGF, respectively) and also the dose-response curves for a gradual increase of both EGF and Wnt at the same level. By introducing three output indexes (i.e., state difference (SD), state transition interval (STI), and effective concentration 50 (EC50)), we quantified the dose-response curves and identified the feedback combinations (i.e., combinations of ‘connected’ and ‘non-connected’ feedback links) that have a significant effect on each output index. To this end, we employed K-map analysis (27) which is a graphical method widely used in engineering circuit analysis for simplification of logic equations representing the Boolean logic relationship between input and output (see Fig. 1 and Materials and Methods for detailed procedures and Supplementary Data B for detailed K-map analysis).
The feedback combinations that have a significant influence on three output indexes and, among those, the most commonly involved feedback combinations are summarized in Table S1 of Supplementary Data C where all these were investigated by gradually increasing EGF for five fixed levels of Wnt and vice versa. Table S1 of Supplementary Data C also shows the feedback combinations that have a significant effect on the output indexes for simultaneous increase of both EGF and Wnt. In particular, the feedback combination was most commonly observed with respect to SD for all levels of Wnt. The logical expression means all possible combinations containing two feedback links F4, F5 while not containing F3. This suggests that network topologies containing this feedback combination would have a significant effect on SD for a gradual increase of EGF with any fixed level of Wnt. The three feedback combinations , and were most commonly observed with respect to STI and the two feedback combinations and were most commonly observed with respect to EC50 for all levels of Wnt. These results suggest that network topologies containing these feedback combinations would have a significant effect on STI and EC50, respectively, for gradual increase of EGF at any fixed level of Wnt. However, there was no commonly observed feedback combination for different levels of EGF with respect to the three output indexes. The commonly observed feedback combinations for gradual increase of both EGF and Wnt were similar to those for gradual increase of EGF. We note, however, that the feedback combinations and significantly affected STI and EC50, respectively, only for the combined stimulation. Furthermore, the network topologies having a significant effect on SD and STI contained the feedback links F4F5 and F2, respectively (Fig. 4A&B), and those affecting EC50 contained the feedback links F2F3 (Fig. 4C). Those feedback links form feedback loops with non-indexed links (see the red line of Fig. 4). Taken together, this implies that the feedback loops containing F2 have two roles in regulating E-cadherin expression: one is to increase EC50 and the other is to shorten STI. This suggests the hypothesis that CPFL mediated by RKIP induce a switch-like behavior of E-cadherin for the gradual increase of EGF irrespective of the levels of Wnt since the increase of EC50 and decrease of STI in a dose-response curve characterizes the switch-like response.
Figure 4. Feedback loops in the ERK and Wnt signaling pathways have different roles in the regulation of E-cadherin.

(A) The positive feedback loop (including F4F5) significantly increases SD where the activity of PKCδ gets increased through inhibition of GSK3β by ERK phosphorylation. (B) The positive feedback loop (including F2) mediated by RKIP significantly increases STI. (C) EC50 is significantly decreased by CFLs (including F2F3) formed by the positive feedback mediated by RKIP and the negative feedback by ERK mediated phosphorylation of the SOS/Grb2 complex.
To verify the hypothesis and further investigate the functional roles of the multiple CFLs included in the signaling network, we distinguished the CFL mediated by RKIP into two PFLs, ERK RKIP MEK → ERK (PFL1) and ERK → Snail RKIP MEK → ERK (PFL2). So, we have 4 PFLs to analyze (Fig. 5). We removed each feedback loop one by one and simulated the resulting model for a gradual increase of EGF. Specifically, PFL1 is removed by removing the RKIP phosphorylation by ERK, PFL2 by removing the RKIP regulation by Snail, PFL3 by removing the PKCδ inhibition by GSK3β, and PFL4 by removing the Ras expression by β-catenin/TCF. Deletion of either PFL1 or PFL2 significantly diminished the switch-like behavior of cellular responses such as E-cadherin expression (Fig. 5A&B), which also supports our hypothesis that the RKIP-mediated PFLs cause a switch-like behavior of E-cadherin expression. In addition, the deletion of PFL3 suggests that this feedback loop also plays an important role in causing a switch-like behavior of the Snail and E-cadherin expression curves. On the other hand, when PFL4 was removed, the Snail and E-cadherin expression curves were shifted to the right but the switch-like shapes were well conserved, which suggests that the crosstalk with the Wnt signaling pathway increases the sensitivity of Snail and E-cadherin expressions to EGF stimulation. Taken together, these simulation results suggest that multiple CPFLs cooperatively induce a switch-like behavior of the cellular responses.
Figure 5. CPFLs cooperatively induce a switch-like behavior of cellular responses and RKIP determines ERK activation and E-cadherin suppression.

(A) Snail expression in response to normalized EGF simulation. (B) E-cadherin expression. ‘Control’ denotes the case when all PFLs are connected and ‘No PFLi’ (i=1,…,4) indicates the case when PFLi is removed. Within a certain range of concentration values of RKIP a switch-like behavior of cellular responses is observed. This switch-like behavior disappears, if one of the CPFLs mediated by RKIP is blocked. (C) The activation profile of ERK. (D) The expression profile of E-cadherin. The right inset shows the illustration of four feedback loops through which the ERK signaling pathway is regulated through: ERK RKIP MEK → ERK (PFL1), ERK →Snail RKIP MEK → ERK (PFL2), ERK GSK3β PKCδ → Ras → ERK (PFL3), ERK GSK3β → Destruction complex β-catenin→ Ras → ERK (PFL4).
RKIP regulation of E-cadherin expression
Most colon cancers have constitutively activated mutations in either or often both of ERK and Wnt signaling pathways (28-31), and the expression level of RKIP was found significantly decreased in various metastatic cancer cells (32-36). This suggests that the basal level of RKIP expression influences the dynamics of a signaling network in different ways depending on whether ERK, Wnt, or both pathways are persistently activated. To probe this, we simulated our model for two different initial conditions of RKIP (high or low) and three different combinations of sustained stimulations (EGF=1&Wnt=1, EGF=1&Wnt=0, or EGF=0&Wnt=1). The simulation results showed that higher RKIP levels delayed the suppression of E-cadherin and the activation of ERK when both the EGF and Wnt signaling pathways (EGF=1&Wnt=1) were persistently stimulated. (Fig. S2 of Supplementary Data C). On the other hand, the suppression of E-cadherin expression for at a higher RKIP level was much slower and less pronounced when only the EGF signaling was active (EGF=1&Wnt=0). This suggests that the crosstalk between ERK and Wnt signaling pathways might have a synergistic role in suppressing the E-cadherin expression. We note, however, that RKIP is not crucial in the regulation of E-cadherin expression when only the Wnt signaling pathway is persistently stimulated (EGF=0&Wnt=1). In summary, we found that the initial level of RKIP and the different combination of constitutively activated mutations in either or both of ERK and Wnt signaling pathways play an important role in shaping the dynamics of ERK activity and determining the E-cadherin expression.
To further investigate the effect of RKIP on cellular responses, we increased the RKIP expression by changing its production rate and simulated the activation of ERK and the expression of Snail, Slug and E-cadherin when both EGF and Wnt were constitutively activated. The simulation results show that the activation of ERK (Fig. 5C), the expression of Snail and Slug (Fig. S3A&B of Supplementary Data C) are decreased and E-cadherin expression is up-regulated as the level of RKIP increases (Fig. 5D). We found that all these responses show switch-like behaviors. In other words, when RKIP increment is below or above a certain threshold range (3-4 in this case), phospho-ERK, Snail, Slug and E-cadherin do not change, but they are dramatically induced (Snail, Slug) or decreased (phospho-ERK, E-cadherin) when RKIP levels cross the narrow threshold range. However, when one of the two PFLs (PFL1&2 in Fig. 5C&D and Fig. S3A&B of Supplementary Data C) mediated by RKIP was blocked, the switch-like behaviors disappeared. This can be explained by the two RKIP-mediated PFLs that cooperatively compensate for the increase of RKIP up to the point where RKIP levels are high enough to sequester Raf completely and thereby block MEK activation and the positive feedback. Insets of Fig. 5C&D show how RKIP determines the system dynamics (see also the insets of Fig. S3 of Supplementary Data C). When RKIP increment is below the threshold range, there was no prominent change for the activation of ERK and the expression of Snail and Slug since the expression of RKIP is suppressed by the high level of Snail and the level of phospho-RKIP is increased by the high level of ERK activity. In contrast, when RKIP accumulates above the threshold range, ERK activation and Snail expression were significantly decreased, since the expression of RKIP is no longer suppressed by Snail and the phospho-RKIP level is decreased by the reduced ERK activity.
DISCUSSION
ERK and Wnt signaling pathways implicated in the EMT process contain multiple CFLs that form a highly interconnected network. However, little is known about the functional role of these feedback loops. In this study, we found that the positive feedback in which activated ERK counteracts the inhibition of PKCδ by GSK3β has a significant role in inducing a large state change of E-cadherin in response to EGF stimulation while the positive feedback mediated by RKIP decreases the state transition interval in the state change of E-cadherin (Fig. 4A&B and Table 1). CFLs composed of the RKIP-mediated PFLs and the negative feedback loop where ERK phosphorylates and inhibits the SOS/Grb2 complex increase EC50 in response to the EGF stimulation (Fig. 4C and Table 1). On the other hand, it turns out that the negative feedback loop where the β-catenin/TCF complex induces Axin dose not have any significant role in the regulation of E-cadherin expression.
In a previous study (18) we found that the positive feedback loop formed by the phosphorylation of RKIP by ERK induces a switch-like behavior of ERK and MEK activities. In this study, we have further revealed that such switch-like behaviors are cooperatively caused by CPFLs through phosphorylation of RKIP by ERK and transcriptional repression of RKIP by Snail (Fig. 5C&D). If one of these feedback loops is blocked, the switch-like behavior becomes weaker or even disappears.
From extensive in silico simulation of the dose-response characteristics to oncogenic stimulation and the Karnaugh-map (K-map) analysis method (Fig. 1), we found that particular feedback combinations had a significant effect on E-cadherin regulation. Some of them were commonly observed for gradual increase of EGF with a fixed level of Wnt, while none was observed for a gradual increase of Wnt with a fixed level of EGF (Table S1 of Supplementary Data C). This suggests that the dose-response of E-cadherin expression to EGF stimulation is mostly not affected by the crosstalk with Wnt, but that Wnt stimulation is significantly affected by the crosstalk with EGF. This result might raise the interesting possibility that the ERK and Wnt signaling pathways play different roles in inducing EMT, which can also be partially supported by other recent experimental results. For instance, both the ERK and Wnt signaling pathways are activated by the growth factors secreted by malignant tumor cells with epithelial phenotype, and they contribute to the EMT process possibly at an initial phase (6). However, in transformed mesenchymal cells, the ERK signaling pathway seems to be strongly activated compared to the Wnt signaling pathway since the mesenchymal cells secrete various growth factors that strongly stimulate ERK, such as fibroblast growth factors (FGF), EGF, and HGF (37). Thus, it seems that both the ERK and Wnt pathways participate in the initial phase of the EMT process, while the ERK pathway takes the major role in the later phase when the epithelial cells are transformed to the mesenchymal cells.
Our simulation results show that RKIP controls the EMT process through regulation of E-cadherin (Fig. 5C&D). A number of recent studies showed a statistically significant inverse relationship between RKIP expression and metastasis and overall survival both in animal models and in human cancer patients (32-36, 38-40). For instance, Fu et al. (33) showed that highly metastatic C4-2B prostate cancer cells lack RKIP expression, and that enforced expression of RKIP decreased cell invasiveness in in vitro assays and suppressed the development of lung metastasis when implanted into mouse prostate without affecting the growth of the primary tumor. In human cancer patients it was shown that the retention of RKIP expression in colorectal tumors correlates with a low risk of metastatic relapse and improved survival (32). Similar results were obtained by Minoo et al. (36) who used colon cancer tissue microarrays to show that the loss of cytoplasmic RKIP is associated with distant metastasis, vascular invasion, and worse survival. Thus, while an important role for RKIP in metastasis suppression especially in colon and prostate cancer is now emerging, the underlying mechanisms how RKIP expression is altered in tumors and how RKIP counteracts the development of metastasis are less well understood. The regulation of RKIP expression seems to involve at least two mechanisms. One is the silencing of the rkip gene promoter by hypermethylation (38), the other is the repression of the rkip gene promoter by Snail (19). The molecular function of RKIP as inhibitor of the ERK pathway that inhibits MEK phosphorylation by Raf is well characterized (16-17). Recent data show that ERK can inactivate RKIP as part of a feedback loop and thereby confer non-linear dynamics on ERK activation (18). However, so far this feedback loop only has been analyzed in isolation. The same is true for most other feedback loops described here.
Our analysis of the combinatorial effects of such feedback loops revealed several new findings. One is that RKIP is an important feedback loop that exerts major control over both STI and EC50, and that RKIP levels are crucial for these effects (Fig. 4B&C). Thus, the CPFLs where ERK phosphorylates RKIP, and Snail transcriptionally represses the expression of RKIP cause a switch-like behavior of E-cadherin expression (Fig. 5D), where the RKIP expression determines the EMT progression by antagonizing the suppression of E-cadherin. Another result is that subtle changes in the combination of feedback links may substantially change the output of the network. This is interesting in light of the observation that β-catenin is found in the nucleus of the mesenchymal-like cells at the invasive front of colorectal tumors, while it is cytosolic in the central epithelial areas of the same tumor (41). These changes were attributed to slightly different microenvironments. Our results suggest that combinatorial changes in the network topology could be the molecular substrate for such dramatic influences of the microenvironment. This flexible topology afforded by different feedback link combinations also would enable the facile transition of cells between epithelial and mesenchymal morphologies as observed in tumors. In summary, our analysis shows that feedback loops formed by combination of feedback links do not only have roles in shaping dynamic response kinetics of networks, but also in the flexible specification and diversification of responses.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank J.-R. Kim, D. Shin, and D.-S. Kim for their helpful discussions and suggestions. This work was supported by the National Research Foundation of Korea (NRF) grants funded by the Korea Government, the Ministry of Education, Science & Technology (MEST) (2009-0086964 and 2010-0017662). The WK lab was supported by the Science Foundation Ireland under Grant No. 06/CE/B1129 and Cancer Research UK.
REFERENCES
- 1.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–81. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Radisky DC. Epithelial-mesenchymal transition. J Cell Sci. 2005;118:4325–6. doi: 10.1242/jcs.02552. [DOI] [PubMed] [Google Scholar]
- 3.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 4.Moustakas A, Heldin CH. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007;98:1512–20. doi: 10.1111/j.1349-7006.2007.00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beavon IR. The E-cadherin-catenin complex in tumour metastasis: structure, function and regulation. Eur J Cancer. 2000;36:1607–20. doi: 10.1016/s0959-8049(00)00158-1. [DOI] [PubMed] [Google Scholar]
- 6.Mimeault M, Batra SK. Interplay of distinct growth factors during epithelial mesenchymal transition of cancer progenitor cells and molecular targeting as novel cancer therapies. Ann Oncol. 2007;18:1605–19. doi: 10.1093/annonc/mdm070. [DOI] [PubMed] [Google Scholar]
- 7.Park KS, Jeon SH, Kim SE, et al. APC inhibits ERK pathway activation and cellular proliferation induced by RAS. J Cell Sci. 2006;119:819–27. doi: 10.1242/jcs.02779. [DOI] [PubMed] [Google Scholar]
- 8.Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci. 2007;120:3327–35. doi: 10.1242/jcs.03485. [DOI] [PubMed] [Google Scholar]
- 9.Jeon SH, Yoon JY, Park YN, et al. Axin inhibits extracellular signal-regulated kinase pathway by Ras degradation via beta-catenin. J Biol Chem. 2007;282:14482–92. doi: 10.1074/jbc.M611129200. [DOI] [PubMed] [Google Scholar]
- 10.Almeida M, Han L, Bellido T, Manolagas SC, Kousteni S. Wnt proteins prevent apoptosis of both uncommitted osteoblast progenitors and differentiated osteoblasts by beta-catenin-dependent and -independent signaling cascades involving Src/ERK and phosphatidylinositol 3-kinase/AKT. J Biol Chem. 2005;280:41342–51. doi: 10.1074/jbc.M502168200. [DOI] [PubMed] [Google Scholar]
- 11.Desbois-Mouthon C, Cadoret A, Blivet-Van Eggelpoel MJ, et al. Insulin and IGF-1 stimulate the beta-catenin pathway through two signalling cascades involving GSK-3beta inhibition and Ras activation. Oncogene. 2001;20:252–9. doi: 10.1038/sj.onc.1204064. [DOI] [PubMed] [Google Scholar]
- 12.Ding Q, Xia W, Liu JC, et al. Erk associates with and primes GSK-3beta for its inactivation resulting in upregulation of beta-catenin. Mol Cell. 2005;19:159–70. doi: 10.1016/j.molcel.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 13.Graham NA, Asthagiri AR. Epidermal growth factor-mediated T-cell factor/lymphoid enhancer factor transcriptional activity is essential but not sufficient for cell cycle progression in nontransformed mammary epithelial cells. J Biol Chem. 2004;279:23517–24. doi: 10.1074/jbc.M314055200. [DOI] [PubMed] [Google Scholar]
- 14.Holnthoner W, Pillinger M, Groger M, et al. Fibroblast growth factor-2 induces Lef/Tcf-dependent transcription in human endothelial cells. J Biol Chem. 2002;277:45847–53. doi: 10.1074/jbc.M209354200. [DOI] [PubMed] [Google Scholar]
- 15.Wang Q, Zhou Y, Wang X, Evers BM. Glycogen synthase kinase-3 is a negative regulator of extracellular signal-regulated kinase. Oncogene. 2006;25:43–50. doi: 10.1038/sj.onc.1209004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yeung K, Janosch P, McFerran B, et al. Mechanism of suppression of the Raf/MEK/extracellular signal-regulated kinase pathway by the raf kinase inhibitor protein. Mol Cell Biol. 2000;20:3079–85. doi: 10.1128/mcb.20.9.3079-3085.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yeung K, Seitz T, Li S, et al. Suppression of Raf-1 kinase activity and MAP kinase signalling by RKIP. Nature. 1999;401:173–7. doi: 10.1038/43686. [DOI] [PubMed] [Google Scholar]
- 18.Shin SY, Rath O, Choo SM, et al. Positive- and negative-feedback regulations coordinate the dynamic behavior of the Ras-Raf-MEK-ERK signal transduction pathway. J Cell Sci. 2009;122:425–35. doi: 10.1242/jcs.036319. [DOI] [PubMed] [Google Scholar]
- 19.Beach S, Tang H, Park S, et al. Snail is a repressor of RKIP transcription in metastatic prostate cancer cells. Oncogene. 2008;27:2243–8. doi: 10.1038/sj.onc.1210860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou BP, Deng J, Xia W, et al. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–40. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- 21.Dong C, Waters SB, Holt KH, Pessin JE. SOS phosphorylation and disassociation of the Grb2-SOS complex by the ERK and JNK signaling pathways. J Biol Chem. 1996;271:6328–32. doi: 10.1074/jbc.271.11.6328. [DOI] [PubMed] [Google Scholar]
- 22.Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172–83. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leung JY, Kolligs FT, Wu R, et al. Activation of AXIN2 expression by beta-catenin-T cell factor. A feedback repressor pathway regulating Wnt signaling. J Biol Chem. 2002;277:21657–65. doi: 10.1074/jbc.M200139200. [DOI] [PubMed] [Google Scholar]
- 24.Kim D, Rath O, Kolch W, Cho KH. A hidden oncogenic positive feedback loop caused by crosstalk between Wnt and ERK pathways. Oncogene. 2007;26:4571–9. doi: 10.1038/sj.onc.1210230. [DOI] [PubMed] [Google Scholar]
- 25.Paul S, Dey A. Wnt signaling and cancer development: therapeutic implication. Neoplasma. 2008;55:165–76. [PubMed] [Google Scholar]
- 26.Lee E, Salic A, Kruger R, Heinrich R, Kirschner MW. The roles of APC and Axin derived from experimental and theoretical analysis of the Wnt pathway. PLoS Biol. 2003;1:E10. doi: 10.1371/journal.pbio.0000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tocci RJ. Digital Systems: Principles and Applications. 4th ed Prentice-Hall International, Inc; Englewood Cliffs, N.J.: 1991. [Google Scholar]
- 28.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–90. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 29.Friday BB, Adjei AA. Advances in targeting the Ras/Raf/MEK/Erk mitogen-activated protein kinase cascade with MEK inhibitors for cancer therapy. Clin Cancer Res. 2008;14:342–6. doi: 10.1158/1078-0432.CCR-07-4790. [DOI] [PubMed] [Google Scholar]
- 30.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 31.van de Wetering M, Sancho E, Verweij C, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–50. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 32.Al-Mulla F, Hagan S, Behbehani AI, et al. Raf kinase inhibitor protein expression in a survival analysis of colorectal cancer patients. J Clin Oncol. 2006;24:5672–9. doi: 10.1200/JCO.2006.07.5499. [DOI] [PubMed] [Google Scholar]
- 33.Fu Z, Smith PC, Zhang L, et al. Effects of raf kinase inhibitor protein expression on suppression of prostate cancer metastasis. J Natl Cancer Inst. 2003;95:878–89. doi: 10.1093/jnci/95.12.878. [DOI] [PubMed] [Google Scholar]
- 34.Hagan S, Al-Mulla F, Mallon E, et al. Reduction of Raf-1 kinase inhibitor protein expression correlates with breast cancer metastasis. Clin Cancer Res. 2005;11:7392–7. doi: 10.1158/1078-0432.CCR-05-0283. [DOI] [PubMed] [Google Scholar]
- 35.Keller ET, Fu Z, Brennan M. The biology of a prostate cancer metastasis suppressor protein: Raf kinase inhibitor protein. J Cell Biochem. 2005;94:273–8. doi: 10.1002/jcb.20169. [DOI] [PubMed] [Google Scholar]
- 36.Minoo P, Zlobec I, Baker K, et al. Loss of raf-1 kinase inhibitor protein expression is associated with tumor progression and metastasis in colorectal cancer. Am J Clin Pathol. 2007;127:820–7. doi: 10.1309/5D7MM22DAVGDT1R8. [DOI] [PubMed] [Google Scholar]
- 37.Guarino M, Rubino B, Ballabio G. The role of epithelial-mesenchymal transition in cancer pathology. Pathology. 2007;39:305–18. doi: 10.1080/00313020701329914. [DOI] [PubMed] [Google Scholar]
- 38.Al-Mulla F, Hagan S, Al-Ali W, et al. Raf kinase inhibitor protein: mechanism of loss of expression and association with genomic instability. J Clin Pathol. 2008;61:524–9. doi: 10.1136/jcp.2007.046987. [DOI] [PubMed] [Google Scholar]
- 39.Granovsky AE, Rosner MR. Raf kinase inhibitory protein: a signal transduction modulator and metastasis suppressor. Cell Res. 2008;18:452–7. doi: 10.1038/cr.2008.43. [DOI] [PubMed] [Google Scholar]
- 40.Keller ET, Fu Z, Yeung K, Brennan M. Raf kinase inhibitor protein: a prostate cancer metastasis suppressor gene. Cancer Lett. 2004;207:131–7. doi: 10.1016/j.canlet.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 41.Brabletz T, Jung A, Reu S, et al. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A. 2001;98:10356–61. doi: 10.1073/pnas.171610498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.