

Abstract
Hepatic ischemia-reperfusion injury (IRI) occurs upon restoration of hepatic blood flow after a period of ischemia. Decreased endogenous nitric oxide (NO) production resulting in capillary luminal narrowing is central in the pathogenesis of IRI. Exogenous NO has emerged as a potential therapy for IRI based on its role in decreasing oxidative stress, cytokine release, leukocyte endothelial-adhesion and hepatic apoptosis. This review will highlight the influence of endogenous NO on hepatic IRI, role of inhaled NO in ameliorating IRI, modes of delivery, donor drugs and potential side effects of exogenous NO.
Keywords: Nitric oxide, Liver, Ischemia-reperfusion injury, Drug delivery
INTRODUCTION
Ischemia-reperfusion injury (IRI) is a series of multifaceted cellular events that takes place on the resumption of oxygen delivery after a period of hypoxia. This injury could be severe enough to lead to a significant morbidity and mortality.
The liver may be involved in IRI in procedures that are associated with sequential vascular impediment and restoration of blood flow; for example hepatic resections and orthotopic liver transplantation. During these procedures, unclamping of the vascular inflow to the liver after a temporary period of cross clamping results in major hepatocellular damage.
Nitric oxide (NO) has various protective effects on cells during IRI. NO has been demonstrated to inhibit oxidative stress, cytokine release, leukocyte endothelial adhesion and apoptosis[1]. On a cellular-signaling level, NO effects are mediated via redox-sensitive sites, and include: inhibition of protein kinase C, activation of tyrosine kinase, inactivation of nuclear factor (NF)-κB and activation of G proteins[2]. Previous studies have demonstrated that a reduction of NO during hepatic IRI, generally via a reduction in endothelial nitric oxide synthase activity, leads to liver injury[3]. Inhaled NO or NO donor drugs are novel treatments that have been used clinically to attenuate liver IRI[4]. This review will discuss the pathophysiology of liver involvement during IRI, and the clinical use of nitric oxide in ameliorating the impact of liver IRI.
BRIEF REVIEW OF THE PATHOPHYSIOLOGY OF IRI
The complex mechanisms of IRI have been revealed by advanced molecular biology[5] (Figure 1). During the ischemic phase, anaerobic metabolism ensues and produces an inadequate amount of high-energy phosphates which are fundamental to most cellular functions. Low levels of high-energy phosphates affect a myriad of cellular functions: homeostasis, signaling interactions, cellular proliferation and processing of the apoptotic death cycle. Adenosine triphosphate (ATP) depletion impairs sodium/potassium ATPase (Na+/K+-ATPase) function, resulting in an impairment of the efflux of sodium from the cell. Additionally, toxic metabolites, which are generated during ischemia, attract free water into ischemic cells and organelles leading to the formation of cellular edema[6]. If the ischemic insult lasts greater than 24 h, it is likely that ATP-synthase activity becomes irreversible after blood restoration, leading to cellular necrosis, apoptosis or neuroapoptosis[7]. Ischemia also causes an increased expression of adhesion molecules that leads to endothelial cell and neutrophil adhesion, resulting in vascular studding and occlusion[8]. Furthermore, disequilibrium between NO and endothelin (ET) induces vasoconstriction and subsequent microcirculatory failure even though blood circulation has been re-established[9]. Re-establishment of blood flow will serve to amplify inflammation with consequent injury that is highly variable but dependent on numerous variables including the extent of mediators produced (i.e. reactive oxygen species), the degree of endothelial and neutrophil adhesive responses and the degree of Kupffer cell activation.
Figure 1.

Multifaceted hepatic ischemia-reperfusion injury. Kupffer and endothelial cells produce cytokines and chemokines, recruiting neutrophils that further accentuate injury. EC: Endothelial cell; KC: Kupffer cell; ATP: Adenosine triphosphate; TNF: Tumor necrosis factor; IL: Interleukin; ICAM: Intercellular adhesion molecule; VCAM: Vascular adhesion molecule; PAF: Platelet activation factor; LTB4: Leukotriene B4; GMS-CSF: Granulocyte macrophage colony stimulating factor; INF: Interferon; ROS: Reactive oxygen species (Courtesy of Dr. Joan Rosello-Catafau, Barcelona, Spain).
PRINCIPAL PARTICIPANTS IN LIVER IRI
Sinusoidal endothelial cells
Injury to these cells is initiated during cold ischemia whereby Ca2+-ATPase results in the accumulation of intracellular calcium[10]. Following this event, a series of actions occur making the endothelium more susceptible to platelet adhesion and reduced sinusoidal flow.
Kupffer cells
Kupffer cells are crucial in liver injury orchestration. Metabolic alterations of these cells occur during no-flow ischemia leading to the formation of reactive oxygen species during early reperfusion[11]. Additionally, at the onset of reperfusion, Kupffer cells undergo further activation by Toll-like receptor 4 signaling and/or by complement. Subsequently, Kupffer cells release pro-inflammatory cytokines such as TNF-α and interleukin-1 which themselves can perpetuate inflammatory injury by such means as leukocyte activation.
Hepatocytes
While major participants in the promotion of injury, during cold ischemia hepatocytes undergo intracellular bioenergetic perturbations that reduce ATP stores due to mitochondrial dysfunction and predispose these cells to injury during reperfusion[12].
Leukocytes and lymphocytes
As a result of IRI, cellular adhesion molecules (i.e. intercellular adhesion molecule-1 or ICAM-1, vascular adhesion molecule-1 or VCAM-1), selectins and integrins are activated and upregulated on the surface of endothelial cells, neutrophils and platelets. The activated neutrophils adhere to endothelial cells at the initial stages of reperfusion, and subsequently transmigrate across the endothelium where they continue to injury orchestration. The accumulation of activated neutrophils contributes to microcirculatory disturbances both locally and remotely. Activated neutrophils release reactive oxygen species, specifically superoxide radical (O2-•), proteases and various cytokines[13]. Monocytes and macrophages are also activated shortly following reperfusion[14]. Recent studies propose an important role for lymphocytes, especially CD4+ T cells, in augmenting injury responses after IRI. However, lymphocytes may also play a protective role, but this is probably dependent on cell type and time course of injury[15].
Reactive oxygen species and reactive nitrogen species
During periods of ischemia, reactive oxygen species (ROS) and reactive nitrogen species (RNS) are generated which can promote intracellular damage. Due to electron transport chain alterations, mitochondrial dysfunction ensues leading to reductions in ATP production and with subsequent loss of inner membrane stability resulting in mitochondrial swelling and rupture. With the reintroduction of oxygen during reperfusion, ROS are produced due to reactions of oxygen introduced during reperfusion with xanthine oxidase. ROS serve to stimulate other cell lines including Kupffer cells to produce proinflammatory cytokines[16]. The major ROS are hydroxyl radical (OH•) and hydrogen peroxide (H2O2). Reactions of ROS such as O2-• with NO yield products such as peroxynitrite (ONOO-), a RNS which can be an extremely aggressive oxidant.
Cytokines
Cytokines play a vital role in IRI, both by inducing and sustaining the inflammatory response, and by modulating IRI severity. Tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1) are the two cytokines most commonly implicated in liver IRI. TNF-α is a pleiotropic cytokine generated by various different cell types in response to inflammatory and immunomodulatory stimuli. TNF-α modulates leukocyte chemotaxis and activation, and induces ROS production in Kupffer cells[17]. Additionally, IL-1 is known to promote production of ROS, induce TNF-α synthesis by Kupffer cells and induce neutrophil recruitment[18].
Complement
The complement system also contributes significantly to IRI and is composed of approximately 30 soluble and membrane-bound proteins. This system can be stimulated in three pathways: (1) the antibody-dependent classical pathway; (2) the alternative pathway; or (3) the mannose-binding lectin pathway[19]. Complement, when activated, acts as a membrane-attacking complex that stimulates the production of proinflammatory cytokines and chemotactic agents. Furthermore, it can regulate adaptive immunity[20].
THE INFLUENCE OF ENDOGENOUS NO ON LIVER IRI
Damage to the liver due to IRI is a culmination of inflammatory cross talk with the principal participants mentioned previously. IRI is the main cause of liver injury in response to vascular clamping during hepatic procedures such as hepatectomy and liver transplantation. This insult on the liver results in disturbances of the sinusoidal microcirculation and the generation of a variety of mediators such as ROS, cytokines, activation of chemokines and other cell signaling molecules previously mentioned.
Hepatic IRI can cause severe hepatocellular injury that contributes to morbidity and mortality after liver surgery. As briefly mentioned previously, reductions of NO during liver IRI occur and are associated with increased liver injury[3]. This is now appreciated to be due to decreases in NO steady state production resulting from low concentrations of endothelium-derived nitric oxide synthase (eNOS). This event coupled with NO inactivation due to reactions with abundant ROS, such as O2-•, results in reduced NO bioavailability. The consequences of this reduced bioavailability include, but are not exclusive to, increased oxidative stress, increased apoptosis, increased leukocyte adhesion, increased microcirculatory tone, and perturbed mitochondrial function. Interestingly, restoration of NO to more “physiologic” concentrations serves to diminish the liver ischemic injury via countering of the adverse actions mentioned previously. Studies have demonstrated findings that are consistent with the premise that eNOS is crucial for minimizing injury during liver IRI. For example, liver injury was demonstrated to be less in wild type mice compared to eNOS knockouts (eNOS-/-)[21] (Figure 2), in addition to the findings that agents given to increase eNOS expression or donate NO afford greater liver IRI protection[22,23]. It is also well established that the NO concentrations during various inflammatory states are significantly increased by increasing expression of inducible nitric oxide synthase (iNOS). However, the influence of iNOS and its true contribution in conferring liver protection (or not) deserves additional studies. In a rat model of liver IRI, iNOS expression was significantly increased correlating with increases in iNOS RNA at 1 and 5 h[24]. This is consistent with other studies measuring iNOS expression in conditions of liver IR. In a porcine model of IRI, intraportal injection of the selective iNOS inhibitor, aminoguanidine, was demonstrated to decrease injury[25]. In an intriguing study, iNOS knockout mice (iNOS-/-) exposed to warm liver IRI demonstrated a much greater magnitude of injury compared to wild type mice. Of notable interest was the finding that even though injury was greater in the iNOS knockout mice, little to no iNOS RNA was detectable in the wild type mice. It would appear that for now, the true influence of iNOS on liver injury during IR remains unclear.
Figure 2.

Increased liver injury as assessed by serum alanine aminotransferase in endothelium-derived nitric oxide synthase knockout mice compared with their wild type controls. aP < 0.05 vs sham-operated controls. cP < 0.05 vs time-matched wild type control. ALT: Alanine aminotransferase; eNOS: Endothelium-derived nitric oxide synthase (Courtesy of Dr. Ianes N. Hines, Chapel Hill, NC).
A number of other endogenous NO-mediated mechanisms thought to confer protection have been published. For example, NO has been shown to inhibit caspase proteases via S-nitrosylation, thereby inhibiting apoptosis[26]. This appears to be somewhat concentration-dependent. Low physiological concentrations of NO may inhibit apoptosis. In contrast, higher concentrations may lead to the formation of toxic products such as ONOO- or other ROS which lead to cell necrosis and apoptosis[27]. Other published mechanisms of NO-mediated protection include inhibition of NF-κB[28], reversible inhibition of mitochondrial complex I, and decreased mitochondrial calcium accumulation[29]. As to be expected, controversy exists concerning “if” and “how” NO exerts cellular protection. For instance, in a study by Jaeschke et al[11], administration of a NO synthase inhibitor did not attenuate or accentuate liver injury during the initial reperfusion period. Inhibition of NO was observed not to influence neutrophil migration to the injured sites. While this contradicts a number of other studies, based on their findings, the authors concluded that NO availability was unlikely to be involved in the post-ischemic oxidant stress and reperfusion injury[30]. Nevertheless, the majority of published literature has demonstrated the beneficial effects of NO during liver IRI. These conflicting results might be explained by the fact that the mechanism of NO-mediated protection varies depending on cell type, quantities supplied, laboratory methods applied, timing and duration of NO exposure.
While iNOS was shown to be protective against hepatic IRI in some studies, it was shown to be deleterious in others. In a rat model of hepatic IRI, Takamatsu et al[31] observed increased hepatic expression of iNOS mRNA, ALT, and plasma iNOS at 3, 12, and 24 h after hepatic reperfusion. Concomitantly, there was evidence of histologic damage and nitrotyrosine formation in the liver sampled post-reperfusion. These changes were absent in the control group given the selective iNOS inhibitor, ONO-1714. The authors concluded that peroxynitrite may be involved in iNOS-mediated hepatic injury following IR[31].
In another model of hepatic IR in rats, Wang et al[32] observed an increase in iNOS protein and mRNA expression on the first day following hepatic reperfusion. Higher levels of iNOS correlated with evidence of increased hepatic injury in the form of elevated serum levels of ALT and AST. Administration of the non-selective nitric oxide synthase (NOS) inhibitor, L-NAME, significantly increased AST and ALT, while administration of the selective iNOS inhibitor, AE-ITU, significantly decreased AST and ALT levels, respectively[32]. The authors postulated that the deleterious effects of L-NAME were due to inhibition of eNOS, while the protective effects of AE-ITU were due to inhibition of injury-provoking iNOS. In a rat model of hepatic IR and small-for-size living-related liver transplantation, Jiang et al[33] observed increased iNOS mRNA and protein expression post-reperfusion from a warm ischemic insult with peak expression at 3 h post-reperfusion. This was accompanied by significant increases in concentrations of AST, ALT, malondialdehyde (MDA) and histologic evidence of damage compared to controls. The authors postulated that iNOS-induced hepatic damage was via significant production of ROS[33]. We summarize some key studies investigating endogenous NO and NOS in hepatic IRI in Table 1[3,21,25,31-37].
Table 1.
Effect of endogenous nitric oxide and nitric oxide synthase on liver ischemia-reperfusion injury
| Species | Experimental methods | Ischemic time (min) | NO or NOS effects | Ref. |
| Pigs | Aminoguanidine, 5 min before ischemia | 120 | NO derived from iNOS, antioxidant | [25] |
| Dogs | FK 409, 30 min before ischemia and15 min before and 45 min after reperfusion | 60 | NO, improves hepatic microcirculation | [34] |
| Rats | L-arginine, 7 d before IRI | 60 | NO, antioxidant | [35] |
| Rats | L-NAME 60 min before ischemia | 30 | NO, antioxidant | [3] |
| Mouse | Gadolinium chloride 24 h before ischemia | 45 | NO derived from eNOS, antioxidant, suppresses Kupffer cell function, regulated basal hepatic blood flow, but did not affect blood flow after reperfusion, attenuated neutrophils infiltration | [21] |
| L-NAME methyl ester 15 min prior to ischemia | ||||
| Rats | L-arginine or Sodium nitroprusside or L-Name prior to ischemia | 60 | NO, improves peripheral liver blood flow after reperfusion, cytoprotective | [36] |
| Male rats | Arginine or L-NAME or 8-bromo guanosine 3′5′-cyclic monophosphate or rat atrial natriuretic peptide (ANP 1-28) 30 min before ischemia | 45 | NO, antioxidant, antiproinflammatory cytokines, improves microcirculation by the cGMP pathway, inhibits neutrophil infiltration and platelet aggregation | [37] |
| Male rats | IRI group: had partial clamping of portal vein and hepatic artery | 90 | iNOS expression peaked at 3 h and diminished at 24 h post reperfusion in IRI and ONO-1714 groups | [31] |
| ONO-1714 group: as above plus ONO-1714 just prior to reperfusion and 6 h thereafter | ONO-1714 significantly inhibited plasma nitrates at 24 h post reperfusion | |||
| Control group: sham operation | ONO-1714 significantly inhibited plasma ALT at 12 h post reperfusion, together with inhibiting histological damage and peroxynitrate expression in liver | |||
| Male rats | Microvessel clamping of portal vein and left hepatic artery L-NAME and AE-ITU given to each of 6 rats exposed to microvessel clamping (time unknown) | 60 | L-NAME worsened, elevated levels of ALT/AST in IRI groups | [32] |
| AE-ITU mildly and significantly decreased levels of AST | ||||
| Male rats | Portal vein, hepatic artery and bile ducts clamped by microvessel clamp followed by reperfusion | 60 | Significant elevation of AST/ALT, MDA/SOD in IRI and small-for-size liver transplantation groups | [33] |
NO: Nitric oxide; NOS: Nitric oxide synthase; iNOS: Inducible nitric oxide synthase; eNOS: Endothelium-derived nitric oxide synthase; cGMP: Cyclic guanosine monophosphate; L-NAME: L-nitroarginine; ANP: Atrial natriuretic peptide; IRI: Ischemia-reperfusion injury; AST: Aspartate aminotransferase; ALT: Alanine aminotransferase; AE-ITU: Aminoethyl-isothiourea; MDA: Malondialdehyde; SOD: Superoxide dismutase.
THE USE OF EXOGENOUS NO ADMINISTRATION IN ATTENUATING HEPATIC IRI
Inhaled nitric oxide
Inhaled NO was approved by the US Food and Drug Administration in December of 1999 for the treatment of persistent hypertension of the newborn. Over the last decade, the primary advantage of inhaled nitric oxide (iNO) was seen to be its ability to selectively decrease pulmonary vascular resistance with minimal effects on systemic blood pressure; however, there is currently much interest in exploring its other benefits, including its antioxidant properties and its cytoprotective abilities[4]. In many animal studies, iNO decreased infarct size and left ventricular dysfunction after IRI, increased coronary artery patency after thrombosis, increased blood flow in brain, kidney and peripheral vasculature, decreased leukocyte adhesion in bowel during ischemia-reperfusion, and decreased platelet aggregation[38]. Date et al[39] reported the use of iNO in 15 out of 32 patients who suffered from immediate severe allograft dysfunction, with iNO administered at 20 to 60 ppm. The mortality was significantly lower in the iNO group (7% and 24%, respectively). The gross benefits reported were that iNO improved oxygenation, decreased pulmonary artery pressure, shortened the period of postoperative mechanical ventilation, and reduced airway complications and mortality[39]. Likewise, a recent retrospective study also presented a picture of improvement of overall respiratory functions. The authors encouraged the administration of iNO for the prevention and treatment of early graft failure in lung transplant recipients[40]. Varadarajan et al[41] were the first group to study the relationship between NO metabolism and IRI in human liver transplantation[41]. From their study, they concluded that reduced bioavailability of eNOS contributed to IRI one hour after portal reperfusion. On the other hand, iNOS did not contribute to early IRI after human liver transplantation. Clinical and mechanistic reports on therapeutic use of iNO demonstrated action well beyond vascular relaxation, subsequently inactivated by oxy- or deoxyhemoglobin in the red blood cells. iNO has various positive effects on extrapulmonary systems. However, how iNO mediates these extrapulmonary effects remains unclear. Evidence supporting stable forms of iNO is probably strongest for S-nitrosothiols (SNOs) and nitrite[38]. In a prospective, blinded, placebo-controlled study, 80 ppm of iNO was administered to patients undergoing orthotropic liver transplantation[42]. Many advantages were reported in the iNO group, including reduced platelet transfusion, an improvement in the rate at which liver function was restored post-transplantation, and a decrease in the length of hospital stay. Most interesting was the finding of an approximate 75% reduction of hepatocellular apoptosis in patients treated with iNO[42] (Figure 3). Possible biochemical intermediates of iNO include plasma and red blood cell nitrate, nitrite, SNOs, C- or N-nitrosamines and red blood cell ferrous nitrosylhemoglobin. In this study, a detailed analysis indicated that the most likely candidate transducer of iNO in liver IRI was nitrite.
Figure 3.
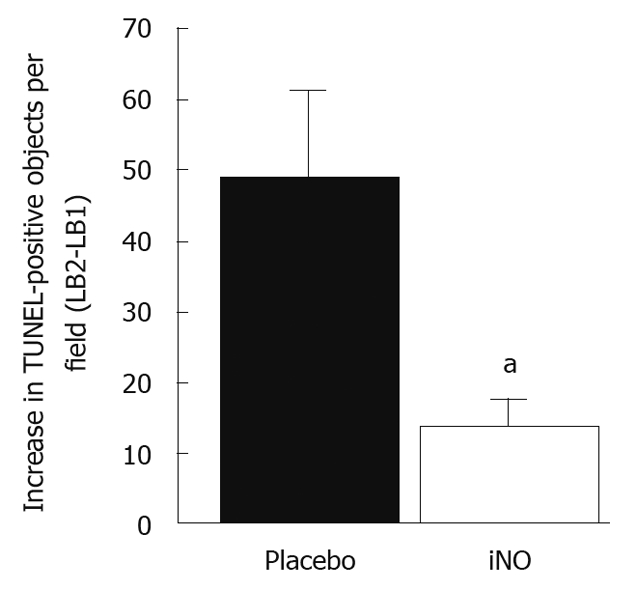
Decreased apoptosis indicated by TUNEL staining in patients treated with inducible nitric oxide compared to controls (Courtesy of John D. Lang, MD, Seattle, WA). aP < 0.05. iNO: Inducible nitric oxide.
iNO delivery systems
An iNO delivery system should allow for constant and accurate measurements of NO and nitrogen dioxide (NO2) concentration in inspired gas, as well as minimization of the contact time between oxygen and NO, in order to decrease the feasibility of producing high NO2 concentrations. The measurement of iNO and NO2 concentrations can be undertaken using chemiluminescence or electrochemical devices. There are some drawbacks of chemiluminescence devices such as cost, the need for a relatively high sample volume, noise and maintenance difficulties[43]. However, an electrochemical analyzer is relatively insensitive, and these measurements may be affected by pressure, humidity, temperature and the presence of other gases in the environment[44]. The delivery system should display the pressure of iNO in the cylinder and should have a backup power supply to avoid sudden discontinuation of iNO. Inhaled NO is usually supplied in nitrogen at various concentrations. The gas mixture concentration should be sampled downstream of the input port just proximal to the patient manifold. iNO also can be administered via nasal cannula, oxygen mask and oxygen hood[45]. Finally, the exhausted gas should be scavenged by passing it through carbon and filters, soda lime or activated charcoal[46].
POTENTIAL TOXICITIES DURING INHALATION
In the presence of high concentrations of O2, NO oxidizes to nitrogen dioxide (NO2). NO2 reacts with the alveolar lining fluid to form nitric acid. NO dissolved in the alveolar lining fluid reacts with O2- yielding OONO,- then decomposes into a hydroxyl anion[47]. Nitration of tyrosine residues of proteins is used as a marker of oxidative stress[48]. The rate at which NO is oxidized to NO2 depends on the square of NO concentration and fractional concentration of oxygen to which it is exposed. The Occupational and Health Administration recommend 5 ppm exposure to NO per 8 h per 24-h-interval as the upper safe limit of human exposure[49]. In order to protect against NO2 toxicity, iNO should be given with the least possible O2 concentration. Inhaled NO and NO2 concentrations should be monitored, exhaled gases should be scavenged, and a soda lime canister should be placed in the inspiratory limb of the breathing circuit.
Nitrite
The simple molecule nitrite had been thought to be just an index of NO production for decades[3]. Recently, a number of lines of evidence suggest that nitrite is a pro-mediator of NO homeostasis[50]. Administration of nitrite at near physiological concentrations (< 5 μg) leads to vasodilatation in animal and human studies[46]. Shiva et al[51] observed that nitrite was metabolized across the peripheral circulation. In addition, nitrite caused an increase in peripheral forearm blood flow when 80 ppm iNO was administered[51]. Under distinct conditions such as hypoxia and acidosis, nitrite can be reduced to NO by a number of deoxyhemeproteins (hemoglobin, myoglobin, neuroglobin and cytoglobin), enzymes (cytochrome P450 and xanthine oxidoreductase), and components of the mitochondrial electron transport chain[4]. Since nitrite can be converted back to NO during hypoxia, nitrite therefore is expected to be utilized during IRI. Furthermore, nitrite shows more potential benefits than NO in terms of safety and ease of administration. In other words, nitrite concentrations administered need only to be a small dose in order to increase plasma and tissue nitrite levels several folds. Routes of administration are oral, intravenous injection or infusion, intraperitoneal, via nebulizer or topical[52]. Nitrite has now been demonstrated to have cytoprotective effects in animal models of ischemia-reperfusion in organs. Duranski et al[52] evaluated the effects of nitrite therapy in in vivo murine models of hepatic and myocardial IRI, and showed that nitrite was associated with cytoprotective effects. In that setting, nitrite reduced cardiac infarct size by 67% and limited elevations of liver enzymes in a dose-dependent manner. These workers also demonstrated that nitrite was reduced to NO regardless of eNOS and heme oxygenase-1 enzyme activities[52]. The exact mechanisms as to how nitrite protects against this particular condition are being explored, but it appears that the benefit is mediated through the modulation of mitochondrial function by involving the posttranslational S-nitrosation of complex I to attenuate reperfusion oxygen radical generation and prevent cytochrome-C release[51].
NO donor drugs
Since nitric oxide is not considered to be an ideal gas for the treatment of IRI, NO donor drugs are now being explored as an alternative to the parent compound. Novel drugs have been developed and used for the delivery of NO in order to compensate for the very short half-life of NO in vivo. However, there are only two types of NO donor drugs that are currently used clinically: organic nitrates and sodium nitroprusside. Organic nitrates are the most commonly used NO donor drug treatment for coronary artery disease and congestive heart failure because the drugs produce clear clinical responses through their vasodilatory effects. Preparations of drugs include slow release oral forms, ointments, transdermal patches, nebulizers and traditional intravenous forms. The main limitation of organic nitrates is the induction of drug tolerance with prolonged continuous use. NO release from nitroglycerin is likely via the enzyme, mitochondrial aldehyde dehydrogenase[53]. The mechanism of NO release from sodium nitroprusside, on the other hand, is more complex, as demonstrated by Yang et al[53] in a murine model of hepatic IRI. Sodium nitroprusside is thought to down-regulate the mRNA expression of several enzymes related to hepatic injury[54]. We summarize other novel NO donor drugs in Table 2[2,34,36,54,55].
Table 2.
Nitric oxide donors
| Model | Drugs | Outcomes | Ref. |
| Canine liver IRI | FK-409 | Promoted hepatic tissue blood flow, decreased serum endothelin-1, cytoprotection | [34] |
| Isolated hepatocytes | S-nitroso-N-acetylpenicillamine | Drug induced the expression of heat shock protein 70 mRNA and protein resulting in cytoprotection from TNFα | [2] |
| Murine liver IRI | Sodium nitroprusside | Promotes hepatic tissue blood flow after reperfusion-cytoprotection | [36] |
| Murine liver IRI | PEG-poly SNO-BSA, a sustained release of NO | Decreased neutrophil accumulation, prevented the excessive production of iNOS | [54] |
| Murine liver IRI | Macromolecule S-nitrosothiols | Prevented hepatocellular injury | [55] |
NO: Nitric oxide; SNO: S-nitrosothiol; iNOS: Inducible nitric oxide synthase; IRI: Ischemia-reperfusion injury; TNF: Tumor necrosis factor.
CONCLUSION
Ischemia-reperfusion injury is a well-defined threat to the liver during periods of interruption and restoration of oxygen delivery, as occurs in certain procedures such as hepatic resections and orthotopic liver transplantations. Relative NO deficiency seems central in the pathogenesis of this injury. Replacing NO per se either by inhalation, nitrate anion or via donor drugs represents a novel means for ameliorating IRI. Further randomized controlled trials are needed to evaluate this therapy in patients undergoing operative procedures causing IRI.
Footnotes
Peer reviewer: Ana J Coito, Associate Professor of Surgery, Department of Surgery, The Dumont, UCLA Transplant Center, 77-120 CHS, Box 957054, Los Angeles, CA 90095-7054, United States
S- Editor Shi ZF L- Editor Logan S E- Editor Ma WH
References
- 1.Phillips L, Toledo AH, Lopez-Neblina F, Anaya-Prado R, Toledo-Pereyra LH. Nitric oxide mechanism of protection in ischemia and reperfusion injury. J Invest Surg. 2009;22:46–55. doi: 10.1080/08941930802709470. [DOI] [PubMed] [Google Scholar]
- 2.Kim YM, de Vera ME, Watkins SC, Billiar TR. Nitric oxide protects cultured rat hepatocytes from tumor necrosis factor-alpha-induced apoptosis by inducing heat shock protein 70 expression. J Biol Chem. 1997;272:1402–1411. doi: 10.1074/jbc.272.2.1402. [DOI] [PubMed] [Google Scholar]
- 3.Köken T, Inal M. The effect of nitric oxide on ischemia-reperfusion injury in rat liver. Clin Chim Acta. 1999;288:55–62. doi: 10.1016/s0009-8981(99)00138-2. [DOI] [PubMed] [Google Scholar]
- 4.Zaky A, Siriussawakul A, Tostenrud RP, Pauldine R, Lang JD Jr. Clinical update on therapeutic use of Nitric oxide. Contemp Crit Care. 2009;7:1–12. [Google Scholar]
- 5.Massip-Salcedo M, Roselló-Catafau J, Prieto J, Avíla MA, Peralta C. The response of the hepatocyte to ischemia. Liver Int. 2007;27:6–16. doi: 10.1111/j.1478-3231.2006.01390.x. [DOI] [PubMed] [Google Scholar]
- 6.Jennings RB, Shen AC, Hill ML, Ganote CE, Herdson PB. Mitochondrial matrix densities in myocardial ischemia and autolysis. Exp Mol Pathol. 1978;29:55–65. doi: 10.1016/0014-4800(78)90026-6. [DOI] [PubMed] [Google Scholar]
- 7.Sammut IA, Burton K, Balogun E, Sarathchandra P, Brooks KJ, Bates TE, Green CJ. Time-dependent impairment of mitochondrial function after storage and transplantation of rabbit kidneys. Transplantation. 2000;69:1265–1275. doi: 10.1097/00007890-200004150-00011. [DOI] [PubMed] [Google Scholar]
- 8.Yadav SS, Howell DN, Gao W, Steeber DA, Harland RC, Clavien PA. L-selectin and ICAM-1 mediate reperfusion injury and neutrophil adhesion in the warm ischemic mouse liver. Am J Physiol. 1998;275:G1341–G1352. doi: 10.1152/ajpgi.1998.275.6.G1341. [DOI] [PubMed] [Google Scholar]
- 9.Montalvo-Jave EE, Escalante-Tattersfield T, Ortega-Salgado JA, Piña E, Geller DA. Factors in the pathophysiology of the liver ischemia-reperfusion injury. J Surg Res. 2008;147:153–159. doi: 10.1016/j.jss.2007.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bigelow DJ, Thomas DD. Rotational dynamics of lipid and the Ca-ATPase in sarcoplasmic reticulum. The molecular basis of activation by diethyl ether. J Biol Chem. 1987;262:13449–13456. [PubMed] [Google Scholar]
- 11.Jaeschke H, Bautista AP, Spolarics Z, Spitzer JJ. Superoxide generation by Kupffer cells and priming of neutrophils during reperfusion after hepatic ischemia. Free Radic Res Commun. 1991;15:277–284. doi: 10.3109/10715769109105223. [DOI] [PubMed] [Google Scholar]
- 12.Kamiike W, Burdelski M, Steinhoff G, Ringe B, Lauchart W, Pichlmayr R. Adenine nucleotide metabolism and its relation to organ viability in human liver transplantation. Transplantation. 1988;45:138–143. doi: 10.1097/00007890-198801000-00030. [DOI] [PubMed] [Google Scholar]
- 13.Teoh NC, Farrell GC. Hepatic ischemia reperfusion injury: pathogenic mechanisms and basis for hepatoprotection. J Gastroenterol Hepatol. 2003;18:891–902. doi: 10.1046/j.1440-1746.2003.03056.x. [DOI] [PubMed] [Google Scholar]
- 14.Ysebaert DK, De Greef KE, Vercauteren SR, Ghielli M, Verpooten GA, Eyskens EJ, De Broe ME. Identification and kinetics of leukocytes after severe ischaemia/reperfusion renal injury. Nephrol Dial Transplant. 2000;15:1562–1574. doi: 10.1093/ndt/15.10.1562. [DOI] [PubMed] [Google Scholar]
- 15.Linfert D, Chowdhry T, Rabb H. Lymphocytes and ischemia-reperfusion injury. Transplant Rev (Orlando) 2009;23:1–10. doi: 10.1016/j.trre.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diesen DL, Kuo PC. Nitric Oxide and Redox Regulation in the Liver: Part II. Redox Biology in Pathologic Hepatocytes and Implications for Intervention. J Surg Res. 2009:Epub ahead of print. doi: 10.1016/j.jss.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Colletti LM, Kunkel SL, Walz A, Burdick MD, Kunkel RG, Wilke CA, Strieter RM. The role of cytokine networks in the local liver injury following hepatic ischemia/reperfusion in the rat. Hepatology. 1996;23:506–514. doi: 10.1002/hep.510230315. [DOI] [PubMed] [Google Scholar]
- 18.Kato A, Gabay C, Okaya T, Lentsch AB. Specific role of interleukin-1 in hepatic neutrophil recruitment after ischemia/reperfusion. Am J Pathol. 2002;161:1797–1803. doi: 10.1016/S0002-9440(10)64456-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qin X, Gao B. The complement system in liver diseases. Cell Mol Immunol. 2006;3:333–340. [PubMed] [Google Scholar]
- 20.Boros P, Bromberg JS. New cellular and molecular immune pathways in ischemia/reperfusion injury. Am J Transplant. 2006;6:652–658. doi: 10.1111/j.1600-6143.2005.01228.x. [DOI] [PubMed] [Google Scholar]
- 21.Hines IN, Harada H, Flores S, Gao B, McCord JM, Grisham MB. Endothelial nitric oxide synthase protects the postischemic liver: potential interactions with superoxide. Bio Pharmacoth. 2005;59:183–189. doi: 10.1016/j.biopha.2005.03.011. [DOI] [PubMed] [Google Scholar]
- 22.Duranski MR, Elrod JW, Calvert JW, Bryan NS, Feelisch M, Lefer DJ. Genetic overexpression of eNOS attenuates hepatic ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2006;291:H2980–H2986. doi: 10.1152/ajpheart.01173.2005. [DOI] [PubMed] [Google Scholar]
- 23.Katsumi H, Nishikawa M, Yamashita F, Hashida M. Prevention of hepatic ischemia/reperfusion injury by prolonged delivery of nitric oxide to the circulating blood in mice. Transplantation. 2008;85:264–269. doi: 10.1097/TP.0b013e31815e902b. [DOI] [PubMed] [Google Scholar]
- 24.Hur GM, Ryu YS, Yun HY, Jeon BH, Kim YM, Seok JH, Lee JH. Hepatic ischemia/reperfusion in rats induces iNOS gene transcription by activation of NF-kappaB. Biochem Biophys Res Commun. 1999;261:917–922. doi: 10.1006/bbrc.1999.1143. [DOI] [PubMed] [Google Scholar]
- 25.Isobe M, Katsuramaki T, Kimura H, Nagayama M, Matsuno T, Yagihashi A, Hirata K. Role of inducible nitric oxide synthase on hepatic ischemia and reperfusion injury. Transplant Proc. 2000;32:1650–1652. doi: 10.1016/s0041-1345(00)01435-4. [DOI] [PubMed] [Google Scholar]
- 26.Maejima Y, Adachi S, Morikawa K, Ito H, Isobe M. Nitric oxide inhibits myocardial apoptosis by preventing caspase-3 activity via S-nitrosylation. J Mol Cell Cardiol. 2005;38:163–174. doi: 10.1016/j.yjmcc.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 27.Kim PK, Zamora R, Petrosko P, Billiar TR. The regulatory role of nitric oxide in apoptosis. Int Immunopharmacol. 2001;1:1421–1441. doi: 10.1016/s1567-5769(01)00088-1. [DOI] [PubMed] [Google Scholar]
- 28.Marshall HE, Hess DT, Stamler JS. S-nitrosylation: physiological regulation of NF-kappaB. Proc Natl Acad Sci USA. 2004;101:8841–8842. doi: 10.1073/pnas.0403034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burwell LS, Brookes PS. Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10:579–599. doi: 10.1089/ars.2007.1845. [DOI] [PubMed] [Google Scholar]
- 30.Jaeschke H, Schini VB, Farhood A. Role of nitric oxide in the oxidant stress during ischemia/reperfusion injury of the liver. Life Sci. 1992;50:1797–1804. doi: 10.1016/0024-3205(92)90064-v. [DOI] [PubMed] [Google Scholar]
- 31.Takamatsu Y, Shimada K, Yamaguchi K, Kuroki S, Chijiiwa K, Tanaka M. Inhibition of inducible nitric oxide synthase prevents hepatic, but not pulmonary, injury following ischemia-reperfusion of rat liver. Dig Dis Sci. 2006;51:571–579. doi: 10.1007/s10620-006-3172-5. [DOI] [PubMed] [Google Scholar]
- 32.Wang LM, Tian XF, Song QY, Gao ZM, Luo FW, Yang CM. Expression and role of inducible nitric oxide synthase in ischemia-reperfusion liver in rats. Hepatobiliary Pancreat Dis Int. 2003;2:252–258. [PubMed] [Google Scholar]
- 33.Jiang WW, Kong LB, Li GQ, Wang XH. Expression of iNOS in early injury in a rat model of small-for-size liver transplantation. Hepatobiliary Pancreat Dis Int. 2009;8:146–151. [PubMed] [Google Scholar]
- 34.Aiba M, Takeyoshi I, Ohwada S, Kawashima Y, Iwanami K, Sunose Y, Yamada T, Tsutsumi H, Matsumoto K, Morishita Y. Novel nitric oxide donor (FK409) ameliorates liver damage during extended liver resection with warm ischemia in dogs. J Am Coll Surg. 2001;193:264–271. doi: 10.1016/s1072-7515(01)01002-x. [DOI] [PubMed] [Google Scholar]
- 35.Chattopadhyay P, Verma N, Verma A, Kamboj T, Khan NA, Wahi AK. L-arginine protects from pringle manoeuvere of ischemia-reperfusion induced liver injury. Biol Pharm Bull. 2008;31:890–892. doi: 10.1248/bpb.31.890. [DOI] [PubMed] [Google Scholar]
- 36.Nilsson B, Delbro D, Wallin M, Friman S. Protective effect of nitric oxide and prostaglandin E(2) in ischemia/reperfusion injury of the liver. Transplant Proc. 2001;33:2518–2520. doi: 10.1016/s0041-1345(01)02083-8. [DOI] [PubMed] [Google Scholar]
- 37.Cottart CH, Nivet-Antoine V, Do L, Al-Massarani G, Descamps G, Xavier-Galen F, Clot JP. Hepatic cytoprotection by nitric oxide and the cGMP pathway after ischaemia-reperfusion in the rat. Nitric Oxide. 2003;9:57–63. doi: 10.1016/j.niox.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 38.McMahon TJ, Doctor A. Extrapulmonary effects of inhaled nitric oxide: role of reversible S-nitrosylation of erythrocytic hemoglobin. Proc Am Thorac Soc. 2006;3:153–160. doi: 10.1513/pats.200507-066BG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Date H, Triantafillou AN, Trulock EP, Pohl MS, Cooper JD, Patterson GA. Inhaled nitric oxide reduces human lung allograft dysfunction. J Thorac Cardiovasc Surg. 1996;111:913–919. doi: 10.1016/s0022-5223(96)70364-1. [DOI] [PubMed] [Google Scholar]
- 40.Yerebakan C, Ugurlucan M, Bayraktar S, Bethea BT, Conte JV. Effects of inhaled nitric oxide following lung transplantation. J Card Surg. 2009;24:269–274. doi: 10.1111/j.1540-8191.2009.00833.x. [DOI] [PubMed] [Google Scholar]
- 41.Varadarajan R, Golden-Mason L, Young L, McLoughlin P, Nolan N, McEntee G, Traynor O, Geoghegan J, Hegarty JE, O'Farrelly C. Nitric oxide in early ischaemia reperfusion injury during human orthotopic liver transplantation. Transplantation. 2004;78:250–256. doi: 10.1097/01.tp.0000128188.45553.8c. [DOI] [PubMed] [Google Scholar]
- 42.Lang JD Jr, Teng X, Chumley P, Crawford JH, Isbell TS, Chacko BK, Liu Y, Jhala N, Crowe DR, Smith AB, et al. Inhaled NO accelerates restoration of liver function in adults following orthotopic liver transplantation. J Clin Invest. 2007;117:2583–2591. doi: 10.1172/JCI31892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mupanemunda RH, Edwards AD. Treatment of newborn infants with inhaled nitric oxide. Arch Dis Child Fetal Neonatal Ed. 1995;72:F131–F134. doi: 10.1136/fn.72.2.f131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Macrae DJ, Field D, Mercier JC, Møller J, Stiris T, Biban P, Cornick P, Goldman A, Göthberg S, Gustafsson LE, et al. Inhaled nitric oxide therapy in neonates and children: reaching a European consensus. Intensive Care Med. 2004;30:372–380. doi: 10.1007/s00134-003-2122-3. [DOI] [PubMed] [Google Scholar]
- 45.Ambalavanan N, St John E, Carlo WA, Bulger A, Philips JB 3rd. Feasibility of nitric oxide administration by oxygen hood in neonatal pulmonary hypertension. J Perinatol. 2002;22:50–56. doi: 10.1038/sj.jp.7210652. [DOI] [PubMed] [Google Scholar]
- 46.Ambalavanan N, El-Ferzli GT, Roane C, Johnson R, Carlo WA. Nitric oxide administration using an oxygen hood: a pilot trial. PLoS One. 2009;4:e4312. doi: 10.1371/journal.pone.0004312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pryor WA, Squadrito GL. The chemistry of peroxynitrite: a product from the reaction of nitric oxide with superoxide. Am J Physiol. 1995;268:L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- 48.Ischiropoulos H. Biological tyrosine nitration: a pathophysiological function of nitric oxide and reactive oxygen species. Arch Biochem Biophys. 1998;356:1–11. doi: 10.1006/abbi.1998.0755. [DOI] [PubMed] [Google Scholar]
- 49.Fullerton DA, McIntyre RC Jr. Inhaled nitric oxide: therapeutic applications in cardiothoracic surgery. Ann Thorac Surg. 1996;61:1856–1864. doi: 10.1016/0003-4975(96)00046-X. [DOI] [PubMed] [Google Scholar]
- 50.Cannon RO 3rd, Schechter AN, Panza JA, Ognibene FP, Pease-Fye ME, Waclawiw MA, Shelhamer JH, Gladwin MT. Effects of inhaled nitric oxide on regional blood flow are consistent with intravascular nitric oxide delivery. J Clin Invest. 2001;108:279–287. doi: 10.1172/JCI12761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shiva S, Gladwin MT. Nitrite mediates cytoprotection after ischemia/reperfusion by modulating mitochondrial function. Basic Res Cardiol. 2009;104:113–119. doi: 10.1007/s00395-009-0009-3. [DOI] [PubMed] [Google Scholar]
- 52.Duranski MR, Greer JJ, Dejam A, Jaganmohan S, Hogg N, Langston W, Patel RP, Yet SF, Wang X, Kevil CG, et al. Cytoprotective effects of nitrite during in vivo ischemia-reperfusion of the heart and liver. J Clin Invest. 2005;115:1232–1240. doi: 10.1172/JCI22493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang SL, Chen LJ, Kong Y, Xu D, Lou YJ. Sodium nitroprusside regulates mRNA expressions of LTC4 synthesis enzymes in hepatic ischemia/reperfusion injury rats via NF-kappaB signaling pathway. Pharmacology. 2007;80:11–20. doi: 10.1159/000102595. [DOI] [PubMed] [Google Scholar]
- 54.Katsumi H, Nishikawa M, Yamashita F, Hashida M. Prevention of hepatic ischemia/reperfusion injury by prolonged delivery of nitric oxide to the circulating blood in mice. Transplantation. 2008;85:264–269. doi: 10.1097/TP.0b013e31815e902b. [DOI] [PubMed] [Google Scholar]
- 55.Katsumi H, Nishikawa M, Yasui H, Yamashita F, Hashida M. Prevention of ischemia/reperfusion injury by hepatic targeting of nitric oxide in mice. J Control Release. 2009;140:12–17. doi: 10.1016/j.jconrel.2009.07.013. [DOI] [PubMed] [Google Scholar]