

Abstract
Two critical requirements for developing methods for the site-specific incorporation of amino acid analogues into proteins in vivo are (i) a suppressor tRNA that is not aminoacylated by any of the endogenous aminoacyl-tRNA synthetases (aaRSs) and (ii) an aminoacyl-tRNA synthetase that aminoacylates the suppressor tRNA but no other tRNA in the cell. Here we describe two such aaRS–suppressor tRNA pairs, one for use in the yeast Saccharomyces cerevisiae and another for use in Escherichia coli. The “21st synthetase–tRNA pairs” include E. coli glutaminyl-tRNA synthetase (GlnRS) along with an amber suppressor derived from human initiator tRNA, for use in yeast, and mutants of the yeast tyrosyl-tRNA synthetase (TyrRS) along with an amber suppressor derived from E. coli initiator tRNA, for use in E. coli. The suppressor tRNAs are aminoacylated in vivo only in the presence of the heterologous aaRSs, and the aminoacylated tRNAs function efficiently in suppression of amber codons. Plasmids carrying the E. coli GlnRS gene can be stably maintained in yeast. However, plasmids carrying the yeast TyrRS gene could not be stably maintained in E. coli. This lack of stability is most likely due to the fact that the wild-type yeast TyrRS misaminoacylates the E. coli proline tRNA. By using error-prone PCR, we have isolated and characterized three mutants of yeast TyrRS, which can be stably expressed in E. coli. These mutants still aminoacylate the suppressor tRNA essentially quantitatively in vivo but show increased discrimination in vitro for the suppressor tRNA over the E. coli proline tRNA by factors of 2.2- to 6.8-fold.
The site-specific incorporation of amino acid analogues into proteins in vitro has been used successfully for a number of applications, and these experiments have demonstrated the wide range of amino acid analogues, which are accepted by the translational machinery (1–4). The most common approach involves the read-through of an amber stop codon by an amber suppressor tRNA, which is chemically aminoacylated with the analogue of choice. Unfortunately, the procedure in general provides only small amounts of protein, and use of semisynthetic methods for the preparation of the aminoacyl-tRNA has limited its general application. Recent improvements to the system include increasing the overall translational efficiency or increasing the suppression-to-termination ratio by the use of extracts containing temperature-sensitive release factors (5–7). Isotopically labeled amino acids and amino acid analogues such as fluorotyrosine that are recognized by one of the aaRSs can also be incorporated site-specifically into proteins through a separate enzymatic aminoacylation step, thus avoiding the need for chemical aminoacylation (ref. 8 and S. Mamaev, J. Olejnik, A.K.K., V. Bergo, U.L.R., and K. J. Rothschild, unpublished observations). Microinjection of suppressor tRNAs aminoacylated with amino acid analogues into Xenopus oocytes has allowed the in vivo structure–function analysis of ion channel proteins (9), but extremely low protein yields limit further work.
The availability of methods to incorporate amino acid analogues site-specifically into proteins in vivo in eukaryotes and in eubacteria would greatly expand the scope and utility of unnatural amino acid mutagenesis. First, there is the potential to synthesize large amounts of the protein, making this a particularly useful technique for preparing material for spectroscopic and crystallographic analyses. Second, potential problems associated with posttranslational modifications and folding may be overcome for some proteins if a eukaryotic system is used. And finally, the availability of an in vivo system opens the door to the study of in vivo structure–function studies, including studies of protein–protein interactions, protein localization, etc., with the use of amino acid analogues that carry photoactivatable groups, fluorescent groups, and other chemically reactive groups.
The basic approach for the in vivo work relies on a suppressor tRNA aminoacylated with an amino acid analogue by a mutant aaRS, to insert the analogue at a specific site in a protein (10). The site of insertion of the analogue is specified by an appropriately placed stop codon within the gene for the protein of interest. This approach has two key requirements: (i) a suppressor tRNA, which cannot be aminoacylated by any of the endogenous aaRSs in a cell, and (ii) an aaRS, which aminoacylates the suppressor tRNA but no other tRNA in the cell. Therefore, an important first goal is to identify such an aaRS–suppressor tRNA pair (“21st synthetase–tRNA pair”). Once such a 21st synthetase–tRNA pair is identified, the next goal is to isolate mutants of the aaRS that will attach the desired amino acid analogue, but not the normal amino acid, to the suppressor tRNA.
In this paper we describe two candidate 21st synthetase–tRNA pairs,
one for use in yeast and another for use in Escherichia
coli. The yeast system represents the only 21st synthetase–tRNA
pair for use in a eukaryote. The suppressor tRNAs (11, 12) are derived
from a tRNA of another eukaryotic organism (human initiator tRNA for
use in yeast) or from a tRNA of the same organism (E. coli
initiator tRNA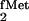 for use in E. coli). The
21st synthetases are E. coli GlnRS for use in yeast and
mutants of yeast TyrRS for use in E. coli. The 21st
synthetase–tRNA pairs that we have developed are different in
principle from the ones described by Schultz and coworkers for use in
E. coli, which are based on the import of both the aaRS and
the suppressor derived from a cognate tRNA from a heterologous organism
(13, 14).
for use in E. coli). The
21st synthetases are E. coli GlnRS for use in yeast and
mutants of yeast TyrRS for use in E. coli. The 21st
synthetase–tRNA pairs that we have developed are different in
principle from the ones described by Schultz and coworkers for use in
E. coli, which are based on the import of both the aaRS and
the suppressor derived from a cognate tRNA from a heterologous organism
(13, 14).
Materials and Methods
General.
Standard E. coli and yeast genetic techniques were used (15, 16). Yeast cells were transformed with the method of Tan et al. (17). Synthetic minimal media for growth and maintenance of yeast cells were supplemented with 2% raffinose + 2% dextrose or 2% raffinose + 2% galactose and amino acids to give SRD or SRG, respectively. Radiolabeled amino acids were from New England Nuclear, oligonucleotides were from Genosys (The Woodlands, TX), anti-myc antibody was from Roche Molecular Biochemicals, and anti-tetra-His antibody was from Qiagen (Chatsworth, CA).
Strains.
E. coli strains DH5α and JM109 were used for plasmid propagation and isolation. E. coli CA274 [lacZ125(am) trp49(am) relA1 spoT1] and the isogenic Su+ strain CA275 (CA274 supF) were used for in vivo screening of stably expressed Saccharomyces cerevisiae TyrRS mutants. S. cerevisiae HEY301–129 [a met8–1am trp1–1am his4–580am leu2–3,112 ura3-1 ade1 can1-100] (18) was used as a tester strain for suppression in yeast.
Plasmids.
The plasmids pETTyrRS, pACamG, and pRSVCATam27 carrying the
U2:A71/U35A36/G72 mutant tRNA gene have been
described previously (19). E. coli UQ27 containing the
plasmid pUCProRS was obtained from W. McClain (Univ. of Wisconsin). The
plasmid pETTyrRS-myc was constructed by replacement of the
StuI-BamHI fragment of pETTyrRS with a
PCR-generated fragment comprising the same 3′ sequence of the gene with
the addition of bases coding for the 11 amino acid c-myc tag. For
overexpression, the three mutant TyrRS genes were cloned into pET3d
(Novagen). The plasmid pUC8pro1, containing the E. coli
tRNA
gene have been
described previously (19). E. coli UQ27 containing the
plasmid pUCProRS was obtained from W. McClain (Univ. of Wisconsin). The
plasmid pETTyrRS-myc was constructed by replacement of the
StuI-BamHI fragment of pETTyrRS with a
PCR-generated fragment comprising the same 3′ sequence of the gene with
the addition of bases coding for the 11 amino acid c-myc tag. For
overexpression, the three mutant TyrRS genes were cloned into pET3d
(Novagen). The plasmid pUC8pro1, containing the E. coli
tRNA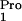 gene, was constructed by PCR amplification of
the proK gene, including the promoter and termination
sequences, from E. coli genomic DNA, and subsequent cloning
into the SmaI site of pUC8.
gene, was constructed by PCR amplification of
the proK gene, including the promoter and termination
sequences, from E. coli genomic DNA, and subsequent cloning
into the SmaI site of pUC8.
pScM, a YEp420-derived 2 μ URA+ plasmid, contains the wild-type human initiator tRNA coding sequence downstream of the yeast tRNAArg 5′-flanking sequence (20). The wild-type initiator tRNA coding sequence was replaced by a fragment carrying the U35A36/U50G51:C63A64/U54/C60 mutations in the tRNA (Fig. 1 Left) to yield the plasmid pScM.M2. For inducible expression of E. coli GlnRS in yeast, a c-His-6-tagged version of the E. coli GlnRS gene was cloned under the control of the GAL1 promoter of the 2 μ LEU+ plasmid pESC-LEU (Stratagene). The pESC-LEU.GlnRSHis6 was constructed by amplifying the gene encoding the c-His-6-tagged E. coli GlnRS from pQE16-GlnRS (21) and inserting a sequence of seven A residues directly upstream of the initiation codon ATG by using PCR. The PCR product was digested with BamHI and HindIII and cloned into the multiple cloning site of pESC-LEU.
Figure 1.

Cloverleaf structure of the human initiator tRNA (Left) and E. coli initiator tRNA (Right). Arrows indicate the sequence changes in these tRNAs for their use as part of the 21st synthetase–tRNA pairs in yeast (Left) and E. coli (Right).
In Vivo Selection and Characterization of Stably Expressed S. cerevisiae TyrRS Mutants.
A library of c-myc-tagged mutant yeast TyrRS genes was prepared by error-prone PCR (22), with the use of pETTyrRS-myc as a template, and cloned between the XmaI and PstI sites of the isopropyl-β-d-thiogalactoside (IPTG)-inducible expression vector pKK223–3 (Amersham Pharmacia). The mixture of plasmids was electroporated into competent CA274 cells carrying the pACamG plasmid, and chloramphenicol-resistant colonies were selected on plates containing carbenicillin (50 μg/ml), tetracycline (10 μg/ml), and chloramphenicol (100 μg/ml) at 37°C. The Su+ phenotype of the chloramphenicol-resistant colonies was confirmed by scoring for growth on M9 minimal plates containing 0.4% lactose and by colony color on MacConkey plates containing carbenicillin, tetracycline, and lactose. Colonies were inoculated into 2× YT medium containing all three antibiotics, grown to midlog phase, and induced with 1 mM IPTG. After an additional 4 h of growth, total tRNAs were isolated, and the extent of aminoacylation of tRNA was determined as described below.
Purification of Wild-Type and Mutant S. cerevisiae TyrRS.
The wild-type, myc-tagged, and three mutant TyrRS proteins were overproduced in E. coli NovaBlue (DE3) and purified by modification of a previously described method (19). Fractions containing TyrRS were pooled and concentrated with the use of an Amicon 30 spin filter, then dialyzed against storage buffer (50 mM potassium phosphate, pH 7.2/5 mM DTT/150 mM KCl/50% glycerol) and stored at −20°C.
Enzyme Assays.
The incubation mixture for aminoacylation contained 30 mM Hepes⋅KOH (pH 7.5), 50 mM KCl, 8 mM MgCl2, 2 mM DTT, 3 mM ATP, 15 μM [3H]tyrosine (specific activity 20–33 μCi/nmol), 0.18 mg/ml BSA, tRNA, and TyrRS. The enzyme was diluted in a solution containing 15 mM potassium phosphate (pH 7.5), 100 mM KCl, 4 mM DTT, 10% glycerol, and 0.18 mg/ml BSA. Kinetic parameters were determined by Lineweaver–Burk blots. The rate of aminoacylation was linear over the enzyme concentrations used, and the extent of tRNA aminoacylation was limited to less than 10%.
Purification of tRNAs.
Yeast tRNATyr was purified from an enriched
mixture of yeast tRNATyr and
tRNAPhe from counter-current distribution of
total yeast tRNA (23) by electrophoresis on nondenaturing 12%
polyacrylamide gels. The suppressor tRNAs derived from E.
coli tRNA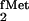 and E. coli
tRNAPro were isolated from E. coli
B105 cells carrying the pRSVCATam27 (U2:A71/U35A36/G72) plasmid and
E. coli JM109 carrying the pUC8pro1 plasmid, respectively,
by phenol extraction and subsequently purified by electrophoresis on
nondenaturing 12% polyacrylamide gels (24). The purity of the tRNAs
was determined to be greater than 90% by aminoacylation, with an
appropriate aaRS preparation in all cases.
and E. coli
tRNAPro were isolated from E. coli
B105 cells carrying the pRSVCATam27 (U2:A71/U35A36/G72) plasmid and
E. coli JM109 carrying the pUC8pro1 plasmid, respectively,
by phenol extraction and subsequently purified by electrophoresis on
nondenaturing 12% polyacrylamide gels (24). The purity of the tRNAs
was determined to be greater than 90% by aminoacylation, with an
appropriate aaRS preparation in all cases.
Extent of Aminoacylation of Suppressor tRNAs in Vivo.
Total tRNA from E. coli was isolated under acidic conditions by the guanidine thiocyanate–phenol–chloroform method (25) (Tri-Reagent; Molecular Research Center, Cincinnati). Total tRNA from yeast was isolated as described (26). The extent of aminoacylation was determined by fractionation of the tRNAs by acid–urea gel electrophoresis, followed by detection of the suppressor tRNAs by Northern blot hybridization with specific oligonucleotide probes (27).
Results
21st Synthetase–tRNA Pair for Use in Yeast.
We reported previously on a mutant human initiator tRNA that can act as an amber suppressor in mammalian COS1 cells (12). This tRNA has the U35A36 mutations in the anticodon sequence, which allows it to read the UAG codon and the U50G51:C63A64 mutations in the TΨC stem, which allows it to participate in elongation. This mutant tRNA is not aminoacylated by any of the mammalian aaRSs, but is aminoacylated by E. coli GlnRS and, therefore, acts as an amber suppressor in mammalian cells expressing E. coli GlnRS. The mutant tRNA to be used as part of the 21st synthetase–tRNA pair in yeast has additional mutations, U54 and C60 in the TΨC loop (Fig. 1 Left), designed to make the tRNA more active in elongation (28).
The gene encoding the human initiator tRNA is not transcribed by yeast RNA polymerase III in the context of its own 5′-flanking sequence (29). Therefore, for expression in yeast, the mutant human initiator tRNA coding sequence was cloned downstream of the 5′-flanking sequence of the yeast tRNAArg gene (20). This construct (pScM.M2) expresses the mutant human initiator tRNA constitutively. To express the E. coli GlnRS in yeast, we used plasmid pESC-LEU.GlnRSHis6, which carries the E. coli GlnRS gene appropriately modified for expression in yeast (see Materials and Methods), under the control of the inducible yeast GAL1 promoter.
The Mutant Human Initiator tRNA Is Aminoacylated Only in Yeast Cells Expressing the E. coli GlnRS.
Transformants of S. cerevisiae HEY301-129 harboring pESC-LEU.GlnRSHis6/pScM.M2 were grown in selective medium in the absence or presence of galactose. Immunoblot analysis showed that E. coli GlnRS was produced only in cells grown in the presence of galactose (data not shown). tRNAs were isolated from cells under acidic conditions, and the extent of in vivo aminoacylation of tRNA was determined by acid–urea gel electrophoresis followed by Northern blot analysis with a probe specific for the M2 mutant tRNA (27). Aminoacylation of the mutant tRNA in yeast is strictly dependent on expression of E. coli GlnRS (Fig. 2A). PhosphorImager analysis of the blot showed that approximately 45% of the tRNA was aminoacylated.
Figure 2.

The human initiator tRNA mutant is aminoacylated in yeast only in the presence of E. coli GlnRS and is active in suppression. (A) Acid–urea gel analysis of the mutant tRNA expressed in S. cerevisiae HEY301–129, grown in SRD (minus E. coli GlnRS) or SRG (plus E. coli GlnRS). (B) Test for suppression of the met8-1 amber allele in S. cerevisiae HEY301-129. URA+LEU+ transformants carrying pScM.M2 (the mutant suppressor tRNA) and pESC-LEU (plus and minus the E. coli GlnRS gene) were streaked on a selective plate (SRG) lacking methionine.
The Mutant Human Initiator tRNA Acts as a Suppressor of Amber Codons Only in Yeast Cells Expressing E. coli GlnRS.
S. cerevisiae HEY301-129 has three suppressible amber alleles (met8-1, trp1-1, his4-580) (18). To test for tRNA-mediated suppression of these amber alleles, HEY301-129 cells harboring pESC-LEU.GlnRSHis6 or the pESC-LEU plasmid without the GlnRS gene were transformed with pScM.M2. URA+LEU+ transformants were isolated and streaked on selective minimal medium plates supplemented with two of the three amino acids. Suppression is indicated by cell growth in the absence of tryptophan, histidine, or methionine. Fig. 2B shows the results obtained for the methionine marker. Expression of the mutant initiator tRNA alone does not result in suppression of the met8-1 allele; however, coexpression of E. coli GlnRS along with the mutant initiator tRNA results in suppression. Suppression of the his4-580 allele was also strictly dependent on the coexpression of E. coli GlnRSHis6 and the mutant tRNA (data not shown). Control experiments showed that coexpression of the wild-type human initiator tRNA and E. coli GlnRS did not result in suppression of the met8-1 or the his4-580 amber alleles. All transformants grew in the absence of tryptophan, indicating suppressor-tRNA independent read-through of the trp1-1 allele, which is known to be a leaky mutation (18).
We also compared the growth rates of HEY301-129 carrying the 21st synthetase–tRNA pair, grown in the SRG minimal medium lacking either methionine or histidine, with that of transformants grown in the presence of methionine, histidine, and tryptophan (complete medium). After an initial lag phase, the doubling times of cells grown in medium lacking methionine or histidine was 360 and 375 min, respectively, compared with the doubling time of 255 min of cells grown in complete medium. These results suggest that although only 45% of the suppressor tRNA is aminoacylated in HEY301-129 (Fig. 2A), the suppressor activity of the tRNA is high enough to produce adequate amounts of the methionine and histidine biosynthesis enzymes for good growth in medium lacking one or the other of these amino acids.
21st Synthetase–tRNA Pair for Use in E. coli.
In the course of studies on E. coli initiator
tRNA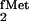 designed to identify elements in the tRNA
important for its function, we previously isolated a mutant tRNA
(U35A36/G72 mutant) that is a suppressor of amber codons in E.
coli (30). This tRNA is aminoacylated by E. coli GlnRS.
Introduction of further mutations (U2:A71) resulted in a tRNA (Fig. 1
Right) that is now an extremely poor amber suppressor in
E. coli, because it is not aminoacylated by any of the other
aaRSs in E. coli (19). In vitro also, this tRNA
is not aminoacylated by purified preparations of E. coli
GlnRS, LysRS, MetRS, TyrRS, or a highly enriched preparation of ProRS
(A.K.K. and U.L.R., unpublished observations). The mutant tRNA contains
the C1⋅G72 base pair, which is one of the critical determinants
for yeast TyrRS recognition (11, 31). Because of this makeup of the
mutant tRNA, it is aminoacylated by yeast TyrRS and is, therefore,
active as an amber suppressor in yeast (11). Based on this observation,
we used a genetic selection system requiring the activity of the
suppressor tRNA in E. coli to clone the gene for yeast TyrRS
in a low copy yeast shuttle vector (19). If the yeast TyrRS does not
aminoacylate any of the endogenous E. coli
tRNAs, the yeast TyrRS and the U2:A71/U35A36/G72 mutant E.
coli tRNA
designed to identify elements in the tRNA
important for its function, we previously isolated a mutant tRNA
(U35A36/G72 mutant) that is a suppressor of amber codons in E.
coli (30). This tRNA is aminoacylated by E. coli GlnRS.
Introduction of further mutations (U2:A71) resulted in a tRNA (Fig. 1
Right) that is now an extremely poor amber suppressor in
E. coli, because it is not aminoacylated by any of the other
aaRSs in E. coli (19). In vitro also, this tRNA
is not aminoacylated by purified preparations of E. coli
GlnRS, LysRS, MetRS, TyrRS, or a highly enriched preparation of ProRS
(A.K.K. and U.L.R., unpublished observations). The mutant tRNA contains
the C1⋅G72 base pair, which is one of the critical determinants
for yeast TyrRS recognition (11, 31). Because of this makeup of the
mutant tRNA, it is aminoacylated by yeast TyrRS and is, therefore,
active as an amber suppressor in yeast (11). Based on this observation,
we used a genetic selection system requiring the activity of the
suppressor tRNA in E. coli to clone the gene for yeast TyrRS
in a low copy yeast shuttle vector (19). If the yeast TyrRS does not
aminoacylate any of the endogenous E. coli
tRNAs, the yeast TyrRS and the U2:A71/U35A36/G72 mutant E.
coli tRNA (Fig. 1 Right) would
comprise a potential 21st synthetase–tRNA pair for use in
E. coli.
(Fig. 1 Right) would
comprise a potential 21st synthetase–tRNA pair for use in
E. coli.
Wild-Type Yeast TyrRS Can Aminoacylate E. coli tRNAPro.
Our initial attempts to clone and express the yeast TyrRS in E. coli with the use of the standard expression vector pKK223-3 yielded a lethal phenotype, which could only partially be overcome by growth of the cells in the presence of the mutant suppressor tRNA and at lower temperatures (22°C or 30°C). The toxicity of the TyrRS gene was most likely caused by misaminoacylation of one of the E. coli tRNAs, with some compensation provided by the presence of another substrate, the suppressor tRNA, for the enzyme to act on. The two major identity determinants for yeast TyrRS are the anticodon bases (shared only by the E. coli tRNATyr, which is not a substrate for the yeast enzyme) and the C1⋅G72 base pair (11, 31, 32). This base pair is found only in tyrosine tRNAs of eukaryotes and archaebacteria and proline tRNAs of E. coli. Therefore, we considered the possibility that yeast TyrRS was misaminoacylating E. coli tRNAPro. Aminoacylation reactions in vitro with purified yeast TyrRS and total E. coli tRNA demonstrated that the yeast enzyme is capable of aminoacylating tRNAPro with tyrosine (Fig. 3). The toxicity of yeast TyrRS to E. coli appears to be dependent at least in part on its intracellular concentration, as the gene was originally isolated by a functional selection, which relied on expression from a low copy yeast shuttle vector. However, for its use as a 21st synthetase, the yeast TyrRS first had to be modified so that it would no longer mischarge the E. coli tRNAPro.
Figure 3.

Acid–urea gel electrophoretic analysis of E. coli tRNAPro in total E. coli tRNA incubated in vitro with purified yeast TyrRS and tyrosine.
In Vivo Selection for Mutants of Yeast TyrRS That Are
Stable in E. coli and Can Still Aminoacylate the Mutant
tRNA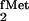 (U2:A71/U35A36/G72).
(U2:A71/U35A36/G72).
The selection originally used (19) for the isolation of the yeast TyrRS gene from a yeast cDNA library was adapted for the purpose of isolating TyrRS mutants, which could be stably maintained in E. coli (indicative of negligible mischarging of the endogenous tRNAPro), while requiring that the enzyme retain its aminoacylation activity with the mutant suppressor tRNA. The selection relied on coexpression of the suppressor tRNA, along with an amber mutant of the chloramphenicol acetyltransferase (CAT) gene, both carried by the pACamG plasmid. The activity of the yeast TyrRS was selected for by requiring growth on chloramphenicol, an indication that functional enzyme was present and was able to aminoacylate the suppressor tRNA. A library of yeast TyrRS mutants was prepared by error-prone PCR (Materials and Methods), cloned into the pKK223–3 plasmid, and used to transform E. coli CA274 carrying the pACamG plasmid. Twelve chloramphenicol-resistant colonies were picked, and the Su+ phenotype of three of them was confirmed further by restreaking on chloramphenicol plates (Fig. 4A) and by screening for colony color on MacConkey lactose plates and scoring for growth on plates containing M9 minimal medium with lactose, which relied on suppression of the two amber mutations in CA274 lacZ(am) and trpA(am) (data not shown). The stability of the three clones carrying the plasmids pKKTyrRS*5, pKKTyrRS*9, and pKKTyrRS*10 was confirmed by continued maintenance of the plasmids in liquid medium. Analysis of the in vivo aminoacylation levels of the mutant suppressor tRNA on acid–urea gels (27) showed that the tRNA was essentially quantitatively aminoacylated in the presence of any of the three yeast TyrRS mutants, in contrast to undetectable levels of aminoacylation in the absence of the enzyme (Fig. 4B).
Figure 4.

The E. coli initiator tRNA mutant is aminoacylated in E. coli only in the presence of the yeast TyrRS mutants, and the aminoacyl-tRNA is active in suppression. (A) Growth of E. coli CA274 transformed with pACamG (carrying the mutant suppressor tRNA gene and the CATam27 gene), and pKK223-3 (plus and minus the mutant TyrRS genes) on a 2× YT plate containing 100 μg/ml chloramphenicol. (B) Acid–urea gel analysis of the mutant tRNA expressed in E. coli in the presence and absence of the TyrRS mutants.
Sequencing revealed that three independent yeast TyrRS mutants had been isolated, with no overlapping mutations other than a serine-to-aspartate mutation found in the C-terminal myc tags of TyrRS*5 and TyrRS*9. TyrRS*10 had a different mutation in the myc tag, a glutamic acid-to-valine change. TyrRS*10 had six additional mutations in the coding sequence, whereas TyrRS*5 had three and TyrRS*9 had only one (Table 1). All mutants also had several silent mutations, none of which appeared to involve poorly used codons in E. coli. This latter result is consistent with the finding by immunoblot analysis that there were no appreciable differences in the levels of the various TyrRS mutants (data not shown). The mutations in TyrRS*10 were scattered throughout the gene, whereas the mutations in TyrRS*5 and TyrRS*9 were primarily in the C-terminal region of the protein, which is the binding domain for the anticodon region of the tRNA. None of the mutations fall in the connective polypeptide 1 region, which has been shown to be important for discrimination of the base pair at positions 1 and 72 in the tRNA (33).
Table 1.
Sequence changes found in yeast TyrRS mutants that are stably expressed in E. coli
| Mutant | Amino acid changes |
|---|---|
| TyrRS*5 | His-285 → Tyr |
| Phe-286 → Tyr | |
| Lys-372 → Met | |
| (Ser-9' → Asp) | |
| TyrRS*9 | Asn-344 → His |
| (Ser-9' → Asp) | |
| TyrRS*10 | Thr-6 → Ser |
| Val-35 → Leu | |
| Asn-113 → Asp | |
| Met-218 → Ile | |
| Lys-381 → Thr | |
| Glu-391 → Gly | |
| (Glu-1' → Val) |
Only sense mutations are listed. Mutations found in the myc tag region are indicated by parentheses, with the first amino acid of the tag numbered as 1'.
In Vitro Characterization of the Yeast TyrRS Mutants.
The mutant TyrRS genes were cloned into the expression vector pET3d, and the TyrRS mutants were purified. The enzymatic activity of the wild-type, the myc-tagged, and the three mutant TyrRS proteins was determined with three substrates: yeast tRNATyr, the mutant suppressor tRNA, and E. coli tRNAPro. Interestingly, the mutations had very little effect on activity toward the natural substrate (yeast tRNATyr) or the desired substrate (the suppressor tRNA) (Table 2). TyrRS*9, containing only one mutation, differed less than 2-fold in kcat/Km from the wild type with both substrates, whereas both TyrRS*5 and TyrRS*10 differed less than 3-fold. With the E. coli tRNAPro as substrate, however, there was a more significant effect, a decrease in kcat/Km from more than 5-fold (TyrRS*5) to as high as 10-fold (TyrRS*10). Some of the decrease in activity with this substrate appears to be a result of the addition of the C-terminal myc tag, which was also seen to have a smaller effect on activity with the mutant suppressor tRNA. However, the myc tag alone is unlikely to be responsible for the selection of these particular mutants, as only a limited number of chloramphenicol-resistant colonies were isolated from the in vivo selection. Most importantly, all three of the mutants show an increase in specificity for the suppressor tRNA over tRNAPro (Table 3), with TyrRS*9 preferring the suppressor tRNA over tRNAPro by a factor of 15-fold.
Table 2.
Kinetic parameters of wild-type and mutant yeast TyrRS†
| tRNA substrate Enzyme | kcat/Km, s−1 M−1 × 106 | Relative kcat/Km |
|---|---|---|
| Yeast tRNATyr | ||
| TyrRS (wt) | 8.5 | 1 |
| TyrRS-myc | 11.8 | 1.2 |
| TyrRS*5 | 3.1 | 0.36 |
| TyrRS*9 | 6.7 | 0.79 |
| TyrRS*10 | 3.6 | 0.54 |
| Suppressor tRNA | ||
| TyrRS (wt) | 0.014 | 1 |
| TyrRS-myc | 0.0067 | 0.48 |
| TyrRS*5 | 0.0077 | 0.55 |
| TyrRS*9 | 0.015 | 1.1 |
| TyrRS*10 | 0.0059 | 0.42 |
| E. coli tRNAPro | ||
| TyrRS (wt) | 0.0061 | 1 |
| TyrRS-myc | 0.0016 | 0.26 |
| TyrRS*5 | 0.0015 | 0.24 |
| TyrRS*9 | 0.0010 | 0.16 |
| TyrRS*10 | 0.00061 | 0.10 |
Kinetic parameters listed are the average of two or more experiments. The tRNA concentrations varied from 0.067 to 5× Km. Enzyme concentrations ranged from 0.05 nM to 730 nM, depending on the tRNA used.
Table 3.
Ratio of specificity constants for the aminoacylation of the U2:A71/U35A36/G72 mutant suppressor tRNA and E. coli tRNAPro by the wild-type and mutant yeast TyrRS
| Enzyme |
kcat/Km
(suppressor tRNA)
|
|---|---|
| kcat/Km (tRNAPro) | |
| TyrRS (wt) | 2.30 |
| TyrRS-myc | 4.19 |
| TyrRS*5 | 5.13 |
| TyrRS*9 | 15.0 |
| TyrRS*10 | 9.70 |
Discussion
The 21st synthetase–tRNA pairs that we have developed for use in eukaryotes and in eubacteria consist, in both cases, of a heterologous aaRS and a suppressor tRNA derived from the initiator tRNA of the same organism or a related organism (11, 12). Besides providing the only example of a 21st synthetase–tRNA pair for use in a eukaryotic system, our approach differs somewhat from that of Schultz and coworkers, in which both the aaRS and the suppressor tRNA are imported from a heterologous organism (13, 14), and it offers certain advantages. For example, because the suppressor tRNAs used are derived from tRNA of the same organism or a related organism, transcripts derived from the suppressor tRNA genes are likely to be processed well to yield functional suppressor tRNAs (34). Moreover, the suppressor tRNAs are likely to carry base modifications that are normally found in the tRNA and are, therefore, more likely to be optimally active in suppression (35). We have shown that the suppressor activity of the human initiator tRNA mutant in yeast is quite high. We have also compared the growth rates of E. coli CA274 lacZ(am) trpA(am) carrying either of the three mutant yeast TyrRS–suppressor tRNA pairs with that of an isogenic Su+ E. coli strain CA275, in a medium requiring suppression of the two amber alleles, and have found that the growth rates are similar (data not shown). Because the endogenous tyrosine suppressor tRNA in E. coli CA275 is a “strong” suppressor (36), the suppression efficiency of the 21st synthetase–tRNA pair must also be quite high. An important potential application of incorporation of amino acid analogues into proteins in vivo is the synthesis of large amounts of proteins carrying chemical or spectroscopic probes or heavy atoms such as iodine at specific sites. This application requires that suppression efficiency at the sites of incorporation of amino acid analogues be high. The systems that we have developed fulfill this requirement.
The approach used by Schultz and coworkers (13, 14), in which both the aaRS and the suppressor tRNA are imported from a heterologous organism, is based on the inability of a few eubacterial aaRSs to aminoacylate the corresponding tRNAs from eukaryotes or archaebacteria and vice versa. For example, the E. coli GlnRS does not aminoacylate yeast tRNAGln, and the yeast GlnRS does not aminoacylate E. coli tRNAGln (37). Therefore, the import of such synthetase–tRNA pairs from a eukaryotic organism to eubacteria or vice versa would, at the outset, appear to provide a simple solution to the identification of potential 21st synthetase–tRNA pairs. However, although the GlnRS–tRNAGln suppressor combination appears to be portable in the case of both eukaryotes (34) and eubacteria (13), this combination cannot be used as a general approach. For example, the fact that yeast tRNATyr is not aminoacylated by E. coli TyrRS does not mean that the suppressor derived from it will not be aminoacylated by other E. coli aaRSs. Initially, we considered the possibility of importing the yeast TyrRS and the suppressor derived from yeast tRNATyr as the 21st synthetase–tRNA pair for use in E. coli. We found, however, that when expressed to moderately high levels, this tRNA was a good suppressor on its own in E. coli, suggesting that it was aminoacylated by one of the E. coli aaRSs (C. M. Chow and U.L.R., unpublished observations). This result agrees with a recent report of Wang et al. (14) but differs from that of Ohno et al. (38). Furter has also reported that an amber suppressor derived from yeast tRNAPhe is aminoacylated in E. coli by LysRS (39).
Similarly, as shown here, the fact that the yeast TyrRS does not
aminoacylate the E. coli tyrosine tRNA does not mean that it
will not aminoacylate any of the other E. coli tRNAs. For
example, although we initially cloned the gene for yeast TyrRS in
E. coli on the basis of its ability to complement the
aminoacylation defect of a suppressor tRNA derived from the E.
coli tRNA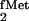 (19), we could not isolate
transformants expressing the yeast TyrRS at moderate levels, presumably
because the yeast TyrRS misaminoacylated E. coli
tRNAPro with tyrosine (Fig. 3) and was,
therefore, toxic to E. coli. It was necessary to isolate
mutants of yeast TyrRS that could be stably expressed in E.
coli, most likely because of their much reduced activity in
misaminoacylation of tRNAPro. The usual approach
to circumventing the problem of toxicity of proteins being expressed in
E. coli is to tightly regulate the expression of the
protein. Such an approach is, however, not useful here because, once
induced, the yeast TyrRS will still misaminoacylate the E.
coli tRNAPro with tyrosine, leading to
misincorporation of tyrosine for proline in proteins. A further problem
with the regulation of expression of the 21st synthetases, which
have the capability of misaminoacylating endogenous tRNAs,
would be the loss of site specificity of incorporation of the amino
acid analogue in the target protein.
(19), we could not isolate
transformants expressing the yeast TyrRS at moderate levels, presumably
because the yeast TyrRS misaminoacylated E. coli
tRNAPro with tyrosine (Fig. 3) and was,
therefore, toxic to E. coli. It was necessary to isolate
mutants of yeast TyrRS that could be stably expressed in E.
coli, most likely because of their much reduced activity in
misaminoacylation of tRNAPro. The usual approach
to circumventing the problem of toxicity of proteins being expressed in
E. coli is to tightly regulate the expression of the
protein. Such an approach is, however, not useful here because, once
induced, the yeast TyrRS will still misaminoacylate the E.
coli tRNAPro with tyrosine, leading to
misincorporation of tyrosine for proline in proteins. A further problem
with the regulation of expression of the 21st synthetases, which
have the capability of misaminoacylating endogenous tRNAs,
would be the loss of site specificity of incorporation of the amino
acid analogue in the target protein.
Kinetic analysis of the three yeast TyrRS mutants with the yeast
tRNATyr, the suppressor tRNA derived from
E. coli tRNA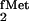 , and E. coli
tRNAPro as substrates, shows that compared with
the wild-type yeast TyrRS, which prefers the suppressor tRNA over
tRNAPro by a factor of about 2, with the mutant
enzymes the corresponding numbers are between 5 and 15 (Table 3). Thus
relatively small decreases in activity of yeast TyrRS toward E.
coli tRNAPro are enough to prevent the
misaminoacylation in vivo of tRNAPro
with tyrosine by yeast TyrRS. This observation is consistent with data
showing that the high fidelity of aminoacylation of tRNAs in
vivo is a result not only of positive and negative interactions of
aaRSs with cognate and noncognate tRNAs, respectively, but is also
achieved by maintaining a delicate balance of levels of enzymes and
tRNAs, which helps to prevent misaminoacylation reactions (40–43).
, and E. coli
tRNAPro as substrates, shows that compared with
the wild-type yeast TyrRS, which prefers the suppressor tRNA over
tRNAPro by a factor of about 2, with the mutant
enzymes the corresponding numbers are between 5 and 15 (Table 3). Thus
relatively small decreases in activity of yeast TyrRS toward E.
coli tRNAPro are enough to prevent the
misaminoacylation in vivo of tRNAPro
with tyrosine by yeast TyrRS. This observation is consistent with data
showing that the high fidelity of aminoacylation of tRNAs in
vivo is a result not only of positive and negative interactions of
aaRSs with cognate and noncognate tRNAs, respectively, but is also
achieved by maintaining a delicate balance of levels of enzymes and
tRNAs, which helps to prevent misaminoacylation reactions (40–43).
Finally, with the requirement for two 21st synthetase–tRNA pairs fulfilled, the next step is isolation of mutants in the 21st synthetases, which aminoacylate the suppressor tRNA with the amino acid analogues of choice instead of with the normal amino acid. This isolation of mutants is likely to be a challenging step, possibly requiring multiple rounds of mutagenesis and selection to obtain a 21st synthetase, which shows an increase in selectivity for the amino acid analogue over the normal amino acid. Among the possible selection schemes are the use of (i) phage display of aaRS mutants (44), with selection based on binding to an affinity matrix consisting of an aminoalkyl-adenylate derived from the amino acid analogue, and (ii) antibodies directed against peptides containing the amino acid analogue in a reporter protein expressed on the cell surface (10, 45) to select for cells carrying the desired aaRS mutant.
Acknowledgments
We thank Annmarie McInnis for patience and care in the preparation of this paper. This work was supported by Grant DAAD19-199-1-0300 from the U.S. Army Research Office and in part by Grant R37GM17151 from the National Institutes of Health.
Abbreviations
- aaRS
aminoacyl-tRNA synthetase
- CAT
chloramphenicol acetyltransferase
- GlnRS
glutaminyl-tRNA synthetase
- TyrRS
tyrosyl-tRNA synthetase
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
See commentary on page 2125.
References
- 1.Noren C J, Anthony-Cahill S J, Griffith M C, Schultz P G. Science. 1989;244:182–188. doi: 10.1126/science.2649980. [DOI] [PubMed] [Google Scholar]
- 2.Bain J D, Glabe C G, Dix T A, Chamberlin A R, Diala E S. J Am Chem Soc. 1989;111:8013–8014. [Google Scholar]
- 3.Baldini G, Martoglio B, Schachenmann A, Zugliani C, Brunner J. Biochemistry. 1988;27:7951–7959. doi: 10.1021/bi00420a054. [DOI] [PubMed] [Google Scholar]
- 4.Ellman J, Mendel D, Anthony-Cahill S, Noren C J, Schultz P G. Methods Enzymol. 1991;202:301–336. doi: 10.1016/0076-6879(91)02017-4. [DOI] [PubMed] [Google Scholar]
- 5.Kim D M, Kigawa T, Choi C Y, Yokoyama S. Eur J Biochem. 1996;239:881–886. doi: 10.1111/j.1432-1033.1996.0881u.x. [DOI] [PubMed] [Google Scholar]
- 6.Steward L E, Collins C S, Gilmore M A, Carlson J E, Ross J B A, Chamberlin A R. J Am Chem Soc. 1997;119:6–11. [Google Scholar]
- 7.Short G F, Golovine S Y, Hecht S M. Biochemistry. 1999;38:8808–8819. doi: 10.1021/bi990281r. [DOI] [PubMed] [Google Scholar]
- 8.Sonar S, Lee C P, Coleman M, Patel N, Liu X, Marti T, Khorana H G, RajBhandary U L, Rothschild K J. Nat Struct Biol. 1994;1:512–517. doi: 10.1038/nsb0894-512. [DOI] [PubMed] [Google Scholar]
- 9.Nowak M W, Kearney P C, Sampson J R, Saks M E, Labarca C G, Silverman S K, Zhong W G, Thorson J, Abelson J N, Davidson N, et al. Science. 1995;268:439–442. doi: 10.1126/science.7716551. [DOI] [PubMed] [Google Scholar]
- 10.Liu D R, Magliery T J, Pasternak M, Schultz P G. Proc Natl Acad Sci USA. 1997;94:10092–10097. doi: 10.1073/pnas.94.19.10092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee C P, RajBhandary U L. Proc Natl Acad Sci USA. 1991;88:11378–11382. doi: 10.1073/pnas.88.24.11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Drabkin H J, Estrella M, RajBhandary U L. Mol Cell Biol. 1998;18:1459–1466. doi: 10.1128/mcb.18.3.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu D R, Schultz P G. Proc Natl Acad Sci USA. 1999;96:4780–4785. doi: 10.1073/pnas.96.9.4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang L, Magliery T J, Liu D R, Schultz P G. J Am Chem Soc. 2000;122:5010–5011. [Google Scholar]
- 15.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning—A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 16.Sherman F, Fink G R, Hicks J B. Methods in Yeast Genetics. Plainview, NY: Cold Spring Harbor Lab. Press; 1986. [Google Scholar]
- 17.Tan S L, Dossett M, Katze M G. BioTechniques. 1998;25:792–796. doi: 10.2144/98255bm07. [DOI] [PubMed] [Google Scholar]
- 18.Edwards H, Schimmel P. Mol Cell Biol. 1990;10:1633–1641. doi: 10.1128/mcb.10.4.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chow C M, RajBhandary U L. J Biol Chem. 1993;268:12855–12863. [PubMed] [Google Scholar]
- 20.Francis M A, RajBhandary U L. Mol Cell Biol. 1990;10:4486–4494. doi: 10.1128/mcb.10.9.4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park H J, RajBhandary U L. Mol Cell Biol. 1998;18:4418–4425. doi: 10.1128/mcb.18.8.4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cadwell R C, Joyce G F. PCR Methods and Applications. Plainview, NY: Cold Spring Harbor Lab. Press; 1992. [Google Scholar]
- 23.RajBhandary U L, Ghosh H P. J Biol Chem. 1969;244:1104–1113. [PubMed] [Google Scholar]
- 24.Mandal N, RajBhandary U L. J Bacteriol. 1992;174:7827–7830. doi: 10.1128/jb.174.23.7827-7830.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 26.Farruggio D, Chaudhuri J, Maitra U, RajBhandary U L. Mol Cell Biol. 1996;16:4248–4256. doi: 10.1128/mcb.16.8.4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Varshney U, Lee C P, RajBhandary U L. J Biol Chem. 1991;266:24712–24718. [PubMed] [Google Scholar]
- 28.Åström S U, von Pawel-Rammingen U, Byström A S. J Mol Biol. 1993;233:43–58. doi: 10.1006/jmbi.1993.1483. [DOI] [PubMed] [Google Scholar]
- 29.Drabkin H J, RajBhandary U L. J Biol Chem. 1985;260:5596–5602. [PubMed] [Google Scholar]
- 30.Seong B L, Lee C P, RajBhandary U L. J Biol Chem. 1989;264:6504–6508. [PubMed] [Google Scholar]
- 31.Fechter P, Rudinger-Thirion J, Theobald-Dietrich A, Giegé R. Biochemistry. 2000;39:1725–1733. doi: 10.1021/bi992276t. [DOI] [PubMed] [Google Scholar]
- 32.Bare L A, Uhlenbeck O C. Biochemistry. 1986;25:5825–5830. doi: 10.1021/bi00367a072. [DOI] [PubMed] [Google Scholar]
- 33.Wakasugi K, Quinn C L, Tao N J, Schimmel P. EMBO J. 1998;17:297–305. doi: 10.1093/emboj/17.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drabkin H J, Park H J, RajBhandary U L. Mol Cell Biol. 1996;16:907–913. doi: 10.1128/mcb.16.3.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singer C E, Smith G R, Cortese R, Ames B N. Nat New Biol. 1972;238:72–74. doi: 10.1038/newbio238072a0. [DOI] [PubMed] [Google Scholar]
- 36.Miller J H, Albertini A M. J Mol Biol. 1983;164:59–71. doi: 10.1016/0022-2836(83)90087-6. [DOI] [PubMed] [Google Scholar]
- 37.Whelihan E F, Schimmel P. EMBO J. 1997;16:2968–2974. doi: 10.1093/emboj/16.10.2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ohno S, Yokogawa T, Fujii I, Asahara H, Inokuchi H, Nishikawa K. J Biochem (Tokyo) 1998;124:1065–1068. doi: 10.1093/oxfordjournals.jbchem.a022221. [DOI] [PubMed] [Google Scholar]
- 39.Furter R. Protein Sci. 1998;7:419–426. doi: 10.1002/pro.5560070223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yarus M. Nat New Biol. 1972;239:106–108. doi: 10.1038/newbio239106a0. [DOI] [PubMed] [Google Scholar]
- 41.Swanson R, Hoben P, Sumner-Smith M, Uemura H, Watson L, Söll D. Science. 1988;242:1548–1551. doi: 10.1126/science.3144042. [DOI] [PubMed] [Google Scholar]
- 42.Hou Y M, Schimmel P. Biochemistry. 1989;28:4942–4947. doi: 10.1021/bi00438a005. [DOI] [PubMed] [Google Scholar]
- 43.Varshney U, RajBhandary U L. J Bacteriol. 1992;174:7819–7826. doi: 10.1128/jb.174.23.7819-7826.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith G P. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 45.Kalo M S. Biochemistry. 1996;35:999–1009. doi: 10.1021/bi951702h. [DOI] [PubMed] [Google Scholar]