

Abstract
Background
Despite incremental improvements in outcomes for patients with acute lymphoblastic leukemia, significant numbers of patients still die from this disease. Mammalian target of rapamycin inhibitors have shown potential in vitro and in vivo as therapeutic agents against a range of tumors including acute lymphoblastic leukemia.
Design and Methods
Flow cytometry was used to evaluate drug-induced cell death in acute lymphoblastic leukemia cell lines and patients’ samples. Human xenografts in immunocompromised mice were used to assess the in vivo effects of selected combinations. Pharmacological inhibitors and lentiviral small interfering ribonucleic acid knock-down of p53 were used to investigate the mechanism of cell killing involved.
Results
Synergistic interactions between RAD001 and cytotoxic agents were demonstrated in vitro and in vivo, with increased caspase-dependent killing. RAD001 suppressed p53 and p21 responses, while suppression of p53 did not prevent killing, indicating p53 independence. RAD001 and cytotoxic agents activated the JUN N-terminal kinase pathway and the combination further increased JUN N-terminal kinase activation. JUN N-terminal kinase inhibition reduced synergistic cell killing by cytotoxic agents and RAD001 in pre-B acute lymphoblastic leukemia cell lines and patients’ samples. Bortezomib and MG132, which activate the JUN N-terminal kinase pathway, also synergized with RAD001 in killing pre-B acute lymphoblastic leukemia cells. Killing was greater when RAD001 was combined with proteasome inhibitors than with cytotoxic drugs.
Conclusions
These observations suggest that combining mammalian target of rapamycin inhibitors with conventional chemotherapy or selected novel agents has the potential to improve clinical responses in patients with pre-B acute lymphoblastic leukemia.
Keywords: acute lymphoblastic leukemia, proteasome inhibitors, RAD001, mTOR inhibitors, JNK
Introduction
Acute lymphoblastic leukemia (ALL) is the most common malignancy in children, and although the majority of patients respond well to treatment, up to 20% of children will relapse and ultimately have a poor prognosis.1 The majority of adult patients relapse following treatment, with the survival rate at 5 years being less than 12%.2 Chemotherapeutic agents remain the foundation of induction, consolidation and salvage therapy for both children and adults with ALL. Increasing the efficacy of current treatments by the addition of new agents is a reasonable strategy for improving patients’ outcomes.
Inhibitors of the mammalian target of rapamycin (mTOR) have recently emerged as potential therapeutic agents for a number of cancers including hematologic malignancies.3–8 mTOR plays a pivotal role in signaling pathways controlling cell proliferation and survival.9 mTOR inhibitors have significant anti-tumor activity as single agents in vitro and in experimental mouse models of ALL.6,10–13 Furthermore, positive interactions have been demonstrated between mTOR inhibitors and standard chemotherapeutics including vincristine, doxorubicin and methotrexate,3,13–15 as well as newer growth factor receptor or tyrosine kinase inhibitors with anti-cancer agents targeting growth factor pathways.16 Indeed we have demonstrated that the survival of mice receiving vincristine and RAD001 is enhanced, while Teachey et al. found similar results with the combination of methotrexate and CCI-779.13,15 There is, however, considerable variation between studies regarding which agents produce synergistic or additive effects when combined with particular mTOR inhibitors, indicating that further clarification of effective combinations is required.
The response to DNA damage and cellular stress resulting from exposure to chemotherapeutic agents is characterized by induction of p53, which is a key regulator of apoptosis, cell cycle arrest and DNA repair.17 Induction of p53 increases the expression of pro-apoptotic proteins, such as Puma18 and Bax,19 and the cell cycle regulator p21.20 The fate of the cell in response to chemotherapy or radiation-induced stress is dependent upon the balance between signals promoting apoptosis and those initiating DNA repair. Alterations in expression of p21 and p53 can change the balance of signal transduction, favoring or inhibiting apoptosis.21,22 The reported inhibition of p21 expression by rapamycin suggests that mTOR inhibitors may disrupt this balance.23
Standard chemotherapeutic agents, as well as a number of newer agents including proteasome inhibitors, also initiate apoptosis through activation of mitogen-activated protein kinase pathways, particularly the JUN N-terminal kinase (JNK) pathway.17,24 The JNK pathway is particularly important for the induction of p53-independent apoptosis by these agents.25 Rapamycin is also known to result in sustained activation of the JNK pathway, suggesting that this pathway may be important for interactions between mTOR inhibitors and chemotherapeutic agents.26 Proteasome inhibitors are a promising new class of agents, currently used to treat a number of hematologic malignancies including myeloma.27 Although no objective responses were seen in children with ALL when bortezomib was used as a single agent,28 it is possible that better responses may occur when this proteasome inhibitor is combined with other agents.
In this study, we examined interactions between the mTOR inhibitor RAD001, standard chemotherapeutic agents, and the proteasome inhibitors bortezomib and MG132 in ALL cells.
Design and Methods
Cells
Human precursor-B ALL cell lines were obtained as follows: NALM6 from the Deutsche Sammlung Von Mikroorganismen und Zellkulturen Gmbh (DSMZ; Braunschweig, Germany), Reh from the American Type Culture Collection (ATCC; Manassas, VA, USA) and LK-63, as a gift, from Professor Andrew Boyd (Queensland Institute of Medical Research, Brisbane, QLD, Australia). Cells were maintained in RPMI medium containing 10% fetal calf serum (complete medium) as previously described.29 Patients’ samples were obtained following informed consent from patients at Westmead Hospital (Sydney, NSW, Australia), with approval from the Sydney West Area Health Service Human Research Ethics Committee. Details of the patients’ samples have been published previously and are provided in Online Supplementary Table S1.13,30,31 Cells from all samples except 1901 were expanded on stroma to provide sufficient material for the study.
Antibodies and reagents
RAD001 (everolimus) was kindly provided by Novartis (Basel, Switzerland). The pan-caspase inhibitor ZVAD-FMK was purchased from Becton Dickinson (North Ryde, NSW, Australia). The JNK inhibitor SP600125, and the proteasome inhibitor MG132 were purchased from Merck (Melbourne, Vic, Australia). The proteasome inhibitor bortezomib (Velcade™) was purchased from Millennium Pharmaceuticals (Cambridge, MA, USA), etoposide phosphate from Bristol-Myers-Squibb (Rydalmere, NSW, Australia) and vincristine sulphate and doxorubicin from Pfizer (Melrose Park, NSW, Australia). Ionizing radiation was delivered using an X-ray irradiator (XRAD320, Precision X-Ray, Inc. East Haven, CT, USA) at a dose rate of 0.91 Gy/min. 7-amino-actinomycin D (7-AAD), 10x annexin V binding buffer, 10x perm/wash buffer, annexin V-fluorescein isothiocyanate (FITC) and streptavidin-allophycocyanin were obtained from Becton Dickinson and annexin V alexa fluor 647 from Invitrogen (Grand Island, NY, USA). The following antibodies to human antigens were purchased: mouse anti-Bcl2-FITC and active caspase-3 FITC or biotin (Becton Dickinson); mouse anti-p53-FITC, p21-FITC, p53, p21 (Calbiochem, Boronia, VIC, Australia); rabbit anti-c-Jun (Ser73), Bax, Puma and XIAP (Cell Signaling Technologies Boston, MA, USA).
Flow cytometry
Viability was assessed using annexin V and 7-AAD staining as previously described, except that the propidium iodide was replaced by 0.25 μg/mL 7-AAD.32 Intracellular staining was performed on cells fixed in ice cold 70% ethanol and blocked in perm/wash (BD) buffer containing 10% human AB serum for 1 h. Cells were labeled for 1 h with appropriate primary antibodies, or isotype control antibody, at room temperature in the dark. Cells were subsequently washed and resuspended in 500 μL phosphate-buffered saline for analysis by flow cytometry. If required cells were incubated with species-specific, fluorophore-conjugated secondary antibodies for 45 min in the dark at room temperature. Cells were analyzed by flow cytometry using a FACSCalibur flow cytometer.
Lentiviral transduction
Lentiviral transduction was performed with Institutional Biosafety Committee approval. Lentiviral particles containing p53, or luciferase, short hairpin (sh) RNA expression vectors (pSIH-HI-coGFP), were generously provided by Dr Helen Rizos (Westmead Millennium Institute, Sydney, Australia).33 Cells (1×106/mL) were transduced in RPMI containing 10% fetal calf serum, 8 μg/mL polybrene and either luciferase (2.5×106 viral particles/mL) or p53 (0.8×106 viral particles/mL) shRNA-expressing viral particles for 48 h. Viral particles were removed and the cells cultured in complete medium in triplicate for a further 24 h in the absence or presence of vincristine. Successfully transduced cells were identified by expression of green fluorescent protein (GFP).
In vivo studies
In vivo studies were performed as previously described.13 Briefly sub-lethally irradiated NOD/SCID mice were engrafted with ALL xenograft 1345 and treatment commenced when 1% ALL cells were detected in the blood and continued for 4 weeks. RAD001 was administered thrice weekly by gavage at a dose of 5 mg/kg, dexamethasone from Monday-Friday by intraperitoneal injection at a dose of 15 mg/kg and etoposide at a dose of 8 mg/kg by weekly intraperitoneal injections. ALL was monitored by weekly tail vein bleeds and animals sacrificed according to humane measures of declining health in line with animal ethics committee approval of the study. All animals had extensive ALL at the time of sacrifice.
Statistical analysis
Normalization, averages and standard deviations were determined using Microsoft Excel. Fold change analysis of mean fluorescence intensity was employed to determine the significance of changes in target antigen expression. Graphical representation of the square root of the mean was used to determine whether relationships between agents tested were antagonistic, synergistic or additive. The data were also compared with estimated marginal means generated by syntax-based univariate analysis of variance, using SPSS graduate pack statistics. Syntax-based univariate analysis of variance was also used to determine the statistical significance (P<0.05) of variance observed with single agent and combination treatments. In vivo studies were analyzed using a Mantel-Cox log-rank test.
Results
RAD001 causes dose-dependent synergistic killing of pre-B acute lymphoblastic leukemia cells when combined with ionizing radiation or chemotherapy agents
mTOR inhibitors have been previously reported to synergize with several chemotherapeutic agents.12,14 We examined the ability of RAD001 to enhance the cytotoxic effects of ionizing radiation, by flow cytometry using annexin V and 7-AAD staining (Figure 1A). Radiation and cytotoxic agents were titrated and suboptimal concentrations selected to facilitate the demonstration of interactive effects. While 2 μM RAD001 had no effect, higher doses significantly increased the cytotoxicity of ionizing radiation on pre-B ALL cell lines (Figure 1B and D). In contrast, we were unable to detect synergy between dexamethasone and RAD001 (Figure 1D). Plots of the square root transformation of the mean viability and univariate analysis of variance statistics demonstrated a clear synergistic interaction between 16 μM, but not 2 μM, RAD001 and irradiation (Figure 1C). Similarly, 16 μM RAD001 enhanced the efficacy of the chemotherapeutic agent vincristine. This was demonstrated by a greater than 1 log reduction in the IC50 of this drug in NALM6 and LK63 cells and a greater than 2.7-fold reduction in REH cells when the effect of 16 μM RAD001 alone had been corrected for (Figure 2A). Synergy was also observed with several chemotherapeutic agents including etoposide and doxorubicin (Figure 2B left panels and Online Supplementary Figure S1). This synergistic effect was also observed when using samples from patients with ALL. Despite the heterogeneity in cytogenetics and sensitivity to individual agents, statistical significance was reached with at least two agents in all samples tested (Figure 2B, right panels and Online Supplementary Figure S1). As we had previously demonstrated for vincristine,13 the combination of the chemotherapeutic agent etoposide and RAD001 further increased the survival of mice with ALL (Figure 2C), while the combination of dexamethasone and RAD001 was no more effective than RAD001 alone.
Figure 1.

RAD001 synergizes with radiation in a dose-dependent manner to increase ALL cell death. (A) Representative flow cytometry dot plots showing the analysis used to determine cell viability. The viability of the LK63 cells in the examples provided is shown in the lower right corner of each dot plot. ALL cells negative for annexin V and 7-AAD were considered viable. (B) LK63 cells were treated with the indicated concentrations of RAD001 for 24 h with or without exposure to 5 Gy of ionizing radiation and analyzed as in (A). Between two and six replicates were examined at each point and the mean and standard deviation are shown. (C) Data obtained in (B) were square root transformed. Divergent lines represent agonistic, parallel lines additive, and convergent lines antagonistic relationships. (D) NALM6, REH and LK63 cells were treated with 16 μM RAD001 and/or the indicated dose of ionizing radiation or 40 ng/mL of dexamethasone. The mean and standard deviation of triplicate determinations are shown. *P<0.05 compared to 16 μM RAD001 or ionizing radiation indicating an agonistic interaction.
Figure 2.

Synergistic killing of ALL cells by 16 μM RAD001 and chemotherapeutic agents. (A) NALM6, LK63 and REH cells were treated with the indicated concentrations of vincristine in the presence (RAD001) or absence (Control) of 16 μM RAD001 and assessed for viability after 24 h. The data are corrected for the viability in the absence of vincristine for each series (i.e. no drug for the Control series and 16 μM RAD001 alone for the RAD001 series). The IC50 value for vincristine for each series is indicated on the graphs. (B) NALM6, REH and LK63 cells (left panels) or indicated patients’ samples (right panels) were either untreated or exposed to RAD001, indicated chemotherapeutic agents, or the combination of RAD001 and indicated chemotherapeutic agents for 24 h. Drug concentrations were as follows: vincristine (0.6 nM), etoposide (100 mM), doxorubicin (10 mM) and RAD001 (16 μM), except for patients’ samples 2032, 1345 and 2070 where 14 μM RAD001 was used. Cell viability was assessed by flow cytometry, and cells negative for annexin V and 7-AAD staining were considered viable. The mean±SD of triplicate determinations is shown. Error bars denote the standard deviation of the mean. *Indicates a significant synergistic interaction between RAD001 and the indicated therapeutic agent (P<0.05). (C) NOD/SCID mice engrafted with a xenograft from patient 1345 were treated with placebo only (P/P), placebo and RAD001 (P/R), placebo and dexamethasone (P/D), placebo and etoposide (P/E), RAD001 and dexamethasone (R/D) or RAD001 and etoposide (R/E). Kaplan-Meier plots of the survival of the mice are shown. The time indicated is from the commencement of treatment and the arrow indicates the completion of treatment. The P value indicates a significant increase in survival of mice receiving RAD001 and etoposide compared to those receiving RAD001 alone.
Combination therapy results in caspase-dependent cell death
Radiation or 16 μM RAD001 alone induced little activation of caspase-3, but when combined, caspase-3 activation was marked (Figure 3A). The pan-caspase inhibitor Z-VAD-FMK (20 μM) completely prevented the activation of caspase-3 in response to the combination of agents (Figure 3B). This dose of Z-VAD-FMK completely prevented the increased killing observed when 16 μM RAD001 was combined with radiation or chemotherapeutic agents (Figure 3C and Online Supplementary Figure S2). This indicates that the synergistic killing by 16 μM RAD001 and radiation or chemotherapy occurs through caspase-dependent apoptosis.
Figure 3.

Synergistic cell death was caspase-dependent and p53-independent. (A) NALM6 and REH cells were either untreated, treated with 16 μM RAD001, exposed to indicated doses of ionizing radiation or both RAD001 and ionizing radiation for 24 h. Cells were labeled for cleaved caspase-3 and analyzed by flow cytometry. Representative graphs from duplicate determinations are shown. (B) REH cells were treated with 16 μM RAD001 and 2.5 Gy of ionizing radiation in the presence or absence of the pan-caspase inhibitor ZVAD-FMK (20 μM). Cells were labeled and data shown as for (A). (C) NALM6 cells were either untreated or exposed to 1 Gy of ionizing radiation (IR) or the indicated chemotherapeutic agents, 16 μM RAD001 or the combination of RAD001 and radiation or chemotherapeutic agents in the absence or presence of 20 μM ZVAD-FMK for 24 h. Chemotherapeutic agents were used at the following concentrations: 10 μM doxorubicin (Dox) or 100 nM etoposide (Etop). Cells negative for annexin V and 7-AAD staining by flow cytometry were considered viable. The mean±SD of triplicate determinations are shown. † Indicates a significant synergistic interaction between RAD001 and the cytotoxic agent (P<0.05). *Indicates a significant reversal of the synergistic killing (P<0.05). (D) NALM6, REH and LK63 cells were untreated or exposed to 10 nM vincristine, in the absence or presence of the p53 inhibitor pifithrin (25 μM) for 12 h. Cells were analyzed by flow cytometry, and those negative for annexin V and 7-AAD staining were considered viable. The mean±SD of triplicate determinations is shown. (E) NALM6 cells were transduced with lentiviral constructs containing either luciferase shRNA-GFP (Control) or p53 shRNA-GFP and treated with vehicle or 5 nM vincristine. Cells were labeled for p53 and analyzed by flow cytometry using GFP to identify successfully transduced cells. The mean±SD of duplicate determinations is shown. *Indicates a significant suppression of p53 induction in response to vincristine. (F) NALM6 cells were transduced with p53 shRNA-GFP or luciferase shRNA-GFP and treated with 5 nM vincristine or vehicle for 12 h. Cells were labeled for cleaved caspase-3 and analyzed by flow cytometry, the percentage of cells lacking cleaved caspase-3 is shown. §P<0.05 compared to the equivalently treated luciferase shRNA-GFP cells.
Synergistic cell death is independent of p53 responses
p53 plays a fundamental role in the response of cells to stress, including apoptosis and cell cycle arrest following exposure to chemotherapeutic agents.17 The pre-B ALL cell lines NALM6, REH and LK63 showed up-regulation of both p53 and p21 expression after exposure to various cytotoxic agents including vincristine, etoposide and doxorubicin, or to ionizing radiation (Table 1). Rapamycin and its derivatives are reported to reduce p21 expression.23,24 Despite having little effect on p53 or p21 expression when used as a single agent, 16 μM RAD001 completely suppressed the up-regulation of p53, and reduced that of p21, after exposure to chemotherapeutic agents (Table 1). This, and the absence of increased p53 expression in pre-B ALL cells exposed to 16 μM RAD001, strongly suggests that the synergistic effects of RAD001 on chemotherapy-induced cell death are independent of p53 responses. The p53 inhibitor, pifithrin-α,35 did not affect the viability of pre-B ALL cells exposed to vincristine, also suggesting that cell death is independent of p53 function (Figure 3D). To further support this point TP53 gene silencing experiments were undertaken using a lentivirus to introduce p53 shRNA-GFP and luciferase shRNA-GFP into NALM6 cells. The induction of p53 in response to vincristine was completely prevented by the introduction of p53 shRNA (Figure 3E). To determine whether p53 was required for vincristine-induced cell death, the induction of apoptosis was assessed in p53 shRNA-transduced cells using caspase-3 activation. Apoptosis was minimal in cells transduced with luciferase shRNA-GFP or p53 shRNA-GFP. However vincristine-induced activation of caspase-3 was not affected by the presence of p53 shRNA (Figure 3F). This suggests that apoptosis in this model is independent of p53.
Table 1.
RAD001 suppresses expression of p53 and p21 induced by cytotoxic agents.

Activation of JNK plays a significant role in the synergistic effects of RAD001 on chemotherapy and radiation-induced cytotoxicity
The prolonged activation of mitogen-activated protein kinases, particularly JNK, is known to be involved in cell death responses to a range of stimuli including cytotoxic agents.17,24 Both 16 μM RAD001 and vincristine significantly activated JNK signaling as demonstrated by elevated expression and phosphorylation of c-Jun. The combination of these agents resulted in the greatest JNK activity (Figure 4A). To determine the role of JNK activation in the synergistic effects of RAD001, pre-B ALL cell lines were either untreated or exposed to radiation or chemotherapy, 16 μM RAD001 or combination therapy in the presence or absence of the JNK inhibitor SP600125. Although JNK inhibition did not influence cell death resulting from treatment with the single agents, the synergistic killing observed with combination therapy was significantly reduced by JNK inhibition in all cell lines tested (Figure 4 and Online Supplementary Figure S3). This suggests that a threshold level of JNK activation may be required before it significantly contributes to cell death. While the inhibition of synergistic killing was statistically significant, it was not complete. This indicates that, while JNK activation is a significant contributor to the mechanism that underpins synergistic killing, additional factors are involved.
Figure 4.
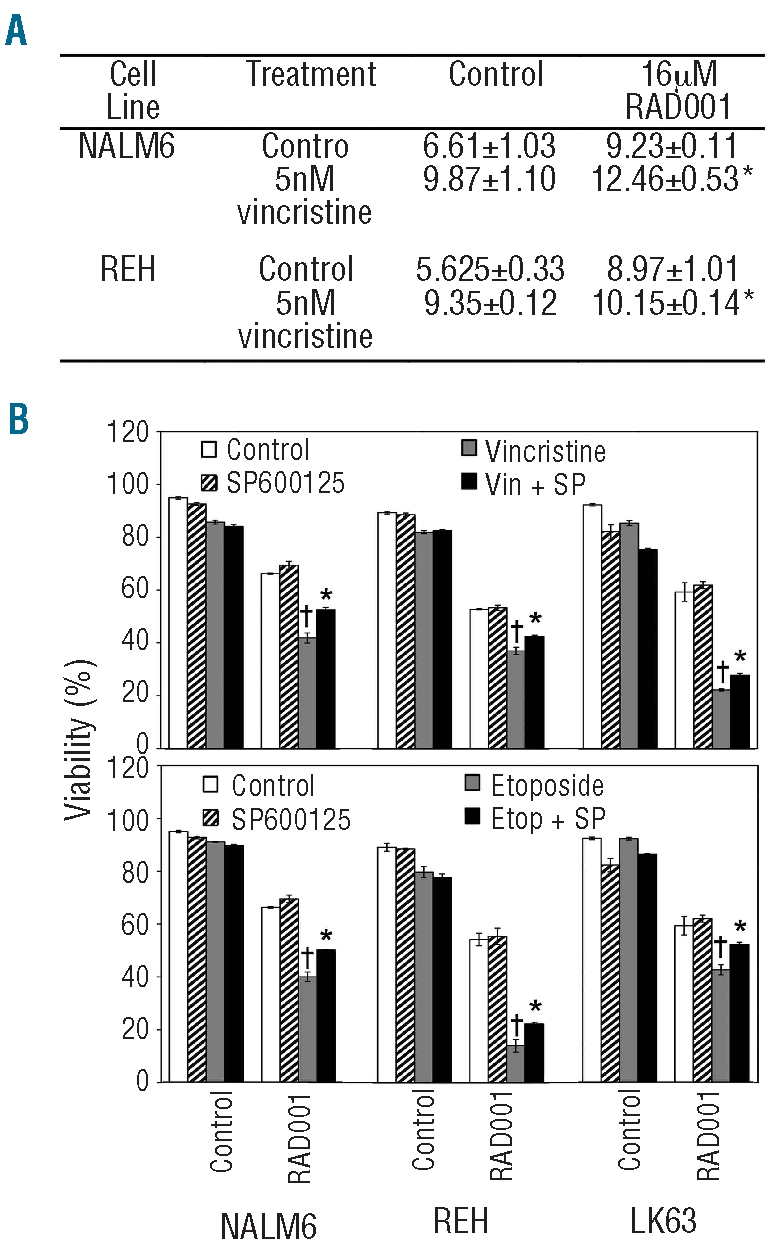
Synergistic cell death was partially mediated by activation of the JNK pathway. (A) Mean fluorescence intensity of flow cytometric analysis of c-Jun phosphorylated Ser73 in ALL cell lines treated with cytotoxic agents with or without the addition of 16 μM RAD001. *P<0.05 compared to cells treated with RAD001 or vincristine as single agents. (B) NALM6, REH and LK63 cells were untreated or exposed to 0.6 nM vincristine or 100 nM etoposide with or without the addition of 16 μM RAD001 in the absence or presence of 5 μM SP600125 for 24 h. Cells were analyzed by flow cytometry and cells lacking annexin V and 7-AAD staining were considered viable. The mean±SD of triplicate determinations is shown. †Indicates a significant synergistic interaction between the chemotherapeutic agent and RAD001 (P<0.05) *Indicates a significant reversal of the synergistic interaction by SP600125 (P<0.05).
Proteasome inhibitors bortezomib and MG132 also synergize with RAD001 to kill pre-B acute lymphoblastic leukemia cells
To determine whether activation of JNK signaling provides a general mechanism to enhance the effects of RAD001 in pre-B ALL cells, the effect of proteasome inhibitors was examined. Proteasome inhibitors are potent activators of the JNK pathway, mediating JNK-dependent apoptosis.24,36 Bortezomib induced JNK activation in ALL cells as expected (data not shown) and this activation was required for optimal killing by this agent (Online Supplementary Figure S4A). RAD001 synergized with bortezomib (Figure 5A) and MG132 (Figure 5B), not only at the 16 μM dose required for interaction with cytotoxic agents, but also at significantly lower doses (Online Supplementary Figure S4B). Similar results were obtained using lower doses of RAD001 and bortezomib in patients’ samples (Figure 5B).
Figure 5.
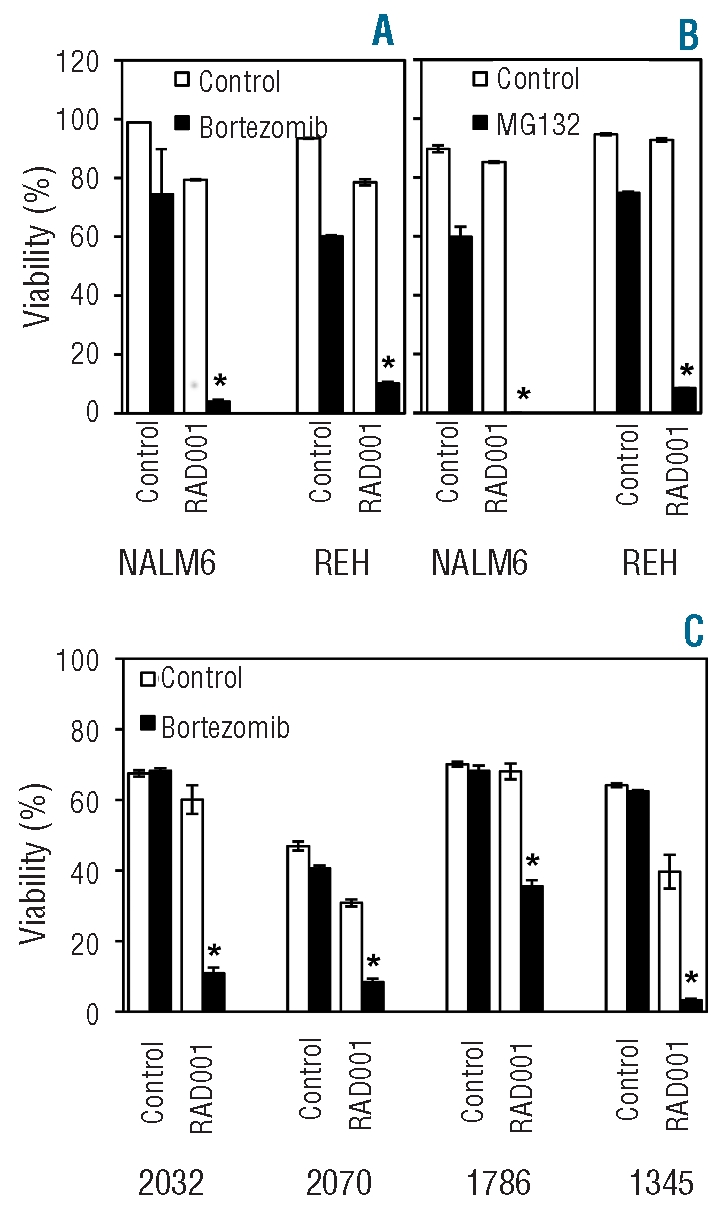
Proteasome inhibitors synergize with RAD001. Indicated cell lines (A and B) were untreated or exposed to 20 nM bortezomib (A) or 500 nM MG132 (B) in the presence or absence of 16 μM RAD001 for 24 h. Cells were analyzed by flow cytometry, and those negative for annexin V and 7-AAD staining were considered viable. The mean±SD of triplicate determinations is shown. *Indicates a synergistic interaction between the two agents being tested (P<0.05). (C) Indicated patients’ samples were treated as for (A) except that RAD001 was used at 8 μM and bortezomib at 10 nM. The mean±SD of triplicate determinations normalized to control is shown. *Indicates a synergistic interaction between the two agents being tested (P<0.05).
Discussion
The poor outcomes of patients with relapsed pre-B ALL37 make novel approaches to therapy necessary. Recent studies indicate that synergy between mTOR inhibitors and chemotherapeutic agents can be achieved in ALL.3,6,13,15 Here, we have confirmed that RAD001 can act synergistically with a broad range of chemotherapeutic agents and ionizing radiation. Unlike in previous studies, we did not observe a synergistic interaction with dexamethasone.38–40 The most likely explanation for this difference is that TALL and Burkitt’s lymphoma cell lines were used as opposed to the B-cell progenitor lines in this study. Indeed REH cells, which are dexamethasone-resistant, were not sensitized by rapamycin in the study by Wei et al., consistent with our findings.
The low micromolar concentrations of RAD0001 required in this study to enhance cell death with conventional chemotherapeutic drugs and irradiation are about 10-fold higher than the concentration of RAD001 required to inhibit the proliferation of pre-B ALL cell lines such as a NALM6 in our laboratory (unpublished data), or the high nanomolar concentrations of rapamycin shown to affect pre-B ALL.6 However, we have demonstrated, using an immunodeficient mouse model and a thrice weekly oral gavage regimen, that low micromolar plasma concentrations of RAD001 are achievable in these animals, and are capable of inhibiting molecular targets of mTOR in xenografted human ALL cells, to produce significant antileukemic effects.13 This is the same scheduling as used in this study. Although trough plasma levels of RAD0001 in humans given doses of 10 mg have been reported to be in the nanomolar range,41 peak levels may be more important for antileukemic activity, and have not been clearly defined in human pharmacokinetic studies. Dosing regimens of up to 70 mg of RAD001 per week, or 10 mg/kg daily, have been used in cancer clinical trials, comparable with doses administered to mice, and are tolerated, thus enabling exploration of RAD001 in clinical trials with selected chemotherapy drugs.
Identifying the mechanism underpinning synergistic killing will facilitate the logical selection of agents to combine with RAD001 to increase tumor cell death in response to chemotherapy. We have previously demonstrated that RAD001 alone does not induce apoptosis in vivo, despite prolonging survival.13 However, cell cycle arrest was observed, as was the induction of autophagy. Autophagy has the potential to reduce cell death by removing damaged organelles and providing essential nutrients, but it has also been suggested that excessive autophagy can result in cell death.42 Since RAD001 treatment resulted in the long-term survival of some mice with ALL, cell death must be occurring in vivo. Whether autophagy contributes to RAD001-induced cell death of ALL cells remains to be determined. We found that synergistic killing by 16 μM RAD001 and chemotherapy was via caspase-dependent apoptosis. Key regulators of DNA damage and stress responses include p53 and p21, the latter inducing G1 cell cycle arrest and DNA repair.43 RAD001 potently inhibited up-regulation of p53 expression, and reduced p21, in response to chemotherapy and ionizing radiation. However, using pharmacological and genetic inhibition of p53 induction, we demonstrated that p53 does not play a major role in chemotherapy-induced death in ALL cells. Unlike in many other malignancies, p53 mutations are rare in newly diagnosed ALL.44 In the context of patients with relapsed disease, in whom complex cytogenetic abnormalities include loss of p53,45,46 the ability to kill pre-B ALL cells independently of p53 activation may prove beneficial. While many cytotoxic agents, including vincristine, are known to induce p53 expression in many cell types,47 the importance of p53 induction in subsequent apoptosis is less clear, with p53-dependent and-independent mechanisms having been reported, including in ALL cells.48–51
mTOR signaling and JNK pathways form an important link in the signal transduction that leads to apoptosis, with suppression of PI-3K signaling being required for JNK-mediated apoptosis.52 In addition, mTOR inhibition is associated with increased JNK pathway activation.26 JNK can enhance apoptotic responses to genotoxic stress,53 with inhibition of JNK being protective.54 In ALL cells, synergistic killing by the Bcl-2 inhibitor ABT-737 and the cytotoxic retinoid N-(4-hydroxyphenyl)retinamide involved JNK signaling.55 Furthermore, suppression of AKT resulted in caspase-dependent apoptosis that was partially mediated by signaling through the JNK pathway in multi-drug resistant T ALL cells.56
The observation that inhibition of JNK led to significant protection from chemotherapy-induced death in pre-B ALL cells provides strong evidence of an inverse relationship between the activation of JNK and chemosensitivity, and indirectly supports JNK activation as a strategy to enhance death by chemotherapy. To determine whether JNK activation could have general applicability for enhancing the efficacy of RAD001 we examined the impact of proteasome inhibitors, which mediate cell death through the JNK pathway.24,36 Bortezomib potently induced JNK activation (data not shown) with a significant proportion of subsequent cell death being JNK-dependent. This observation is consistent with those of studies in other malignancies,57,58 and confirms that JNK activation can contribute significantly to cell death in pre-B ALL cells under certain conditions. The findings support JNK activation as a strategy to enhance cell death by chemotherapeutic agents in pre-B ALL. RAD001 synergized with bortezomib or MG132 to significantly enhance pre-B ALL cell death. The synergy between RAD001 and bortezomib indicates a potentially novel strategy for patients with chemoresistant pre-B ALL. Further evaluation of this strategy, including in an in vivo model, is warranted.
In conclusion, we demonstrated that, at sufficiently high doses, RAD001 synergized with a range of chemotherapeutic agents and ionizing radiation to kill pre-B ALL cells. Synergistic killing was mediated by caspase-dependent apoptosis and was partially dependent on JNK signaling. These finding are important for the further development of mTOR inhibitors as therapeutic agents for the treatment of ALL and their optimal incorporation into clinical use. The identification of synergistic killing by combining RAD001 with a broad range of chemotherapeutic agents has important implications for the design of novel protocols for the treatment of ALL. RAD001 may be an important agent for patients in whom current treatments fail and has the potential to enhance outcomes for patients receiving first-line induction and consolidation therapy in pre-B ALL. Finally the common factor in the synergy between RAD001 and cytotoxic agents, and proteasome inhibitors is activation of JNK signaling. This suggests that developing agents that activate JNK signaling may be a useful therapeutic strategy. Clinical trials of RAD001 in ALL patients with relapsed disease will be required to further evaluate the potential of this strategy.
Acknowledgments
we would like to thank Dr Karen Byth for assistance with the statistical analysis of the data.
Footnotes
Funding: this work was supported by a University of Sydney Postgraduate Award, NHMRC project grant No.352326 and the Leukemia and Lymphoma Society Translational Program Grant No. 6105-08. RAD001 (everolimus) was kindly provided by Novartis (Basel, Switzerland).
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures
The information provided by the authors about contributions from persons listed as authors and in acknowledgments is available with the full text of this paper at www.haematologica.org.
Financial and other disclosures provided by the authors using the ICMJE (www.icmje.org) Uniform Format for Disclosure of Competing Interests are also available at www.haematologica.org.
References
- 1.Nguyen K, Devidas M, Cheng S, La M, Raetz E, Carroll W, et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: a Children’s Oncology Group study. Leukemia. 2008;22(12):2142–50. doi: 10.1038/leu.2008.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tavernier E, Boiron J, Huguet F, Bradstock K, Vey N, Kovacsovics T, et al. Outcome of treatment after first relapse in adults with acute lymphoblastic leukemia initially treated by the LALA-94 trial. Leukemia. 2007;21(9):1907–14. doi: 10.1038/sj.leu.2404824. [DOI] [PubMed] [Google Scholar]
- 3.Avellino R, Romano S, Parasole R, Bisogni R, Lamberti A, Poggi V, et al. Rapamycin stimulates apoptosis of childhood acute lymphoblastic leukemia cells. Blood. 2005;106(4):1400–6. doi: 10.1182/blood-2005-03-0929. [DOI] [PubMed] [Google Scholar]
- 4.Mabuchi S, Altomare D, Cheung M, Zhang L, Poulikakos P, Hensley H, et al. RAD001 inhibits human ovarian cancer cell proliferation, enhances cisplatin-induced apoptosis, and prolongs survival in an ovarian cancer model. Clin Cancer Res. 2007;13(14):4261–70. doi: 10.1158/1078-0432.CCR-06-2770. [DOI] [PubMed] [Google Scholar]
- 5.Peponi E, Drakos E, Reyes G, Leventaki V, Rassidakis G, Medeiros L. Activation of mammalian target of rapamycin signaling promotes cell cycle progression and protects cells from apoptosis in mantle cell lymphoma. Am J Pathol. 2006;169(6):2171–80. doi: 10.2353/ajpath.2006.051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teachey D, Obzut D, Cooperman J, Fang J, Carroll M, Choi J, et al. The mTOR inhibitor CCI-779 induces apoptosis and inhibits growth in preclinical models of primary adult human ALL. Blood. 2006;107 (3):1149–55. doi: 10.1182/blood-2005-05-1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yee K, Zeng Z, Konopleva M, Verstovsek S, Ravandi F, Ferrajoli A, et al. Phase I/II study of the mammalian target of rapamycin inhibitor everolimus (RAD001) in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res. 2006;12 (17):5165–73. doi: 10.1158/1078-0432.CCR-06-0764. [DOI] [PubMed] [Google Scholar]
- 8.Zeng Z, Sarbassov dos D, Samudio I, Yee K, Munsell M, Jackson C, et al. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood. 2007;109(8):3509–12. doi: 10.1182/blood-2006-06-030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dann S, Thomas G. The amino acid sensitive TOR pathway from yeast to mammals. FEBS Lett. 2006;580(12):2821–9. doi: 10.1016/j.febslet.2006.04.068. [DOI] [PubMed] [Google Scholar]
- 10.Yu K, Toral-Barza L, Discafani C, Zhang W, Skotnicki J, Frost P, et al. mTOR, a novel target in breast cancer: the effect of CCI-779, an mTOR inhibitor, in preclinical models of breast cancer. Endocr Relat Cancer. 2001;8(3):249–58. doi: 10.1677/erc.0.0080249. [DOI] [PubMed] [Google Scholar]
- 11.Khariwala S, Kjaergaard J, Lorenz R, Van Lente F, Shu S, Strome M. Everolimus (RAD) inhibits in vivo growth of murine squamous cell carcinoma (SCC VII) Laryngoscope. 2006;116(5):814–20. doi: 10.1097/01.mlg.0000210544.64659.35. [DOI] [PubMed] [Google Scholar]
- 12.Ito D, Fujimoto K, Mori T, Kami K, Koizumi M, Toyoda E, et al. In vivo antitumor effect of the mTOR inhibitor CCI-779 and gemcitabine in xenograft models of human pancreatic cancer. Int J Cancer. 2006;118(9):2337–43. doi: 10.1002/ijc.21532. [DOI] [PubMed] [Google Scholar]
- 13.Crazzolara R, Cisterne A, Thien M, Hewson J, Baraz R, Bradstock KF, et al. Potentiating effects of RAD001 (everolimus) on vincristine therapy in child-hood acute lymphoblastic leukemia. Blood. 2009;113(14):3297–306. doi: 10.1182/blood-2008-02-137752. [DOI] [PubMed] [Google Scholar]
- 14.Grünwald V, DeGraffenried L, Russel D, Friedrichs W, Ray R, Hidalgo M. Inhibitors of mTOR reverse doxorubicin resistance conferred by PTEN status in prostate cancer cells. Cancer Res. 2002;62(21):6141–5. [PubMed] [Google Scholar]
- 15.Teachey D, Sheen C, Hall J, Ryan T, Brown V, Fish J, et al. mTOR inhibitors are synergistic with methotrexate: an effective combination to treat acute lymphoblastic leukemia. Blood. 2008;112(5):2020–3. doi: 10.1182/blood-2008-02-137141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goudar R, Shi Q, Hjelmeland M, Keir S, McLendon R, Wikstrand C, et al. Combination therapy of inhibitors of epidermal growth factor receptor/vascular endothelial growth factor receptor 2 (AEE788) and the mammalian target of rapamycin (RAD001) offers improved glioblastoma tumor growth inhibition. Mol Cancer Ther. 2005;4(1):101–12. [PubMed] [Google Scholar]
- 17.Roos W, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12(9):440–50. doi: 10.1016/j.molmed.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 18.Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7(3):673–82. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- 19.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80(2):293–9. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 20.Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55 (22):5187–90. [PubMed] [Google Scholar]
- 21.Forster K, Obermeier A, Mitina O, Simon N, Warmuth M, Krause G, et al. Role of p21(WAF1/CIP1) as an attenuator of both proliferative and drug-induced apoptotic signals in BCR-ABL-transformed hematopoietic cells. Ann Hematol. 2008;87(3):183–93. doi: 10.1007/s00277-007-0400-9. [DOI] [PubMed] [Google Scholar]
- 22.Fisher DE. Turning p53 on or off: either way may treat cancer. Drug Resist Updat. 2000;3(2):77–9. doi: 10.1054/drup.2000.0128. [DOI] [PubMed] [Google Scholar]
- 23.Beuvink I, Boulay A, Fumagalli S, Zilbermann F, Ruetz S, O’Reilly T, et al. The mTOR inhibitor RAD001 sensitizes tumor cells to DNA-damaged induced apoptosis through inhibition of p21 translation. Cell. 2005;120(6):747–59. doi: 10.1016/j.cell.2004.12.040. [DOI] [PubMed] [Google Scholar]
- 24.Hideshima T, Mitsiades C, Akiyama M, Hayashi T, Chauhan D, Richardson P, et al. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood. 2003;101(4):1530–4. doi: 10.1182/blood-2002-08-2543. [DOI] [PubMed] [Google Scholar]
- 25.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103(2):239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 26.Huang S, Shu L, Dilling M, Easton J, Harwood F, Ichijo H, et al. Sustained activation of the JNK cascade and rapamycin-induced apoptosis are suppressed by p53/p21(Cip1) Mol Cell. 2003;11(6):1491–501. doi: 10.1016/s1097-2765(03)00180-1. [DOI] [PubMed] [Google Scholar]
- 27.Cortes J, Thomas D, Koller C, Giles F, Estey E, Faderl S, et al. Phase I study of bortezomib in refractory or relapsed acute leukemias. Clin Cancer Res. 2004;10(10):3371–6. doi: 10.1158/1078-0432.CCR-03-0508. [DOI] [PubMed] [Google Scholar]
- 28.Horton TM, Pati D, Plon SE, Thompson PA, Bomgaars LR, Adamson PC, et al. A phase 1 study of the proteasome inhibitor bortezomib in pediatric patients with refractory leukemia: a Children’s Oncology Group study. Clin Cancer Res. 2007;13(5):1516–22. doi: 10.1158/1078-0432.CCR-06-2173. [DOI] [PubMed] [Google Scholar]
- 29.Juarez JG, Thien M, Dela Pena A, Baraz R, Bradstock KF, Bendall LJ. CXCR4 mediates the homing of B cell progenitor acute lymphoblastic leukaemia cells to the bone marrow via activation of p38MAPK. Br J Haematol. 2009;145(4):491–9. doi: 10.1111/j.1365-2141.2009.07648.x. [DOI] [PubMed] [Google Scholar]
- 30.Juarez J, Baraz R, Gaundar S, Bradstock K, Bendall L. Interaction of interleukin-7 and interleukin-3 with the CXCL12-induced proliferation of B-cell progenitor acute lymphoblastic leukemia. Haematologica. 2007;92(4):450–9. doi: 10.3324/haematol.10621. [DOI] [PubMed] [Google Scholar]
- 31.Gaundar SS, Bradstock KF, Bendall LJ. p38MAPK inhibitors attenuate cytokine production by bone marrow stromal cells and reduce stroma-mediated proliferation of acute lymphoblastic leukemia cells. Cell Cycle. 2009;8(18):2975–83. [PubMed] [Google Scholar]
- 32.Juarez J, Bradstock K, Gottlieb D, Bendall L. Effects of inhibitors of the chemokine receptor CXCR4 on acute lymphoblastic leukemia cells in vitro. Leukemia. 2003;17 (7):1294–300. doi: 10.1038/sj.leu.2402998. [DOI] [PubMed] [Google Scholar]
- 33.Rizos H, Scurr L, Irvine M, Alling N, Kefford R. p14ARF regulates E2F-1 ubiquitination and degradation via a p53-dependent mechanism. Cell Cycle. 2007;6:1741–7. doi: 10.4161/cc.6.14.4428. [DOI] [PubMed] [Google Scholar]
- 34.Yazbeck V, Buglio D, Georgakis G, Li Y, Iwado E, Romaguera J, et al. Temsirolimus downregulates p21 without altering cyclin D1 expression and induces autophagy and synergizes with vorinostat in mantle cell lymphoma. Exp Hematol. 2008;36(4):443–50. doi: 10.1016/j.exphem.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 35.Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science. 1999;285(5434):1733–7. doi: 10.1126/science.285.5434.1733. [DOI] [PubMed] [Google Scholar]
- 36.Dai Y, Rahmani M, Grant S. Proteasome inhibitors potentiate leukemic cell apoptosis induced by the cyclin-dependent kinase inhibitor flavopiridol through a SAPK/JNK-and NF-kappaB-dependent process. Oncogene. 2003;22(46):7108–22. doi: 10.1038/sj.onc.1206863. [DOI] [PubMed] [Google Scholar]
- 37.Fielding A, Richards S, Chopra R, Lazarus H, Litzow M, Buck G, et al. Outcome of 609 adults after relapse of acute lymphoblastic leukaemia (ALL); an MRC UKALL12/ECOG 2993 study. Blood. 2007;109(3):944–50. doi: 10.1182/blood-2006-05-018192. [DOI] [PubMed] [Google Scholar]
- 38.Bonapace L, Bornhauser BC, Schmitz M, Cario G, Ziegler U, Niggli FK, et al. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J Clin Invest. 2010;120(4):1310–23. doi: 10.1172/JCI39987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beesley AH, Firth MJ, Ford J, Weller RE, Freitas JR, Perera KU, et al. Glucocorticoid resistance in T-lineage acute lymphoblastic leukaemia is associated with a proliferative metabolism. Br J Cancer. 2009;100(12):1926–36. doi: 10.1038/sj.bjc.6605072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei G, Twomey D, Lamb J, Schlis K, Agarwal J, Stam R, et al. Gene expression-based chemical genomics identifies rapamycin as a modulator of MCL1 and glucocorticoid resistance. Cancer Cell. 2006;10(4):331–42. doi: 10.1016/j.ccr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 41.Tabernero J, Rojo F, Calvo E, Burris H, Judson I, Hazell K, et al. Dose-and schedule-dependent inhibition of the mammalian target of rapamycin pathway with everolimus: a phase I tumor pharmacodynamic study in patients with advanced solid tumors. J Clin Oncol. 2008;26(10):1603–10. doi: 10.1200/JCO.2007.14.5482. [DOI] [PubMed] [Google Scholar]
- 42.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.He G, Siddik Z, Huang Z, Wang R, Koomen J, Kobayashi R, et al. Induction of p21 by p53 following DNA damage inhibits both Cdk4 and Cdk2 activities. Oncogene. 2005;24(18):2929–43. doi: 10.1038/sj.onc.1208474. [DOI] [PubMed] [Google Scholar]
- 44.Wada M, Bartram CR, Nakamura H, Hachiya M, Chen DL, Borenstein J, et al. Analysis of p53 mutations in a large series of lymphoid hematologic malignancies of childhood. Blood. 1993;82(10):3163–9. [PubMed] [Google Scholar]
- 45.Marks DI, Kurz BW, Link MP, Ng E, Shuster JJ, Lauer SJ, et al. High incidence of potential p53 inactivation in poor outcome child-hood acute lymphoblastic leukemia at diagnosis. Blood. 1996;87(3):1155–61. [PubMed] [Google Scholar]
- 46.Wattel E, Preudhomme C, Hecquet B, Vanrumbeke M, Quesnel B, Dervite I, et al. p53 mutations are associated with resistance to chemotherapy and short survival in hematologic malignancies. Blood. 1994;84 (9):3148–57. [PubMed] [Google Scholar]
- 47.Fritsche M, Haessler C, Brandner G. Induction of nuclear accumulation of the tumor-suppressor protein p53 by DNA-damaging agents. Oncogene. 1993;8(2):307–18. [PubMed] [Google Scholar]
- 48.Shinwari Z, Manogaran PS, Alrokayan SA, Al-Hussein KA, Aboussekhra A. Vincristine and lomustine induce apoptosis and p21(WAF1) up-regulation in medulloblastoma and normal human epithelial and fibroblast cells. J Neurooncol. 2008;87(2):123–32. doi: 10.1007/s11060-007-9502-4. [DOI] [PubMed] [Google Scholar]
- 49.Li YX, Lin ZB, Tan HR. Wild type p53 increased chemosensitivity of drug-resistant human hepatocellular carcinoma Bel7402/5-FU cells. Acta Pharmacol Sin. 2004;25(1):76–82. [PubMed] [Google Scholar]
- 50.Lam V, Findley HW, Reed JC, Freedman MH, Goldenberg GJ. Comparison of DR5 and Fas expression levels relative to the chemosensitivity of acute lymphoblastic leukemia cell lines. Leuk Res. 2002;26(5):503–13. doi: 10.1016/s0145-2126(01)00162-x. [DOI] [PubMed] [Google Scholar]
- 51.Vayssade M, Faridoni-Laurens L, Benard J, Ahomadegbe JC. Expression of p53-family members and associated target molecules in breast cancer cell lines in response to vincristine treatment. Biochem Pharmacol. 2002;63(9):1609–17. doi: 10.1016/s0006-2952(02)00917-6. [DOI] [PubMed] [Google Scholar]
- 52.Molton S, Todd D, Cook S. Selective activation of the c-Jun N-terminal kinase (JNK) pathway fails to elicit Bax activation or apoptosis unless the phosphoinositide 3′-kinase (PI3K) pathway is inhibited. Oncogene. 2003;22(30):4690–701. doi: 10.1038/sj.onc.1206692. [DOI] [PubMed] [Google Scholar]
- 53.Yoshida K, Yamaguchi T, Natsume T, Kufe D, Miki Y. JNK phosphorylation of 14–3–3 proteins regulates nuclear targeting of c-Abl in the apoptotic response to DNA damage. Nat Cell Biol. 2005;7(3):278–85. doi: 10.1038/ncb1228. [DOI] [PubMed] [Google Scholar]
- 54.Brantley-Finley C, Lyle C, Du L, Goodwin M, Hall T, Szwedo D, et al. The JNK, ERK and p53 pathways play distinct roles in apoptosis mediated by the antitumor agents vinblastine, doxorubicin, and etoposide. Biochem Pharmacol. 2003;66(3):459–69. doi: 10.1016/s0006-2952(03)00255-7. [DOI] [PubMed] [Google Scholar]
- 55.Kang M, Wan Z, Kang Y, Sposto R, Reynolds C. Mechanism of synergy of N(4-hydroxyphenyl)retinamide and ABT-737 in acute lymphoblastic leukemia cell lines: Mcl-1 inactivation. J Natl Cancer Inst. 2008;100(8):580–95. doi: 10.1093/jnci/djn076. [DOI] [PubMed] [Google Scholar]
- 56.Chiarini F, Del Sole M, Mongiorgi S, Gaboardi G, Cappellini A, Mantovani I, et al. The novel Akt inhibitor, perifosine, induces caspase-dependent apoptosis and downregulates P-glycoprotein expression in multidrug-resistant human T-acute leukemia cells by a JNK-dependent mechanism. Leukemia. 2008;22(6):1106–16. doi: 10.1038/leu.2008.79. [DOI] [PubMed] [Google Scholar]
- 57.Dasmahapatra G, Lembersky D, Rahmani M, Kramer L, Friedberg J, Fisher R, et al. Bcl-2 antagonists interact synergistically with bortezomib in DLBCL cells in association with JNK activation and induction of ER stress. Cancer Biol Ther. 2009;8(9):808–19. doi: 10.4161/cbt.8.9.8131. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 58.Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Dunner K, Jr, Huang P, et al. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res. 2005;65(24):11658–66. doi: 10.1158/0008-5472.CAN-05-2370. [DOI] [PubMed] [Google Scholar]