

Abstract
Recent etiological studies have revealed that molecular mimicry between the lipo-oligosaccharide (LOS) component of Campylobacter jejuni and gangliosides of peripheral nervous system plays an important role in the pathogenesis of Guillain-Barré syndrome (GBS). Previously, we demonstrated GD3 ganglioside molecular mimicry in a model of GBS in Lewis rats by sensitization with GD3-like LOS (LOSGD3) from C. jejuni. Since the neuropathophysiological consequences were due largely to the anti-GD3-like antibodies, we subsequently focused our effort upon eliminating the pathogenic antibodies using several strategies to mimic GD3 in this model. Here, we have validated this strategy by the use of peptide glycomimics based on epitopic mimicry between carbohydrates and peptides. We treated rats by i.p. administration of phage-displayed GD3-like peptides. One GD3-like peptide (PGD3-4; RHAYRSMAEWGFLYS) induced in treated rats a remarkable restoration of motor nerve functions, as evidenced by improved histopathology, rotarod performance, and motor nerve conduction velocity. PGD3-4 effectively decreased the titer of anti-GD3/anti-LOSGD3 antibodies and ameliorated peripheral nerve dysfunction in the sera of treated rats. The data suggest that peptide glycomimics of ganglioside may be potential powerful reagents for therapeutic intervention in GBS by neutralizing specific pathogenic anti-ganglioside antibodies.
Keywords: Campylobacter jejuni, ganglioside GD3, Guillain–Barré syndrome, lipo-oligosaccharide, molecular mimicry, phage display peptide
Guillain-Barré syndrome (GBS) is an immune-mediated disorder of the peripheral nervous system, and is the most frequent cause of acute flaccid paralysis in humans occurring with an annual incidence of 1–2 cases/100 000 (Hughes and Rees 1997). It is classified as an acute inflammatory demyelinating polyneuropathy with a variant form designated as acute motor axonal neuropathy (Hughes and Cornblath 2005). Anti-ganglioside antibodies have frequently been proposed as contributors to GBS pathogenesis (Kaida et al. 2009; Ariga and Yu 2005). Gangliosides are abundantly expressed in human nerves (Yu et al. 2004) and generally believed to have important roles as mediators of cell adhesion and modulators of signal transduction (Regina Todeschini and Hakomori 2008). Recently, molecular mimicry between microbial lipo-oligosaccharide (LOS) antigens and endogenous ganglioside GM1 has been proposed as an etiological mechanism for GBS because of the findings that the autoantibodies for GM1 [Galβ1-3GalNAcβ1-4(Neu-Acα2–3)Galβ1–4Glcβ1-1′Cer] can often be elicited by preceding infections by Campylobacter jejuni (Aspinall et al. 1992, 1994; Yuki et al. 1993). In addition to GM1-like LOS (LOSGM1), antibodies to GD1a, GT1a, and GD1c are also elicited by LOS antigens of C. jejuni neuritis-causing strains (Aspinall et al. 1994; Goodyear et al. 1999; Koga et al. 2005). Salloway et al. (1996) reported GD3-like LOS (LOSGD3) in a C. jejuni strain from a patient with Miller Fisher syndrome. In our previous study, we demonstrated that elevated titers of circulating antibodies to GD3 ganglioside [NeuAcα2-8NeuAcα2-3Galβ1-4Glcβ1-1′Cer] occurred in some patients with inflammatory demyelinating polyneuropathies. We have identified in strain HS19 of C. jejuni the presence of an LOSGD3 with a tetrasaccharide epitope [NeuAc-NeuAc-Gal-Hep] (Usuki et al. 2006) that has a terminal trisaccharide structure identical to GD3. This carbohydrate antigen causes LOSGD3-initiated nerve dysfunction in Lewis rats including interfering with ion channels essential for nerve conduction, and this is associated with increased anti-GD3 antibody (Usuki et al. 2006).
Recently, we have initiated development of gangliosidemimic therapy targeting specific pathogenic antibodies with the goal of ameliorating the disease. This approach could prove superior to current GBS treatments, such as plasma-pheresis, intravenous administration of Ig, and immunosuppressive chemotherapy; all of which target both pathogenic and non-pathogenic antibodies. In our first experiment, the efficacy of neutralizing anti-GD3 antibody by intraperitoneal administration of anti-idiotype monoclonal antibody BEC2 specifically directed to the anti-GD3 antibody (Usuki et al. 2010) was examined. Our successful use of BEC2 to inhibit and neutralize circulating anti-GD3 and anti-LOSGD3 antibodies in the treated animals prompted us to seek additional and simpler epitope-neutralization therapies. Designing a 3D conformational epitope structural mimic common to these carbohydrates and peptides, we decided to test using peptide mimics that can be synthesized easily for eventual clinical application. GD3-like peptides were selected by panning of a phage peptide library using an anti-GD3 monoclonal antibody (mAb) (Willers et al. 1999; Popa et al. 2006). In this study, we tested several phage-displayed GD3-like peptides for treatment of our established rat model of LOSGD3-induced neuropathy. The peptide treatment thus designed improved peripheral nerve function, and in this model was most likely a consequence of neutralizing and blocking the pathogenic activity of the elevated anti-LOSGD3/anti-GD3 antibodies.
Materials and methods
The following items were purchased: high-performance thin-layer chromatographic plates coated with silica gel 60 (aluminum-backed sheets) from E. Merck (Darmstadt, Germany), and a mouse hybridoma cell line for mAb R24 from American Type Culture Collection (ATCC, Rockville, MD, USA). The GD3-like peptides were synthesized in the W. M. Keck Biotechnology Resource Center, Yale University (New Haven, CT, USA), based on the peptide sequences reported previously (Willers et al. 1999; Popa et al. 2006), shown in Table 1. The peptide sequences of PGD3-1, PGD3-2, PGD3-3, and PGD3-4 were initially reported by Popa et al. (2006). Two other peptides, PGD3-5 and PGD3-6, were synthesized based on data reported by Willers et al. (1999).
Table 1.
GD3-like peptide
| Peptide | Amino acid sequence |
|---|---|
| PGD3-1 | LAPPRPRSELVFLSV |
| PGD3-2 | PHFDSLLYPCELLGC |
| PGD3-3 | GLAPPDYAERFFLIS |
| PGD3-4 | RHAYRSMAEWGFLYS |
| PGD3-5 | ACTPYAMLPGCK |
| PGD3-6 | SVAVPPPADDPSWRY |
Gangliosides (GM1, GM2, GD1a, GD1b, GT1b, and GQ1b) for an ELISA were prepared from bovine brain tissues in our laboratory. GD3 ganglioside was prepared from bovine buttermilk (Ren et al. 1992). The nomenclature of gangliosides is based on that of Svennerholm (1964).
Preparation of mAb R24 specific to GD3
See Appendix S1.1.
Biotinylation of PGD3-4
One of the GD3-like peptides (PGD3-4) was biotinylated and used as bPGD3-4. See Appendix S1.2.
Preparation of LOSGD3
Campylobacter jejuni ATCC-43446 (serotype HS19) was grown in Brucella broth with gentle shaking (100–150 rpm) for 48 h at 37°C under microaerobic conditions. The cells were recovered by centrifugation at 7 000 g for 30 min, and washed twice with saline. The LOS fraction (LS fraction) was extracted from the cell pellets by hot phenol–water, followed by step-wise silica gel column chromatography (Usuki et al. 2010). The LOSGD3 was purified from the LS fraction by mAb R24-linked affinity purification as previously reported (Usuki et al. 2006, 2010).
Pharmacokinetic analysis
See Appendix S1.3.
Streptavidin-coated ELISA for determination of plasma bPGD3-4 levels
Plasma containing bPGD3-4 was attached to streptavidin-coated 96-well polystyrene plates obtained from Pierce (No.15121, Rockford, IL, USA), and ELISA was performed according to the manufacturer’s instructions. The efficacy of binding of the anti-GD3 antibody to immobilized bPGD3-4 was determined using mAb R24, followed by an anti-mouse horseradish peroxidase-conjugated secondary antibody and colorimetric development. Briefly, the plasma samples from the single-dose administration of bPGD3-4 were applied to the streptavidin-coated ELISA plates at serial double dilutions in 1% bovine serum albumin/phosphate buffered-saline (BSA/PBS) solution. The plate was incubated for 1 h at room temperature, and after washing with 1% BSA/PBS buffer, each well of the plate was treated with the mAb R24 (1 μg/mL in 1% BSA/PBS buffer). After washing with 1% BSA/PBS buffer, each well was treated with an anti-mouse IgG horseradish peroxidase-conjugated antibody (Jackson ImmunoResearch Lab, West Grove, PA, USA). This secondary antibody was pre-treated with rat IgG to eliminate contamination from any anti-rat IgG binding activity. Finally, the bound secondary antibody was visualized by a color-generating reagent (OPD Peroxidase Substrate in PBS; Sigma, St Louis, MO, USA). Half-saturation absorbance values for plasma samples were estimated by serial plasma dilution curves of ELISA. The absorbances were then converted into values of plasma concentration using a standard serial dilution curve of rat serum containing authentic bPGD3-4.
ELISA assay for IgG antibodies to gangliosides and LOSGD3
Anti-ganglioside antibody activity was evaluated for GM1, GM2, GD1a, GD1b, GT1b, GQ1b, GD3, and LOSGD3 by conventional ELISA as previously described (Usuki et al. 2005). To measure half-maximal inhibitory concentrations (IC50s) of GD3-like peptides for binding of GD3 to mAb R24, each well of the ELISA plate (Immunlon 1B, Lab System, Franklin, MA, USA) was coated with 0.1 μg of GD3, and prior to ELISA assay was treated by 1% BSA/PBS solution. The purified mAb R24 IgG (10 μg/mL by 1% BSA/PBS solution) was incubated with various concentrations of GD3-like peptides (PGD3-1, PGD3-2, PGD3-3, PGD3-4, PGD3-5, and PGD3-6) for 30 min. After incubation, the reaction mixture was passed through a 0.22-lm syringe filter (Millipore Corp., Bedford, MA, USA), and 100 μL of the mixture was added to each well and incubated for 1 h at room temperature. A secondary antibody (horseradish peroxidase-conjugated anti-mouse IgG, 1: 5000 dilution in 1% BSA/PBS solution) was applied, incubated for 1 h at room temperature, and visualized by addition of a color-generating reagent (OPD Peroxidase Substrate). The absorbance was measured at 492 nm with a microplate spectrophotometer (BioRad, Hemel Hempstead, UK).
Animal experimental protocol
Immunization was performed according to our established procedure (Usuki et al. 2006) and the scheme is shown in Fig. 1. Twelve-week-old female Lewis rats, weighing 200–220 g each, were used. One hundred micrograms of LOSGD3 was dissolved in 50 mM PBS buffer as vehicle, and with 0.05 mL of keyhole limpet hemocyanin (2 mg/mL) emulsified with an equal volume of complete Freund’s adjuvant. A single subcutaneous injection of 0.1 mL of inoculum (or vehicle) was made into the rats’ shoulders and hind limb footpads. Additional booster injections were administered similarly to LOSGD3- (or vehicle-) treated animals at 2-week intervals with 100 μg of LOSGD3 (or no additive) and 2 mg/mL of keyhole limpet hemocyanin in PBS emulsified with an equal volume of incomplete Freund’s adjuvant after 6 weeks. Forty-four experimental animals were grouped into 10 treatment paradigms (n = 4 each): (i) LOSGD3, (ii) LOSGD3/PGD3-3, (iii) LOSGD3/PGD3-4, (iv) LOSGD3/PGD3-5, (v) LOSGD3/PGD3-6, (vi) vehicle, (vii) vehicle/PGD3-3, (viii) vehicle/PGD3-4, (ix) vehicle/PGD3-5, and (x) LOSGD3/PGD3-6. The remaining four animals comprised the untreated control group (n = 4). As shown in the fourth stepladder of Fig. 1, the rats were subjected to daily i.p. injections of a peptide: 1000 nmol/kg, three times from 8, 11, and 14 weeks for 3 weeks, including a 2-week rest.
Fig. 1.

Timing of treatment schedule of animal experimentation. The timing of treatment and performance tests between week-0 and week-18 in the experimental period are shown. Forty-four experimental animals were grouped into 10 treatment groups (n = 4 each): (i) LOSGD3, (ii) LOSGD3/PGD3-3, (iii) LOSGD3/PGD3-4, (iv) LOSGD3/PGD3-5, (v) LOSGD3/PGD3-6, (vi) vehicle, (vii) vehicle/PGD3-3, (viii) vehicle/PGD3-4, (ix) vehicle/PGD3-5, and (x) LOSGD3/PGD3-6. The remaining four animals were included as the untreated control group (n = 4). As shown on the Bottom line, rats were subjected to i.p. daily injections of a test peptide; 1000 nmol/kg, three times from 8, 11, and 14 weeks for 3 weeks, with 2-week rest intervals. Blood samples were collected retroorbitally by bleeding with a capillary tube at every 4-week intervals (second line below Top). Electrophysiological examination was performed by NCV measurement at week-0 and week-16 (third line below Top). The motor-behavioral performance was performed by rotarod test at week-17 (third line from Top). All animals were killed at the end of week-18.
Before injection of the GD3-like peptide mimics, any endotoxin that might be included in PGD3-3, PGD3-4, PGD3-5, or PGD3-6 was removed by ToxinEraser™ Endotoxin Removal Kit (GenScript Corp., Piscataway, NJ, USa), and the endotoxin remaining was detected by the Limulus Amebocyte Lysate test (ToxinSensorTM Gel Clot Endotoxin Assay Kit; GenScript Corp.).
Blood samples were drawn retro-orbitally by bleeding with a capillary tube at 0, 4, 8, 12, 16, and 18 weeks after the primary immunization.
All experimental animals were weighed weekly and assessed for clinical signs of peripheral nerve abnormalities. Electrophysiological measurements of nerve conduction velocity (NCV) were performed at 0 and 16 weeks, and motor behavioral-performance by the rotarod test at 17 weeks after the primary immunization. All animals were allowed free access to water and food. The use of these animals was approved by the Medical College of Georgia’s Institutional Animal Care and Use Committee. Treatment of these animals was conducted according to approved procedures.
Nerve conduction velocity
NCVs (m/s) were measured in the rat tail nerve using a Nicolet VikingQuest EMG machine (NeuroCare Group, Madison, WI, USA) according to the method described previously (Usuki et al. 2006).
Rotarod motor test
The rotarod motor test was performed as previously reported (Dunham and Miya 1957; Jones and Roberts 1968) and according to the procedure described by Rogers et al. (1997).
Neuromuscular junction (NMJ) activity
A spinal cord-muscle co-culture was performed according to the method of Taguchi et al. (2004). Briefly, muscle and spinal cord explant cells were prepared from muscle tissue and spinal cord tissue (containing dorsal root ganglia) of 17-day-old fetal rats. Muscle cells and spinal cord explants were co-cultured and maintained for up to 1 week in Dulbecco’s modified Eagle’s medium supplemented by fetal calf serum and growth factors. Spontaneous muscle action potential frequency was recorded by a glass microelectrode (Ag/AgCl, 30–40 MΩ) and a recording electrode (3 M KCl). The recording system consisted of a Microelectrode Amplifier MEZ-8301, Memory Oscilloscope VC-11 (Nihon Kohden, Tokyo, Japan) and an A-D Converter DigiData 1200 Interface (Axon Instruments, Inc., Union City, CA, USA). The spontaneous muscle action potential was low-pass filtered at 1 kHz. A rat antiserum solution (10 μL) was delivered directly with a micropipette near the innervated muscle cells.
Histological/morphological examination
Pathological examination was undertaken to correlate changes with clinical manifestations in the animals. At the endpoint (week 18) of animal experimentation (Fig. 1), the animals were killed. After drawing a blood sample from the heart of each animal, the animals were perfused with 4% paraformaldehyde in PBS buffer via a needle inserted into the arcus aortae. After perfusion, the lumbar spinal cord was dissected and sectioned into 4–6 transverse segments spaced 1 mm apart. The right sciatic nerve was carefully dissected from its origin (5-mm distal to the gluteus maximus) through the distal branch point at the peroneal and tibial nerves and stretching carefully avoided. Nerve sections were placed at 4°C overnight in a fixative solution containing 4% paraformaldehyde, 2% glutaraldehyde, and 0.1 M sodium cacodylate buffer (pH 7.4). The nerves were washed thrice in cacodylate buffer (pH 7.4), and then post-fixed at 4°C with 2% osmium tetroxide/0.1 M sodium cacodylate buffer (pH 7.4) for 60 min, dehydrated in graded ethanol, stained at 4°C with 2% uranyl acetate/70% ethanol for 30 min, and embedded in epoxy resin (Poly/Bed 812, Polysciences, Inc., Warrington, PA, USA). One-micrometer-thick cross-sections of fascicle were stained with toluidine blue for histological examination using an Axiophot photomicroscope equipped with an Axiocam (Carl Zeiss, Jena, Germany). Images were stored and analyzed using AxioVision. The total number of myelinated fibers in each fascicle was assessed by visual counting, and the myelin to myelinated-area ratio was calculated using Scion Image software (beta 4.0.3). The total fiber area and the total myelin area were marked, and quantitated automatically by the program. Five images were analyzed per cross-section, and the percentage of myelinated area was determined using the following formula: myelin % = (myelinated area/total area) × 100.
For electron microscopy, ultra-thin sections were prepared and examined by a high-performance, high-contrast, 40–120 kV transmission electron microscope (JEOL JEM-1230, JEOL Ltd, Tokyo, Japan).
Molecular docking of GD3-like peptides
Conformational analyses of the carbohydrate head group of GD3 and GD3-like peptides were performed using the program Dynamic Molecules (http://www.md-simulation.de/manager/). The 3D structure of the B chain of mAb R24 was extracted from the atomic coordinates of PDB code 1r24 using a Swiss-PdbViewer 4.0 (Guex and Peitsch 1997). Molecular docking was performed using Autodock 3.0 (Morris et al. 1996), and the docking results were collated by MGL Tools (Sanner 1999) or PyMOL (DeLano 2002).
Statistical analysis
Statistical analyses were performed using the GraphPad Prism 2.01 software package (GraphPad, San Diego, CA, USA). One-way or two-way ANOVA was performed for differences in treatment results between experimental animal groups, and was examined by Tukey’s multiple comparison test. Differences among groups including the LOSGD3-treated group were analyzed by Dunnett’s multiple comparison test (*p < 0.01, in Fig. 5). After the data were subjected to two-way ANOVA, statistical difference was compared by unpaired t-test of Student (*p < 0.01, **p < 0.001).
Fig. 5.

Effects of peptide treatment on nerve conduction velocity, behavioral tests, and muscle action potential. (a) Effect of treatment of PGD3-3, PGD3-4, PGD3-5, or PGD3-6 on NCV with or without LOSGD3-treatment. NCVs were measured at week-0 (□) and at week-16 (■) for each of the 10 groups. Assessment of the NCV of the four peptide treatment groups was performed using the immunized groups with LOSGD3 and vehicle groups as shown in the upper bar graph: LOSGD3, LOSGD3/PGD3-3, LOSGD3/PGD3-4, LOSGD3/PGD3-5, and LOSGD3/PGD3-6, and in the lower bar graph: vehicle, vehicle/PGD3-3, vehicle/PGD3-4, vehicle/PGD3-5, and vehicle/PGD3-6. Values are mean ± SD for four animals. The values were analyzed by one-way anova and Dunnet’s multiple comparison test (*p < 0.01). (b) Performance of the rotarod test at week-17 after initial sensitization with LOSGD3. The graph bars show retention time of rat on the rotating rod. Control group rats maintained balance on the rotating rod at 10 rpm for more than 20 s. The motor disability in LOSGD3-treated rats reflected in duration time decreased by 10 s. PGD3-4 treatment normalized motor performance with a statistically significant difference from the LOSGD3-treated group (*p < 0.01). (c) Inhibitory effects of rat antisera on muscle action potential frequencies in the spinal cord-muscle co-cultured system. The frequencies were recorded every 5 s. Arrows show addition of 10 μL of rat serum and wash out. Each serum sample of LOSGD3 sera and LOSGD3/PGD3-4 sera was obtained from four rats at the endopoint of the experiment, respectively, in two groups of LOSGD3-treated and LOSGD3/PGD3-4-treated animals. Top graph shows effect on muscle action potential after addition of LOSGD3 serum; the middle shows that of LOSGD3 serum treated by immunoabsorption by GD3. The bottom graph shows the effect on action potential from treatment with LOSGD3/PGD3-4, indicating no inhibitory activity in that serum.
Results
Purification of LOSGD3 from the LOS fraction of C. jejuni
The crude LS fraction was prepared from 30 g, wet weight, of a cell pellet of C. jejuni, strain HS19, according to our method (Usuki et al. 2006). Isolation and purification of LOSGD3 from the LS fraction was achieved by silica gel column and mAb R24 affinity column chromatographies to obtain 130 mg of the final product that was used for experiments.
Competitive inhibition assay
GD3-like peptide mimics (PGD3-1 to PGD3-6) were tested by ELISA for competitive inhibition of GD3 reactivity to mAb R24. Four peptides (PGD3-3, PGD3-4, PGD3-5, and PGD3-6) revealed remarkable inhibitory activities (Fig. 2); the IC50 values were 300 pmol/mL for PGD3-3, 150 pmol/mL for PGD3-4, 100 pmol/mL for PGD3-5, and 250 pmol/mL for PGD3-6. In contrast, PGD3-1 and PGD3-2 had little or no inhibitory activity. The active peptides showed full inhibition of GD3 binding to mAb R24 at a concentration of more than 600 pmol/mL and were used for further study.
Fig. 2.

Effect of GD3-like peptides on binding of R24 mAb to GD3 by ELISA. Each of the GD3-peptide mimics in solution (PGD3-1, PGD3-2, PGD3-3, PGD3-4, PGD3-5, or PGD3-6) was incubated with R24 mAb for 30 min at room temperature at various concentrations as indicated in the horizontal axis. An aliquot of the mixtures (100 μL), was added to each well of an ELISA plate coated with 0.1 μg of GD3. The remaining binding activity of R24 mAb was measured by ELISA. Values are mean ± SEM, n = 6 individual experiments.
Pharmacokinetic study
See Appendix S1.4.
Experimental animals
During the course of animal experimentation, the LOSGD3-sensitized animals showed mild clinical signs of neurological dysfunction, including a remarkable slowness of movement and noticeable weight loss between 13 and 18 weeks post-treatment as compared to the corresponding vehicle treatment group (Fig. 3). Among the four peptide treatment paradigms, animals treated with PGD3-4 revealed a remarkable clinical improvement with lowering of the serum anti-GD3 antibody level, reversal of weight loss and slowing of NCV, and improvement of rotarod performance, as compared with animals treated with PGD3-3, PGD3-5, and PGD3-6. Body weight changes in the three treatment groups (vehicle, LOSGD3, and LOSGD3/PGD3-4) are shown in Fig. 3 (data of the PGD3-3, PGD3-5, and PGD3-6 treatment groups not shown). Differences between the LOSGD3/PGD3-4 treatment groups and the LOSGD3 treatment group showed statistically significant differences during the period of 13–18 weeks after primary immunization with the LOSGD3/PGD3-4 animals showing better protection.
Fig. 3.
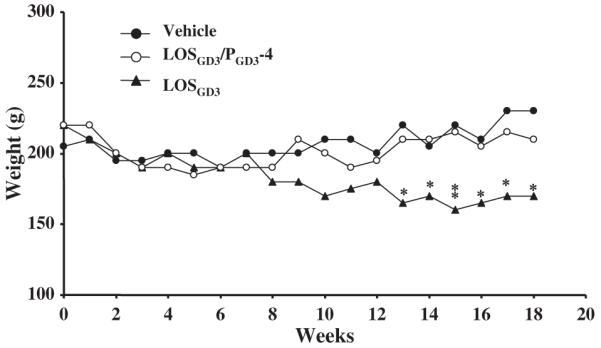
Changes in body weight after sensitization by LOSGD3. The graph shows body weight after vehicle-treatment (●), LOSGD3/PGD3-4-treatment (○), and LOSGD3-treatment (▲) of the week-12 rats. According to two-way ANOVA and Tukey’s test, there is a significant reduction in body weights of the LOSGD3-treated group compared with the vehicle-treated group from week-13 to -18 post-treatment. PGD3-4 treatment maintained weight at the vehicle-treated group level. There was significant difference of body weight compared LOSGD3-treated group by unpaired t-test of Student (*p < 0.05, **p < 0.01).
At the end of the experiment, all animals were killed and the tissues analyzed. There was no adhesive peritonitis or ascites due to infection in animals as a result of administering multiple i.p. doses of the four peptides.
Time course of anti-GD3 and anti-LOSGD3 antibody production
Anti-GD3 and anti-LOSGD3 antibody responses following induction of experimental neuritis using LOSGD3 were examined periodically during the 18 weeks of experimentation by serum ELISA (samples diluted 1–200 with 1% BSA/PBS buffer) (Fig. 4). Other anti-ganglioside antibodies, including anti-GM1, -GM2, -GD1a, -GD1b, -GT1b, and -GQ1b, were not detected in the sera during the entire course of the experiment (data not shown). The anti-GD3 IgG antibody titer was elevated many fold 8 weeks post-inoculation, in parallel with elevation of the anti-LOSGD3 antibody titer (Fig. 4a). The anti-GD3 IgG antibody titer attained and remained at a plateau level (Fig. 4a). There were no IgG antibody responses for GD3 in the vehicle-treated group or in the untreated group (Fig. 4b). In the sera of the animals treated with GD3-like peptides, suppressive effects upon the serum anti-GD3 antibody level varied; the most profound effects were decreased titers to LOSGD3/PGD3-4 and LOSGD3/PGD3-5 treatment groups. Two other peptides (PGD3-4 and PGD3-5) were effective in suppressing serum anti-GD3 antibody activity (Fig. 4), whereas no suppression of either anti-LOSGD3 or anti-GD3 antibodies was found after chronic treatment with two other peptides (PGD3-3 and PGD3-6). None of the peptide mimics had any effect on the serum anti-GD3 titer in the vehicle treatment groups.
Fig. 4.

Serum levels of anti-LOSGD3 and anti-GD3 antibodies. Animal experimental schedule shown in Fig. 1. Animals bled from week-0 to week-18 at every 4-week interval for serum samples for testing anti-GD3 antibody (●) and anti-LOSGD3 antibody (○) using ELISA. Values are mean ± SD for four animals.
NCV changes
NCVs were measured at week 0 and week 16 post-inoculation. As shown in Fig. 5a, control rats showed slightly increased NCV within the experimental period, presumably because of an age-related change. At week 16, the NCV of the group sensitized by LOSGD3 decreased significantly when compared with the corresponding vehicle group (p < 0.01). Among the LOSGD3/peptide-treated groups, PGD3-4 and PGD3-5 showed less attenuation of NCV, although there was not a statistically significant difference between the treatment and LOSGD3/PGD3-5 treatment groups. No change of NCV was observed among the treatment groups with vehicle/peptides (Fig. 5a).
Rotarod test
Animals underwent a rotarod test at week 17, and there was no statistically significant difference in retention time between the vehicle- and untreated-control groups (Fig. 5b). LOSGD3-treated animals had muscle weakness and significantly shorter retention times for all of these experimental groups. Among the peptide-treatment groups, a statistically significant difference was found only between the LOSGD3/PGD3-4 treatment group and the LOSGD3 treatment group (p < 0.01). The PGD3-3, PGD3-5, or PGD3-6 groups did not show any statistically significant differences following LOSGD3 treatment (data not shown). Similarly, there were no statistically significant differences among the vehicle, vehicle/PGD3-4, and control groups (data not shown).
Inhibition of NMJ activity
To determine the nature of the anti-GD3 antibody produced in LOSGD3-sensitized rats that manifest neuromuscular weakness, spontaneous muscle action potential frequencies were examined by addition of the serum to a co-culturing system of spinal cord-muscle cells (Usuki et al. 2005). Sera from rats sensitized by LOSGD3 showed a strong blockade of NMJ action potential frequencies immediately after addition of the serum sample (Fig. 5c, Top). Immunoabsorption [LOSGD3 (+ absorption)] removed the NMJ blockade; the NMJ action potential, however, did not return to the original frequency level (Fig. 5c, Middle). On the other hand, the LOSGD3/PGD3-4 serum did not block the NMJ action potential (Fig. 5c, bottom).
Morphological analysis of motor neurons and motor axons
Structural alterations and pathological changes associated with motor spinal cord neurons and sciatic nerves of the LOSGD3-treated rats were assessed. A representative micrograph of each specific pathological feature from each of the animal groups is shown in Fig. 6. Within the lumbar spinal cord, there were no overt structural differences between the anterior horn cells in the untreated control group and the LOSGD3-treated group (Fig. 6a–g).
Fig. 6.

Histopathological examination. All animals were killed at the endpoint (week 18) of experiment by cardiac puncture, followed by perfusion with 4% paraformaldehyde in PBS buffer. Procedure for dissection and preparation of nerve tissue sections is in Materials and methods. Examples of toluidine blue stained lumbar motor neurons of anterior horn in transverse spinal cord sections (a–g) and transverse sections of sciatic nerves (h–n). Sciatic nerves were examined by electron microscopy (o–u). Scale-bars: a–g (25 μm), h-n (10 μm) and o–u (5 μm). Control rat (a, h, o); vehicle rat (b, i, p); LOSGD3 rat (c, j, q);, LOSGD3/PGD3-3 rat (d, k, r); LOSGD3/PGD3-4 rat (e, l, s); LOSGD3/PGD3-5 rat (f, m, t); LOSGD3/PGD3-5 rat (g, n, u).
Profile counts (numbers of large cells in the anterior horn cross-section) indicated similar densities of lumbar motor neurons in the experimental groups of the control, the LOSGD3, and the LOSGD3/peptide groups (a, 21 ± 3; b, 26 ± 4; c, 23 ± 4; d, 24 ± 2; e, 25 ± 6; f, 18 ± 6; g, 23 ± 5 neurons/section; n = 4, respectively). There were no statistically significant differences among the experimental groups in the neuronal profile area (a, 295 ± 22 μm2; b, 290 ± 22 μm2; c, 252 ± 25 μm2; d, 248 ± 31 μm2; e, 262 ± 25 μm2; f, 288 ± 31 μm2; g, 282 ± 29 μm2; n = 4).
To assess morphologic alterations, if any, corresponding to motor dysfunction or muscle weakness evaluated by NCV measurement and rotarod tests, we examined distal motor nerves near the tibial branch of the sciatic nerve; a nerve containing predominantly myelinated motor fibers that serve skeletal muscle fibers of the lower distal leg. Cross-sections of sciatic nerves were prepared and examined by toluidine blue staining (Fig. 6h–n) and by electron microscopy (Fig. 6o–u). Control and vehicle treatments showed normal myelin thickness by both light and electron microscopic examination (Fig. 6h, i, o and p). The sciatic nerves from the LOSGD3-treatment groups manifested obvious axonal destruction, and the remaining axons were surrounded by a very thin myelin sheath with disorganized and disconnected myelin layers (Fig. 6j). LOSGD3/PGD3-3, -4, -5, or -6 treatment induced axonal disappearance as a result of destroyed myelin or dissociated myelin debris as shown by light microscopic examination (Fig. 6k, l, m, n). The LOSGD3/PGD3-4 treatment group, however, exhibited only a moderate loss of myelinated fibers (Fig. 6l). Upon ultrastructural examination, myelin-axonal degeneration was commonly found in the LOSGD3 treatment and LOSGD3/peptide treatment groups as revealed by the presence of empty and enlarged axons surrounded by a very thin myelin sheath (Fig. 6q, r, s, t, u). On the other hand, the LOSGD3/PGD3-4 treatment group showed a remarkable restoration of myelin thickness (Fig. 6s).
As expected, there was no statistically significant difference in myelin thickness between the control and vehicle groups (Fig. 6o and p). A noticeable change of myelin thickness and a statistically significant difference exist, however, in the percentage of myelinated area in the myelinated (i.e. thickness of the average myelin sheath) between the LOSGD3 and control/vehicle groups (o, 30 ± 4.2%; p, 31.5 ± 3.2%; q, 11.8 ± 3.8%; p < 0.01) (Fig. 6). Among the peptide-treated groups, only the PGD3-4 treatment group benefited so that there was no statistical difference in the percentage of myelinated area (r, 30 ± 4.2%; t, 31.5 ± 3.2%; u, 26.8 ± 3.8%).
Molecular docking of GD3-like peptides
In this study, the tetrasaccharide head group of ganglioside GD3 was found to dock into the binding pocket of mAb R24 that is formed by the complementarity-determining regions (CDRs): CDR1, 2, and 3 with a docking energy of −4.2 kcal/mol (Fig. S2a). Our data indicate that CDR2 binds the galactose and glucose residues and that CDR3 has a role anchoring the two sialic acid residues. PGD3-1 and PGGD3-2 failed to dock into the binding pocket on the B chain of mAb R24, while peptides PGD3-3, PGD3-4, PGD3-5, and PGD3-6 successfully docked into the binding pocket with docking energies from −3.5 to −4.5 kcal/mol (Fig. S2b–e).
Discussion
GBS is an autoimmune disease in which the immune system mistakenly attacks myelin or axons (Hahn 1998). Campylobacteriosis is a frequent antecedent event in GBS; the triggering agent in this case is microbial infection with C. jejuni and occurs with certain ‘neuritogenic’ strains of the microbe. Other frequently encountered agents associated with GBS are cyotomegalovirus, Epstein–Barr virus, and Mycoplasma pneumoniae (Hughes et al. 1999). Rarely, GBS and Helicobacter pylori infection have been associated with the acute inflammatory demyelinating polyradiculoneuropathy of GBS. Molecular mimicry between microbial LOS antigens and endogenous gangliosides is an important pathogenetic mechanism related to GBS, and this concept has gained increasing support (Yu et al. 2006; Yuki 2007). GM1 ganglioside mimicry was reported by Yuki et al. (1990), who successfully induced a rabbit model of GBS using LOSGM1 or GM1 (Yuki et al. 2004; Yu et al. 2006). Direct evidence has also been provided by our studies showing that rats sensitized by LOSGD3-immunization had elevated anti-GD3 antibody titers and manifested nerve degeneration and dysfunction (Usuki et al. 2006). The GD3 antibody, similar to other anti-ganglioside antibodies, produced NMJ-blockade in a muscle/spinal cord co-culture system and inhibited the generation of muscle action potentials (Taguchi et al. 2004; Usuki et al. 2006), thus contributing to the pathophysiology. Based on those findings, we previously tested the efficacy of a novel treatment strategy to remove the pathogenic anti-GD3 antibody using a monoclonal anti-idiotype antibody, BEC2. The idiotype network theory posits that serial idiotype antigens generate antibodies with a serologically unique structure designated as an anti-idiotype (Shoenfeld 2004). BEC2 was generated against the R24 monoclonal antibody and mimics GD3 ganglioside (Chapman and Houghton 1991). Thus, we predicted that BEC2 could effectively neutralize the anti-GD3 antibody titers in our experimental animals, and this turned out to be the case. We then demonstrated that i.p. injection of BEC2 ameliorated the clinical symptoms with a concomitant decrease in the anti-GD3 antibody titers in LOSGD3-induced neuropathy in Lewis rats (Usuki et al. 2010).
In addition to the anti-idiotype monoclonal antibody, in the current study we used a similar approach but employed peptides that have GD3-epitope structural mimicry. These peptides are alternative antigens to GD3, and are recognized by anti-GD3 monoclonal antibodies. They can be easily derived from cloning and isolation by panning phage-displayed peptide libraries. To isolate peptide glycomimics in this way, we used anti-GD3 mAb 4F6 for PGD3-1, PGD3-2, PGD3-3, PGD3-4 (Popa et al. 2006), and anti-GD3 mAb MG21 and mAb MG22 for PGD3-5 and PGD3-6, respectively (Willers et al. 1999). Although those peptides as well as GD3 itself may be monospecific for each mAb, two (PGD3-3, PGD3-4) bound mAb R24 as well as mAb 4F6. The other two, PGD3-5 and PGD3-6, had high affinity with mAb R24 as well as with mAb MG22. As shown in Fig. 2, four peptides (PGD3-3, PGD3-4, PGD3-5, and PGD3-6) were potent inhibitors of GD3 binding to mAb R24 with IC50 values in the range of 100–300 pmol/mL as assessed by competitive inhibition ELISA assay. Peptides PGD3-1 and PGD3-2 had little or no affinity for mAb R24, and were not further tested.
To investigate the binding specificity of the peptides and mAb R24, the mode of interaction between the B chain of mAb R24 (1r24) and the peptides was analyzed in silico by the molecular docking method., It was previously suggested that CDR2 had a role in binding of the two terminal sialic acid residues by docking into the binding pocket of mAb R24 (Yan et al. 1996). In contrast to this report (Yan et al. 1996), our data revealed that CDR2 binds the galactose and glucose residues and CDR3 has a role in anchoring the sialic acid residues of the GD3. PGD3-1 and PGD3-2 failed to dock into the binding pocket on the B chain of mAb R24, whereas PGD3-3, PGD3-4, PGD3-5, and PGD3-6 could successfully dock there (Fig. S2.). The molecular docking results suggest that mAb R24 could recognize and bind the peptides with similar binding affinity (Fig. 2). We wonder as well if the peptides could interact with similar structures formed in the CDRs of the variable regions of other anti-GD3 mAbs, in addition to mAb R24.
According to pharmacokinetic analyses using bPGD3-4, treatment for the chronic polyneuropathy variant was designed by consideration of IC50 values and planning doses and timing to maintain effective plasma concentrations. As shown in Fig. S1a, administration of bPGD3-4 gave rise to a short circulatory half-life (t1/2β), and for this reason, multiple dosings are necessary to maintain effective plasma concentrations (Fig. S1b). This strategy turned out successfully and the other three peptides (PGD3-4, PGD3-5, and PGD3-6) were also reduced anti-GD3/anti-LOSGD3 antibody titers.
PGD3-4 was the most effective inhibitor of anti-GD3/anti-LOSGD3 antibodies: the improvement was statistically significant as well for NCV maintenance and longer duration rotarod performance. The other three peptide glycomimics had variable effectiveness in improving NCV or motor behavioral performance and not significant. In comparison to the other molecular mimicry based-therapy using an BEC2, peptide glycomimic therapy using PGD3-4 had a relatively short half-life of 2.45 h as compared with 161.3 h for BEC2 that effectively improved assay in the rats of NCV and rotarod performance (Fig. 6). Consistent with these findings, pathological examination revealed that changes began in the terminal axons and evolved proximally with concomitant myelin degradation in the sciatic nerve. No motor neuronal degeneration in the spinal cord was observed (Fig. 6c), suggesting that the neuromuscular weakness was caused by structural deterioration of the distal nerve, NMJ, and myelin of the motor neurons. The pathological lesions did not cause frank paralysis, but to abnormalities of motor performances as demonstrated by the rotarod and NCV measurements. More remarkably, PGD3-4 treatment facilitated remyelination based on EM analysis (Fig. 6s). Decreased body weight and elevation of serum anti-GD3/anti-LOSGD3 antibody titers also correlated (Fig. 3).
Inhibitory effects upon NMJ function could also be demonstrated in an in vitro spontaneous muscle action potential assay using rat sera with high and low anti-LOSGD3/anti-GD3 antibody activities (Fig. 5c). NMJ-inhibiting antibody activities have been observed in patients with GBS and is thought to contribute to the mechanisms of neuromuscular weakness (Buchwald et al. 1998). In animals sensitized by LOSGD3, sera with anti-GD3 antibody activities exerted a remarkable inhibitory effect upon spontaneous muscle action potential activity and this could be abolished by immunoabsorption with GD3 to eliminate the anti-GD3 antibody reactivity. In contrast, sera in rats treated with PGD3-4 did not inhibit NMJ activity implying a superiority for PGD3-4 treatment compared with anti-idiotype antibody BEC2 treatment in that the latter had residual NMJ inhibition (Usuki et al. 2010).
Most remarkably, PGD3-4 treatment not only neutralized anti-GD3 antibody, but also restored peripheral nerve dysfunctions with morphology suggesting a facilitation of remyelination. A 15-mer peptide mimicking the HNK-1 epitope reportedly promotes neurite outgrowth (Simon-Haldi et al. 2002). The HNK-1 carbohydrate epitope [GlcA(3-sulfate)β1-3Galβ1-4GlcNAc] is present on myelin-associated glycoprotein as well as sulfoglucuronosyl glycolipids: a carbohydrate epitope carried by many neural adhesion molecules and involved in neural cell interaction during development, regeneration in the peripheral nervous system, synaptic plasticity, and autoimmune neuropathies such as chronic inflammatory demyelinating polyneuropathy that are related to antibodies against sulfoglucuronosyl paragloboside (Yamawaki et al. 1996). We speculate that the tri-saccharide epitotpe (NeuAcα2-8NeuAcα2-3Gal) of GD3 may also be carried by neural adhesion molecules and be involved in neural cell interaction during development and regeneration in the peripheral nervous system. This raises an intriguing possibility that certain GD3-like peptides may mimic important functions of peripheral nervous system-related molecules possessing the trisaccharide structure. Clearly more experiments are needed to examine this possibility.
In conclusion, in our unique rat model of peripheral neurodegeneration rendered by an anti-ganglioside antibody via sensitization of the animal by a specific LOS of C. jejuni, we have shown that this animal model is suitable for testing the therapeutic efficacy of peptide glycomimics, which were designed as molecular mimics for ameliorating disease symptoms. As neuropathies associated with monoclonal gammopathy have neurological symptoms resembling those found in polyneuropathies including GBS (Lozeron and Adams 2007), pathogenic monoclonal anti-ganglioside antibodies are a common feature and appear to be involved in the disease and disability. The current strategy offers a superior approach to ‘neutralize’ the specific monospecific antibodies without affecting other antibodies in the circulation and having more general or systemic effects. Additionally, our strategy is relatively non-invasive and safe, without much hazard of complications found with available treatment paradigms, such as plasmapheresis and intravenous administration of Ig. It must be said although that inhibition of a monospecific epitope may not be sufficient for the complete therapy of GBS, which has a very complex etiology, including cell-mediated immunity components, and the possibility of antibodies against ganglioside complexes in some GBS cases (Kaida et al. 2007, 2009). The presence of anti-ganglioside complexes might exclude the use of a single reagent for a monospecific epitope. One can expect, there-fore, this can be overcome by the use of multiple reagents. Further studies on the application of conformational mimics for the treatment of GBS and related anti-glycolipid antibody-induced neurodegenerative diseases deserve serious consideration.
Supplementary Material
Acknowledgements
This work was supported in main by NIH grants (NS26994 and NS11853) to RKY. We thank Dr Edward Hogan, at IMMAG, Medical College of Georgia, Augusta, GA for helpful discussions and Ms Diana Westbrook for editorial assistance.
Abbreviations used
- BEC2
anti-idiotype monoclonal antibody
- BSA
bovine serum albumin
- CDR
complementarity-determining region
- GBS
Guillain-Barré syndrome
- LOS
lipo-oligosaccharide
- mAb
monoclonal antibody
- NCV
nerve conduction velocity
- NMJ
neuromuscular junction
- PBS
phosphate buffered-saline
Footnotes
Supporting information
Additional Supporting Information may be found in the online version of this article.
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer-reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
References
- Ariga T, Yu RK. Antiglycolipid antibodies in Guillain-Barré syndrome and related diseases: Review of clinical features and antibody specificities. J. Neurosci. Res. 2005;80:1–17. doi: 10.1002/jnr.20395. [DOI] [PubMed] [Google Scholar]
- Aspinall GO, McDonald AG, Raju TS, Pang H, Mills SD, Kurjanczyk LA, Penner JL. Serological diversity and chemical structures of Campylobacter jejuni low-molecular-weight lipopolysaccharides. J. Bacteriol. 1992;174:1324–1332. doi: 10.1128/jb.174.4.1324-1332.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspinall GO, McDonald AG, Pang H, Kurjanczyk LA, Penner JL. Lipopolysaccharides of Campylobacter jejuni serotype O:19: structures of core oligosaccharide regions from the serostrain and two bacterial isolates from patients with the Guillain-Barré syndrome. Biochemistry. 1994;33:241–249. doi: 10.1021/bi00167a032. [DOI] [PubMed] [Google Scholar]
- Buchwald B, Toyka KV, Zielasek J, Weishaupt A, Schweiger S, Dudel J. Neuromuscular blockade by IgG antibodies from patients with Guillain-Barré syndrome: a macro-patch-clamp study. Ann. Neurol. 1998;44:913–922. doi: 10.1002/ana.410440610. [DOI] [PubMed] [Google Scholar]
- Chapman PB, Houghton AN. Induction of IgG antibodies against GD3 ganglioside in rabbits by an anti-idiotypic monoclonal antibody. J. Clin. Invest. 1991;88:186–192. doi: 10.1172/JCI115276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL Molecular Graphics System. DeLano Scinetific; Palo Alto, CA, USA: 2002. Available at: http://www.pymol.org. [Google Scholar]
- Dunham NW, Miya TS. A note on a simple apparatus for detecting neurological deficit in rats and mice. J. Am. Pharm. Assoc. Am. Pharm. Assoc. (Baltim) 1957;46:208–209. doi: 10.1002/jps.3030460322. [DOI] [PubMed] [Google Scholar]
- Goodyear CS, O’Hanlon GM, Plomp JJ, et al. Monoclonal antibodies raised against Guillain-Barré syndrome-associated Campylobacter jejuni lipopolysaccharides react with neuronal gangliosides and paralyze muscle-nerve preparations. J. Clin. Invest. 1999;104:697–708. doi: 10.1172/JCI6837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Hahn AF. Guillain-Barré syndrome. Lancet. 1998;352:635–641. doi: 10.1016/S0140-6736(97)12308-X. [DOI] [PubMed] [Google Scholar]
- Hughes RA, Cornblath DR. Guillain-Barré syndrome. Lancet. 2005;366:1653–1666. doi: 10.1016/S0140-6736(05)67665-9. [DOI] [PubMed] [Google Scholar]
- Hughes RA, Rees JH. Clinical and epidemiologic features of Guillain-Barré syndrome. J. Infect. Dis. 1997;176(Suppl 2):S92–S98. doi: 10.1086/513793. [DOI] [PubMed] [Google Scholar]
- Hughes RA, Hadden RD, Gregson NA, Smith KJ. Pathogenesis of Guillain-Barré syndrome. J. Neuroimmunol. 1999;100:74–97. doi: 10.1016/s0165-5728(99)00195-2. [DOI] [PubMed] [Google Scholar]
- Jones BJ, Roberts DJ. The quantiative measurement of motor inco-ordination in naive mice using an acelerating rotarod. J. Pharm. Pharmacol. 1968;20:302–304. doi: 10.1111/j.2042-7158.1968.tb09743.x. [DOI] [PubMed] [Google Scholar]
- Kaida K, Morita D, Kanzaki M, Kamakura K, Motoyoshi K, Hirakawa M, Kusunoki S. Anti-ganglioside complex antibodies associated with severe disability in GBS. J. Neuroimmunol. 2007;182:212–218. doi: 10.1016/j.jneuroim.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Kaida K, Ariga T, Yu RK. Antiganglioside antibodies and their pathophysiological effects on Guillain-Barré syndrome and related disorders – a review. Glycobiology. 2009;19:676–692. doi: 10.1093/glycob/cwp027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga M, Gilbert M, Li J, Koike S, Takahashi M, Furukawa K, Hirata K, Yuki N. Antecedent infections in Fisher syndrome: a common pathogenesis of molecular mimicry. Neurology. 2005;64:1605–1611. doi: 10.1212/01.WNL.0000160399.08456.7C. [DOI] [PubMed] [Google Scholar]
- Lozeron P, Adams D. Monoclonal gammopathy and neuropathy. Curr. Opin. Neurol. 2007;20:536–541. doi: 10.1097/WCO.0b013e3282ef79e3. [DOI] [PubMed] [Google Scholar]
- Morris GM, Goodsell DS, Huey R, Olson AJ. Distributed automated docking of flexible ligands to proteins: parallel applications of AutoDock 2.4. J. Comput. Aided Mol. Des. 1996;10:293–304. doi: 10.1007/BF00124499. [DOI] [PubMed] [Google Scholar]
- Popa I, Ishikawa D, Tanaka M, Ogino K, Portoukalian J, Taki T. GD3-replica peptides selected from a phage peptide library induce a GD3 ganglioside antibody response. FEBS Lett. 2006;580:1398–1404. doi: 10.1016/j.febslet.2006.01.063. [DOI] [PubMed] [Google Scholar]
- Regina Todeschini A, Hakomori SI. Functional role of glycosphingolipids and gangliosides in control of cell adhesion, motility, and growth, through glycosynaptic microdomains. Biochim. Biophys. Acta. 2008;1780:421–433. doi: 10.1016/j.bbagen.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren S, Scarsdale JN, Ariga T, Zhang Y, Klein RA, Hartmann R, Kushi Y, Egge H, Yu RK. O-Acetylated gangliosides in bovine buttermilk. Characterization of 7-O-acetyl, 9-O-acetyl, and 7,9-di-O-acetyl GD3. J. Biol. Chem. 1992;267:12632–12638. [PubMed] [Google Scholar]
- Rogers DC, Campbell CA, Stretton JL, Mackay KB. Correlation between motor impairment and infarct volume after permanent and transient middle cerebral artery occlusion in the rat. Stroke. 1997;28:2060–2065. doi: 10.1161/01.str.28.10.2060. discussion 2066. [DOI] [PubMed] [Google Scholar]
- Salloway S, Mermel LA, Seamans M, Aspinall GO, Nam Shin JE, Kurjanczyk LA, Penner JL. Miller-Fisher syndrome associated with Campylobacter jejuni bearing lipopolysaccharide molecules that mimic human ganglioside GD3. Infect. Immun. 1996;64:2945–2949. doi: 10.1128/iai.64.8.2945-2949.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanner MF. Python: a programming language for software integration and development. J. Mol. Graph. Model. 1999;17:57–61. [PubMed] [Google Scholar]
- Shoenfeld Y. The idiotypic network in autoimmunity: antibodies that bind antibodies that bind antibodies. Nat. Med. 2004;10:17–18. doi: 10.1038/nm0104-17. [DOI] [PubMed] [Google Scholar]
- Simon-Haldi M, Mantei N, Franke J, Voshol H, Schachner M. Identification of a peptide mimic of the L2/HNK-1 carbohydrate epitope. J. Neurochem. 2002;83:1380–1388. doi: 10.1046/j.1471-4159.2002.01247.x. [DOI] [PubMed] [Google Scholar]
- Svennerholm L. The gangliosides. J. Lipid Res. 1964;5:145–155. [PubMed] [Google Scholar]
- Taguchi K, Ren J, Utsunomiya I, Aoyagi H, Fujita N, Ariga T, Miyatake T, Yoshino H. Neurophysiological and immunohistochemical studies on Guillain-Barré syndrome with IgG anti-GalNAc-GD1a antibodies-effects on neuromuscular transmission. J. Neurol. Sci. 2004;225:91–98. doi: 10.1016/j.jns.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Usuki S, Sanchez J, Ariga T, Utsunomiya I, Taguchi K, Rivner MH, Yu RK. AIDP and CIDP having specific antibodies to the carbohydrate epitope (-NeuAcalpha2-8NeuAcalpha2-3Galbe-ta1-4Glc-) of gangliosides. J. Neurol. Sci. 2005;232:37–44. doi: 10.1016/j.jns.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Usuki S, Thompson SA, Rivner MH, Taguchi K, Shibata K, Ariga T, Yu RK. Molecular mimicry: sensitization of Lewis rats with Campylobacter jejuni lipopolysaccharides induces formation of antibody toward GD3 ganglioside. J. Neurosci. Res. 2006;83:274–284. doi: 10.1002/jnr.20717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usuki S, Taguchi K, Thompson SA, Chapman PB, Yu RK. Novel anti-idiotype antibody therapy for lipooligosaccharide-induced experimental autoimmune neuritis: use relevant to Guillain-Barré Syndrome. J. Neurosci. Res. 2009 doi: 10.1002/jnr.22330. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willers J, Lucchese A, Kanduc D, Ferrone S. Molecular mimicry of phage displayed peptides mimicking GD3 ganglioside. Peptides. 1999;20:1021–1026. doi: 10.1016/s0196-9781(99)00095-9. [DOI] [PubMed] [Google Scholar]
- Yamawaki M, Vasquez A, Ben Younes A, Yoshino H, Kanda T, Ariga T, Baumann N, Yu RK. Sensitization of Lewis rats with sulfoglucuronosyl paragloboside: electrophysiological and immunological studies of an animal model of peripheral neuropathy. J. Neurosci. Res. 1996;44:58–65. doi: 10.1002/(SICI)1097-4547(19960401)44:1<58::AID-JNR8>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Yan X, Evans SV, Kaminki MJ, Gillies SD, Reisfeld RA, Houghton AN, Chapman PB. Characterization of an Ig VH idiotope that results in specific homophilic binding and increased avidity for antigen. J. Immunol. 1996;157:1582–1588. [PubMed] [Google Scholar]
- Yu RK, Bieberich E, Xia T, Zeng G. Regulation of ganglioside biosynthesis in the nervous system. J. Lipid Res. 2004;45:783–793. doi: 10.1194/jlr.R300020-JLR200. [DOI] [PubMed] [Google Scholar]
- Yu RK, Usuki S, Ariga T. Ganglioside molecular mimicry and its pathological roles in Guillain-Barré syndrome and related diseases. Infect. Immun. 2006;74:6517–6527. doi: 10.1128/IAI.00967-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuki N. Ganglioside mimicry and peripheral nerve disease. Muscle Nerve. 2007;35:691–711. doi: 10.1002/mus.20762. [DOI] [PubMed] [Google Scholar]
- Yuki N, Yoshino H, Sato S, Miyatake T. Acute axonal polyneuropathy associated with anti-GM1 antibodies following Campylobacter enteritis. Neurology. 1990;40:1900–1902. doi: 10.1212/wnl.40.12.1900. [DOI] [PubMed] [Google Scholar]
- Yuki N, Taki T, Inagaki F, Kasama T, Takahashi M, Saito K, Handa S, Miyatake T. A bacterium lipopolysaccharide that elicits Guillain-Barré syndrome has a GM1 ganglioside-like structure. J. Exp. Med. 1993;178:1771–1775. doi: 10.1084/jem.178.5.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuki N, Susuki K, Koga M, et al. Carbohydrate mimicry between human ganglioside GM1 and Campylobacter jejuni lipooligosaccharide causes Guillain-Barré syndrome. Proc. Natl. Acad. Sci. U S A. 2004;101:11404–11409. doi: 10.1073/pnas.0402391101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.