

Abstract
Obesity is associated with a variety of disorders and is a significant health problem in developed countries. One factor controlling the level of adiposity is the differentiation of cells into adipocytes. Adipocyte differentiation requires expression of peroxisome proliferator-activated receptor γ (PPARγ), which is activated by ligands to regulate expression of genes involved in adipocyte differentiation. Although 15-deoxy-Δ(12,14)-prostaglandin (PG) J2 (15d-PGJ(2)) has long been known to be a potent activator of PPARγ, the importance of its synthesis in adipose tissue in vivo is not clear. The current study utilized mice deficient in cyclooxygenase-2 (COX-2) to examine the role of COX-2-derived PGs as in vivo modulators of adiposity. As compared with strain- and age-matched wild-type controls, the genetic deficiency of COX-2 resulted in a significant reduction in total body weight and percent body fat. Although there were no significant differences in food consumption between groups, COX-2-deficient mice showed increased metabolic activity. Epididymal adipose tissue from wild-type mice produced a significantly greater level of 15d-PGJ(2), as compared with adipose tissue isolated from mice deficient in COX-2. Furthermore, production of the precursor required for 15d-PGJ(2) formation, PGD(2), was also significantly reduced in COX-2-deficient adipose tissue. The expression of markers for differentiated adipocytes was significantly reduced in adipose tissue from COX-2-deficient mice, whereas preadipocyte marker expression was increased. Macrophage-dependent inflammation was also significantly reduced in adipose tissue of COX-2-deficient mice. These findings suggest that reduced adiposity in COX-2-deficient mice results from attenuated PPARγ ligand production and adipocyte differentiation.
Keywords: Adipose Tissue, Eicosanoid, Eicosanoids Function, Inflammation, Prostaglandins
Introduction
In developed countries, obesity is a significant health problem that is becoming more prevalent. The increase in adiposity, or the percentage of total body mass comprised of lipid stores, correlates with the development of a variety of disorders including, glucose intolerance, dyslipidemia, and hypertension (1). Recent evidence suggests that in addition to being an energy storage depot, adipose tissue may contribute to disease by producing adipokines, chemokines, and other bioactive substances (2). The level of adiposity is regulated by the amount of lipid taken into individual adipocytes and the number of adipocytes in the body. Adipocyte number is regulated by the differentiation of cells into adipocytes, and understanding this process is important for identifying novel methods for controlling obesity.
Adipocyte differentiation requires expression of the nuclear hormone receptor peroxisome proliferator-activated receptor (PPAR)2 γ (3, 4). When activated by its ligand, PPARγ functions as a transcription factor to induce numerous genes involved in adipogenesis. Identification of the endogenous ligand for PPARγ has been controversial, but treatment with the prostanoid 15-deoxy-Δ12,14-prostaglandin (PG) J2 (15d-PGJ2) has long been known to activate PPARγ and induce adipocyte differentiation following exogenous addition (5, 6). 15d-PGJ2 is derived from nonenzymatic conversion of PGJ2, which in turn is formed from nonenzymatic dehydration of PGD2 (7, 8). PGD2 is formed by PGD2 synthases utilizing the substrate PGH2, which is synthesized by the prostaglandin G/H synthases, also known as cyclooxygenase (COX)-1 and -2 (9). PGD2 synthase expression has been shown to increase with the differentiation of adipocytes in vitro, and disruption of expression of the synthase limits the complete differentiation of these cells (10).
There have been conflicting reports as to the significance of endogenous production of 15d-PGJ2 during adipocyte differentiation in vitro. One report has shown that despite an increase in COX-2 expression, the levels of 15d-PGJ2 detected in the medium of adipocyte cultures are not altered during differentiation (11). In contrast, another report examining this same cell type does show an increase in 15d-PGJ2 production during differentiation, and this increase in 15d-PGJ2 is attenuated by COX inhibition (12). Furthermore, this report shows that COX inhibition reduces expression of adipocyte-specific genes, an effect that is reversed by the addition of exogenous 15d-PGJ2 (12). Administration of the COX-2 inhibitor rofecoxib has also been shown to reduce adipose tissue mass development in mice without affecting the amount of diet consumed, although the PG responsible for these effects is not known (13). Recently, COX-2 has been shown to contribute to the differentiation of brown adipocytes in mice, following cold or adrenergic stimulation (14, 15). The current study utilized mice of different ages that were deficient in COX-2 to examine the role of COX-2-derived PGs as modulators of adipocyte differentiation in white adipose tissue.
EXPERIMENTAL PROCEDURES
Animals
COX-2-deficient mice (COX-2 (−/−)) and their matching wild-type littermate controls (COX-2 (+/+)) were a gift from R. Langenbach, National Institute of Environmental Health Sciences. These COX-2-deficient and wild-type control mice were produced by crossing the COX-2 heterozygous mutant line that has been maintained for more than 40 generations on a hybrid C57BL/6J × 129/Ola (C57/129) genetic background, as originally described (16). Male COX-2 (+/+) and COX-2 (−/−) mice were housed under barrier conditions with food (Harlan-Teklad Rodent Diet 2918) and water provided ad libitum. The body weight of the mice was measured at 4, 6, 9, 11, and 12 months of age. At the time points where a significant difference in weight was observed between wild-type and COX-2-deficient mice, additional studies were performed to determine percent body fat composition. Body fat composition of mice was determined by dual-energy x-ray absorptiometry with the Piximus densitometer (GE Medical Systems, Madison, WI). Indirect calorimetry was used to measure oxygen consumption in conscious mice by placing in a metabolic chamber. Oxygen levels were determined from desiccated air after siphoning the chamber using an oxygen analyzer (AEI Technologies, Pittsburgh, PA). The cumulative oxygen consumption of each mouse was recorded and the plateau level was used for calculating oxygen consumption. Food intake was determined by weighing pelleted diet daily for 3 months prior to the termination of the study at 12 months of age. All studies were conducted under the approval of the University of Kentucky Institutional Animal Care and Use Committee and conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85-23).
Tissue Collection
Mice were euthanized by intraperitoneal injection of a lethal dose of ketamine (100 mg/kg) and xylazine (10 mg/kg), followed by thoracotomy. Blood was collected by cardiac puncture for serum preparation. Non-fasting glucose, total serum cholesterol, and triglyceride levels were determined from serum obtained at completion of the studies using individual assay kits (Wako Chemicals) for each parameter. Epididymal fat tissues were frozen in liquid nitrogen and stored at −80 °C for measuring mRNA expression by real time PCR and protein expression by immunoblot.
Mouse Primary Adipocyte Culture
Primary culture of epididymal adipose tissue was performed as described previously (17, 18). Briefly, ∼0.2 g of the epididymal adipose tissue from each mouse was rinsed with DMEM:F-12, minced to achieve pieces of ∼0.5 mm in diameter, suspended in collagen matrix, and plated in 1-ml volumes in a six-well plate (18). Following collagen solidification at 37 °C, additional DMEM:F-12 (2 ml) was added to each well of the cultures.
Real Time Polymerase Chain Reaction
Quantitative gene expression was performed by two-step RT-PCR (reverse transcription-polymerase chain reaction) using Superscript II (Invitrogen) and fluorogenic 5′ nuclease chemistry (TaqMan). The primer/probe real time PCR assays for PPARγ, LPL, pref-1, resistin, C/EBPβ, C/EBPδ, CD68, MCP-1, TNFα, and adiponectin were from Applied Biosystems (Foster City, CA). mRNA expression was determined using the relative standard curve method and normalized to the housekeeping gene hypoxanthine-phosphoribosyltransferase.
Gel Electrophoresis and Immunoblot
Protein was isolated by standard protein lysis buffer containing protease inhibitor and quantified using a BCA protein assay kit (Pierce). Ten μg of protein was loaded into each well and run on a 4–12% BisTris gel with MES buffer (Invitrogen), transferred to a PVDF membrane, and blocked in 5% nonfat milk for 1 h prior to overnight primary antibody incubation (anti-adipocyte fatty acid-binding protein (A-FABP) from R&D Systems, anti-tumor necrosis factor α (TNFα) from Sigma). An antibody against calnexin (Assay Designs) or β-actin (Sigma) was used as an internal control. For serum samples, 2 μl of serum was diluted with 18 μl of reducing sample buffer (Invitrogen), heated at 70 °C for 10 min, and run for immunoblot as described above.
Prostaglandin Measurements
The 24-h medium collected from the primary cultures of the adipose tissue was used to measure PGD2, PGE2, 6-keto-PGF1α, or PGF2α with immunological assay kits from Cayman Chemical. The analysis of serum or medium from primary adipose tissue cultures for 15d-PGJ2 utilized an immunological assay kit from Assay Designs.
Histological Analysis
Epididymal adipose tissue was fixed in 10% neutral buffered formalin overnight and stored in 70% ethanol at 4 °C until further use. Adipose tissue was processed in a dehydrating ethanol gradient, followed by xylene incubation and paraffin embedding. Serial 8-μm sections were used for hematoxylin and eosin (H&E) staining, or for immunohistochemical analysis for Mac-3 (primary antibody from BD Pharmingen). Antibody detection was performed with Vectastain Elite ABC kit (Vector Laboratories) and diaminobenzidine reagent using the manufacturer's instructions. Immunohistochemical sections were counterstained with hematoxylin.
Statistical Analyses
For analysis of adipose tissue directly from the mice, one tissue sample isolated from one individual mouse was considered n of 1. For analysis of culture medium from primary adipocyte cultures, a single sample taken from one well of the culture dish was considered n of 1. For analysis of serum, one sample from one individual mouse was considered n of 1. A minimum n of 6 was analyzed for each experiment and all experiments were repeated a minimum of two separate times with similar results. Significant differences among groups were determined using unpaired two-tailed t test, with differences being considered statistically significant at p < 0.05.
RESULTS
Genetic Deficiency of COX-2 Reduces Adiposity in Mice
We examined the effect of COX-2 deficiency on total body weight by comparing the weight of COX-2-deficient and matched wild-type littermate control mice at multiple stages of development. A comparison of total body weight at 2, 4, and 6 months of age did not detect a significant difference between wild-type and COX-2-deficient mice (Fig. 1A). In contrast, at 9 months of age, mice deficient in COX-2 showed a significantly reduced total body weight, as compared with wild-type control mice (Fig. 1A). The total body weights of wild-type and COX-2-deficient mice remained significantly different at both 11 and 12 months of age (Fig. 1A).
FIGURE 1.

Effect of COX-2 deficiency on the level of adiposity and oxygen consumption of mice. A, average body weight of wild-type and COX-2-deficient mice at 2, 4, 6, 9, 11, and 12 months of age. Percent body fat of wild-type and COX-2-deficient mice at 9 months of age (B), 11 months of age (C), and 12 months of age (D), as determined by x-ray absorptiometry. E, oxygen consumption of 1-year-old mice determined by indirect calorimetry. F, food intake was compared between COX-2 wild-type and COX-2-deficient mice. The mean + S.E. is shown for each group. p values were determined using an unpaired two-tailed t test. n ≥ 10; ** indicates p < 0.01; *** indicates p < 0.001.
With the observation of reduced total body weight in aged COX-2-deficient mice, we determined whether alterations in percent of body fat accounted for the observed reduction in body weight. Mice deficient in COX-2 showed significant reductions in total body fat at 9, 11, and 12 months of age when compared with wild-type control mice (Fig. 1, B–D). The measurement of oxygen consumption with use of a metabolic chamber is a method of indirectly determining metabolic activity (19). In mice that were 1 year of age, metabolic activity as determined by oxygen consumption was significantly greater in COX-2-deficient mice, as compared with wild-type controls (Fig. 1E). There was no difference in food consumption that could account for the reduced body weight and percent body fat in COX-2-deficient mice (Fig. 1F). Furthermore, there were no significant differences in the levels of serum glucose, serum triglyceride, or total serum cholesterol between COX-2 wild-type and COX-2-deficient mice (Table 1). Therefore, in aged mice, the deficiency of COX-2 results in a significant reduction in total body weight and percent body fat that is associated with increased metabolic activity.
TABLE 1.
Non-fasting serum glucose, serum triglyceride, and total serum cholesterol in COX-2 (+/+) and COX-2 (−/−) mice
Data represent mean ± S.E. (n ≥ 10).
| Parameter | Serum level |
|
|---|---|---|
| COX-2 (+/+) mice | COX-2 (−/−) mice | |
| mg/dl | ||
| Glucose | 355 ± 11 | 328 ± 12 |
| Triglyceride | 65 ± 7 | 53 ± 8 |
| Total cholesterol | 173 ± 6 | 153 ± 12 |
The Deficiency of COX-2 Attenuates Adipose Production of PGD2 and 15d-PGJ2
Although exogenous addition of different PGs has been shown to either promote or inhibit adipogenesis in vitro, the physiological importance of PGs produced by adipose tissue is less clear. To determine the PG(s) produced in adipose tissue that are dependent on the activity of COX-2, we compared the levels of different PGs produced by explanted epididymal adipose tissue isolated from COX-2 wild-type and COX-2-deficient mice. Following 24 h of culture, adipose tissue from COX-2 wild-type mice produced a significantly greater level of the PPARγ ligand 15d-PGJ2, as compared with epididymal adipose tissue isolated from mice deficient in COX-2 (Fig. 2A). With the finding of reduced adipose tissue production of 15d-PGJ2, we compared serum levels of this prostanoid in wild-type and COX-2-deficient mice. However, there was no significant difference in serum levels of 15d-PGJ2 between wild-type and COX-2-deficient mice (Fig. 2B). Analysis of the explant culture medium did show a significant reduction in PGD2 production from adipose tissue obtained from COX-2-deficient mice, as compared with that from wild-type mice (Fig. 2C).
FIGURE 2.

COX-2 deficiency reduces 15d-PGJ2 and PGD2 production from adipose tissue explants. Levels of PGs were detected in the culture media (A, C, E, F, and G) or serum (B) from adipose tissue explants isolated from wild-type or COX-2-deficient mice. Epididymal adipose tissue from wild-type or COX-2-deficient mice was analyzed for COX-1 mRNA expression (D) by real time PCR. The mean + S.E. is shown for each group. p values were determined using an unpaired two-tailed t test. n ≥ 10; * indicates p < 0.05.
The deficiency of COX-2 has been shown to result in a compensatory increase in COX-1 expression in various tissues of mice, and has been shown to result in a paradoxical increase in COX-1-dependent PG production (20–22). However, in the current study, there was no significant effect of COX-2 deficiency on adipose expression of COX-1 mRNA (Fig. 2D). In addition, there was no significant difference between adipose tissue from COX-2 wild-type and COX-2-deficient mice in the levels of PGE2, PGF2α, or 6-keto-PGF1α (PGI2 metabolite) detected in medium following 24 h of culture (Fig. 2, E–G). These findings indicate that the deficiency of COX-2 selectively attenuated adipose tissue production of the PPARγ ligand 15d-PGJ2 and of its precursor PGD2, without resulting in a compensatory increase in COX-1-dependent PG production.
The Deficiency of COX-2 Reduces Expression of Markers Characteristic of Adipocyte Differentiation
With 15d-PGJ2 known to induce adipocyte differentiation and our finding of COX-2-dependent 15d-PGJ2 production, we examined the effect of COX-2 deficiency on expression of markers of adipose differentiation. Analysis of A-FABP expression, a marker of fully differentiated mature adipocytes, detected a significantly lower level in the epididymal adipose tissue of COX-2-deficient mice, as compared with COX-2 wild-type mice (Fig. 3, A and B). Although A-FABP is primarily produced by adipocytes, it has been shown to be released from adipose tissue and to enter the circulation (23). In the current report, we determined that as compared with wild-type controls, A-FABP levels were significantly reduced in the serum of COX-2-deficient mice (Fig. 3C). Furthermore, examination of medium from explant cultures of adipose tissue isolated from COX-2-deficient mice also showed significant reduction in A-FABP levels, as compared with medium from cultured adipose tissue of wild-type mice (Fig. 3D). Therefore, the deficiency of COX-2 results in reduced production of A-FABP by epididymal adipose tissue and reduced circulating levels of A-FABP in the mice.
FIGURE 3.

The deficiency of COX-2 attenuates adipocyte fatty acid-binding protein expression. A, representative immunoblot of A-FABP expression in adipose tissue of wild-type and COX-2-deficient mice. Calnexin is used as a loading control. B, densitometry quantitation of the immunoblot bands of A-FABP expression in adipose tissue isolated from wild-type and COX-2-deficient mice. C, densitometry quantitation of A-FABP expression from immunoblots in serum isolated from wild-type and COX-2-deficient mice. D, quantitation of A-FABP expression in culture medium from adipose tissue explants from wild-type and COX-2-deficient mice determined by densitometry of immunoblots. The mean + S.E. is shown for each group. p values were determined using an unpaired two-tailed t test. n ≥ 10; ** indicates p < 0.01; * indicates p < 0.05.
We examined mRNA expression of additional genes that are induced or suppressed during adipocyte differentiation. The expression of PPARγ is a characteristic of the adipocyte phenotype and the inhibition of adipocyte differentiation reduces PPARγ expression (24). Our current findings indicate that PPARγ mRNA expression is significantly reduced in epididymal adipose tissue from mice deficient in COX-2 (Fig. 4A). Lipoprotein lipase (LPL) is expressed early in adipocyte differentiation and expression is induced by PPARγ agonists (25). As compared with wild-type mice, mRNA expression of LPL was reduced in the epididymal adipose tissue from COX-2-deficient mice (Fig. 4B). Preadipocyte factor-1 (pref-1) is a marker of cells prior to the complete differentiation into adipocytes (26). Epididymal adipose tissue of COX-2-deficient mice expressed a significantly greater level of pref-1 than adipose from COX-2 expressing wild-type mice (Fig. 4C). In mice, the expression of resistin by adipose tissue has been shown to be reduced by PPARγ ligands (27). In the current report, we found that in COX-2-deficient mice, epididymal adipose tissue expressed a significantly greater level of resistin than that from COX-2 wild-type mice (Fig. 4D). Therefore, the altered gene expression observed in adipose tissue of COX-2-deficient mice is consistent with attenuated differentiation resulting from reduced ligand-dependent activation of PPARγ.
FIGURE 4.

The deficiency of COX-2 alters expression of genes characteristic of reduced adipose differentiation. mRNA expression of peroxisome proliferator-activated receptor γ (A), lipoprotein lipase (B), preadipocyte factor-1 (C), and resistin (D) in adipose tissue of wild-type and COX-2-deficient mice as determined by quantitative PCR. The mean + S.E. is shown for each group. p values were determined using an unpaired two-tailed t test. n ≥ 8; * indicates p < 0.05; ** indicates p < 0.01; *** indicates p < 0.001.
In addition to 15d-PGJ2, PGI2 has also been shown to induce adipocyte differentiation, and COX-2 is a potential source of PGI2 (28). However, unlike 15d-PGJ2, PGI2 produces this effect by activating a G-protein-coupled receptor that leads to increased expression of the transcription factors CCAAT/enhancer-binding protein (C/EBP) β and C/EBPδ (29). In the current report, the deficiency of COX-2 did not significantly alter mRNA expression of either C/EBPβ or C/EBPδ in epididymal adipose tissue (Fig. 5, A and B). These findings further suggest that attenuated adipose differentiation resulting from the deficiency of COX-2 may not result from the reduced production of PGI2.
FIGURE 5.
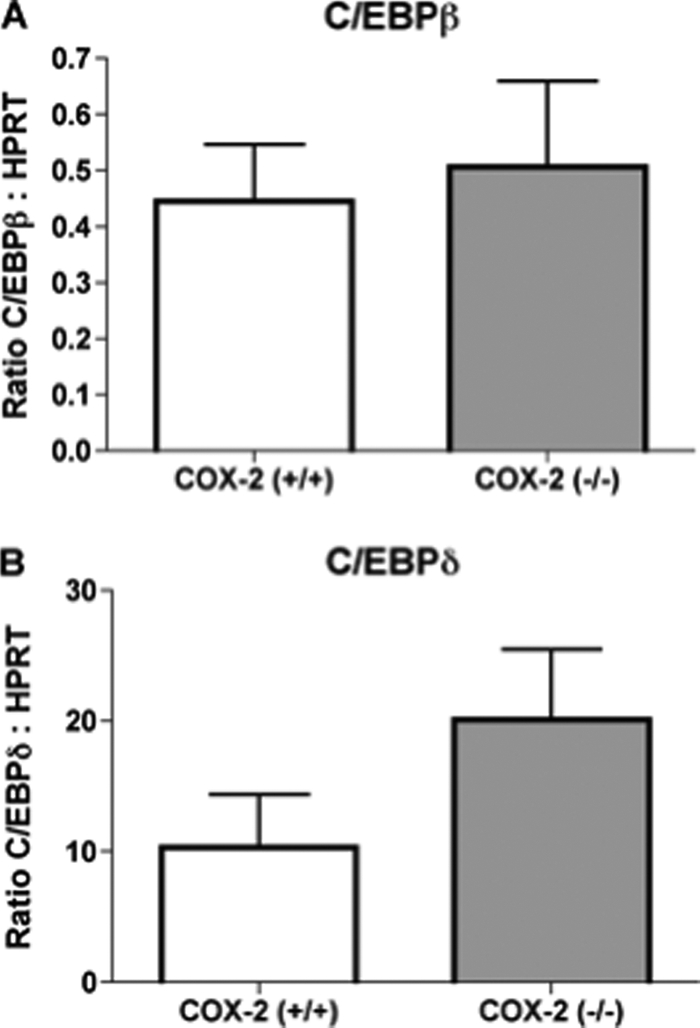
The deficiency of COX-2 does not significantly alter gene expression of CCAAT/enhancer-binding protein β or δ in adipose tissue. Real time PCR quantitation of mRNA expression of (A) C/EBPβ or (B) C/EBPδ in adipose tissue of wild-type and COX-2-deficient mice, n = 10. The mean + S.E. is shown for each group.
COX-2 Deficiency Attenuates Adipose Tissue Inflammation
Obesity is associated with increased inflammation within adipose tissue. The current study examined the effect of COX-2 deficiency on the levels of inflammatory markers produced within epididymal adipose tissue. Histological analysis of epididymal adipose tissue from wild-type mice showed numerous areas that were infiltrated by inflammatory cells (Fig. 6A), without evidence of similar inflammation in adipose tissue from COX-2-deficient mice (Fig. 6B). The size of the adipocytes was determined by measuring the average diameter of the cells in histological sections of epididymal adipose tissue obtained from wild-type and COX-2-deficient mice. As shown in the inset of Fig. 6B, the average diameter of adipocytes in adipose tissue of COX-2-deficient mice was significantly reduced, as compared with that of wild-type mice. Immunohistochemical analysis of adipose tissue from wild-type mice detected significant numbers of Mac-3-positive macrophages surrounding individual adipocytes (Fig. 6C), which were absent in adipose tissue from COX-2-deficient mice (Fig. 6D). Comparison of the macrophage marker CD68 shows a significantly greater level of mRNA expression in adipose tissue from COX-2 expressing mice, as compared with COX-2-deficient mice (Fig. 6E). There was also a reduction in the mRNA expression of the macrophage chemokine monocyte chemoattractant protein-1 (MCP-1) (Fig. 6F). Therefore, the expression of COX-2 was associated with increased macrophage infiltration into epididymal adipose tissue.
FIGURE 6.

Attenuated expression of inflammatory markers in adipose tissue of COX-2-deficient mice. Representative sections of hematoxylin and eosin-stained epididymal adipose tissue from wild-type (A) and COX-2 (B)-deficient mice (B, inset), Histological sections were used for determining average adipocyte diameter (μm). C and D, immunohistochemistry with the macrophage marker Mac-3 of representative sections of epididymal adipose tissue from wild-type (C) and COX-2 (D)-deficient mice. Brown staining indicates detection of Mac-3 antibody by diaminobenzidine reagent. Sections are counterstained with hematoxylin (blue). Comparison of mRNA expression of CD68 (E) and MCP-1 (F) in adipose tissue of wild-type and COX-2-deficient mice as determined by quantitative PCR. The mean + S.E. is shown for each group. n ≥ 8; * indicates p < 0.05; ** indicates p < 0.01.
The activation of macrophages results in the increased production of TNFα, an inflammatory cytokine known to alter adipocyte function. The expression of TNFα mRNA was significantly greater in epididymal adipose tissue of wild-type mice, as compared with COX-2-deficient mice (Fig. 7A). Quantitation of TNFα protein expression in epididymal adipose tissue (Fig. 7B) or serum (Fig. 7C) by Western blot detected a similar decrease in expression resulting from the deficiency of COX-2. Adiponectin is an adipocyte-derived protein with anti-inflammatory properties whose expression is attenuated by TNFα (30). We found that expression of adiponectin mRNA was significantly increased in epididymal adipose from COX-2-deficient mice (Fig. 7D). Therefore, mice deficient in COX-2 show altered expression of markers consistent with reduced adipose tissue inflammation.
FIGURE 7.

Altered expression of pro- and anti-inflammatory cytokine production in adipose tissue of COX-2-deficient mice. A, TNFα gene expression in epididymal adipose tissue of wild-type and COX-2-deficient mice. B, densitometry quantitation determined from immunoblot bands of TNFα protein expression. C, TNFα protein levels in serum of wild-type and COX-2-deficient mice determined from immunoblot densitometry. D, adiponectin mRNA expression in epididymal adipose tissue of wild-type and COX-2-deficient mice. The mean + S.E. is shown for each group. n ≥ 8; * indicates p < 0.05; ** indicates p < 0.01; *** indicates p < 0.001.
DISCUSSION
Our current report shows that in aged mice a significant reduction in total body mass and adiposity resulted from the deficiency of COX-2. The reduced adiposity in COX-2-deficient mice occurred without a reduction in food intake and was associated with increased metabolic activity as determined by oxygen consumption. These findings suggest that prostanoid synthesis dependent on the activity of COX-2 is important for in vivo regulation of adipose tissue levels. Although COX-2 is responsible for production of a variety of different prostanoids, those which have been shown to produce the greatest effects on adipocytes are the J-series PGs. Of the J-series PGs, 15d-PGJ2 is the most potent inducer of adipocyte differentiation in vitro, where exogenous addition to the culture converts fibroblasts into adipocytes (5, 6). 15d-PGJ2 is formed from the spontaneous dehydration of the cyclopentenone prostaglandin PGJ2, which in turn is formed by nonenzymatic dehydration of PGD2 (31). PGD2 formation requires the activity of a PGD2 synthase utilizing PGH2, which is produced by the COX enzymes (9). Our current findings show that adipose tissue isolated from COX-2-deficient mice produced significantly lower levels of both PGD2 and 15d-PGJ2. However, PGD2 has been shown to not affect adipocyte differentiation at physiologic concentrations (32). Therefore, the reduced adiposity that we observe in COX-2-deficient mice may result from reduced adipocyte differentiation as a consequence of attenuated formation of 15d-PGJ2.
A well described mechanism for the adipogenic effects of 15d-PGJ2 results from its activity as a ligand of PPARγ (5, 6). Ligand activation of PPARγ induces the expression of genes characteristic of the adipocyte phenotype. A-FABP expression is increased by PPARγ ligand-induced maturation of adipocytes and is a well characterized marker of fully differentiated adipocytes (29, 33). In the current studies, we compared the expression of A-FABP in epididymal adipose tissue from wild-type and COX-2-deficient mice. The expression level of A-FABP was significantly lower in adipose tissue from COX-2-deficient, as compared with wild-type controls. Although traditionally considered to function as a cytosolic protein, cultured adipocytes secrete significant levels of A-FABP into the medium (23). Therefore, we also compared the levels of A-FABP in culture medium from explanted epididymal tissue of wild-type and COX-2-deficient mice. Adipose tissue that was deficient in COX-2 secreted significantly lower levels of A-FABP than COX-2 expressing tissue. A-FABP that is released from adipose tissue has been shown to be detected in the circulation (23). Thus, in the current report, we compared serum levels of A-FABP between wild-type and COX-2-deficient mice. There was a significantly greater level of A-FABP in the serum of mice expressing COX-2. These findings suggest that the deficiency of COX-2 significantly attenuates adipose expression and secretion of A-FABP, thereby resulting in reduced levels in the circulation.
The expression and activation of PPARγ is a key event in determining the adipocyte phenotype. PPARγ is the only known factor with the capacity to induce the differentiation of fibroblasts into adipocytes in the absence of forced expression of any other gene (34). Furthermore, in mature adipocytes the inactivation of PPARγ produces a regression of the differentiation process (35). Thus, increased expression of PPARγ is characteristic of differentiated adipocytes, whereas attenuated expression represents a pre-adipocyte phenotype. In the current report, we compared the levels of adipose tissue expression of PPARγ between COX-2 expressing and COX-2-deficient mice. As compared with wild-type controls, PPARγ mRNA expression was significantly attenuated in adipose tissue of COX-2-deficient mice. The expression of LPL is increased by PPARγ ligands and is an early marker of differentiated adipocytes (25). In contrast, pref-1 expression decreases following adipocyte differentiation and PPARγ ligands are known to down-regulate resistin expression (26, 27). Our current findings show that although the deficiency of COX-2 significantly reduces expression of LPL, the expression of pref-1 and resistin are significantly greater in adipose tissue from COX-2-deficient mice. These findings of increased expression of preadipocyte markers and attenuated expression of adipocyte markers further suggest that the deficiency of COX-2 results in attenuated adipose differentiation.
In addition to the effects of 15d-PGJ2, other prostanoids may enhance adipocyte differentiation through mechanisms that are independent of PPARγ. The majority of prostanoids produce their biological effects through their respective G-protein-coupled receptors that alter the intracellular levels of cAMP. Of the G-protein-coupled prostanoid receptors, it is primarily the PGI2 receptor that has been shown to promote adipocyte differentiation (28). PGI2 receptor activation enhances differentiation of cultured preadipocytes, in part by increasing intracellular cAMP levels (36). This increased cAMP induces expression of C/EBPβ and C/EBPδ, which is an early step in the processes leading to adipocyte differentiation (29, 37). Our current findings indicate that the deficiency of COX-2 did not affect adipose tissue production of PGI2, as determined by detection of the stable PGI2 metabolite 6-keto-PGF1α. Furthermore, we could identify no significant effect of COX-2 deficiency on mRNA expression of C/EBPβ and C/EBPδ. Therefore, the reduced adiposity that we observed in COX-2-deficient mice is not associated with attenuated production of PGI2 or altered expression of adipogenic factors known to be induced by this prostanoid.
In addition to promoting differentiation, prostanoids have also been shown to inhibit development of the adipocyte phenotype. PGE2 and PGF2α acting through their respective G-protein-coupled receptors have been shown to inhibit adipocyte differentiation, which suggests that reduced adiposity may result from increasing levels of these prostanoids (32, 38). The deficiency of COX-2 has the potential to alter the prostanoids being produced, as the result of a compensatory increase in COX-1 expression, or increased substrate availability for COX-1-dependent prostanoid synthesis (20, 22, 39). Thus, in the current report, we examined the effect of COX-2 deficiency on adipose tissue expression of COX-1 mRNA. The deficiency of COX-2 did not significantly affect mRNA expression of COX-1 in adipose tissue. Furthermore, there was no significant difference between wild-type and COX-2-deficient mice in the levels of PGE2 or PGF2α produced by adipose tissue. These findings suggest that the reduced adiposity that we observed in COX-2-deficient mice was not the result of increased COX-1-dependent production of PGE2 or PGF2α.
Adipose tissue inflammation is considered a causative factor contributing to the development of a variety of diseases associated with obesity. The severity of adipose tissue inflammation resulting from macrophage infiltration correlates with the level of adiposity in both humans and mice (40, 41). These previous reports would suggest that our current finding of reduced adiposity in COX-2-deficient mice would be associated with attenuated adipose inflammation. However, 15d-PGJ2 has been shown to produce anti-inflammatory effects (42), suggesting that reduced production of 15d-PGJ2 resulting from COX-2 deficiency could be expected to increase adipose tissue inflammation. Thus, we compared the level of inflammatory markers expressed by adipose tissue from COX-2-deficient and wild-type mice. mRNA expression of the macrophage marker CD68 was significantly reduced in epididymal adipose tissue isolated from COX-2-deficient mice, as compared with wild-type controls. Immunohistochemistry detected abundant expression of Mac-3-positive cells within the adipose tissue of wild-type mice that was absent in COX-2-deficient adipose tissue. mRNA expression of the macrophage chemokine MCP-1 was also significantly reduced in adipose tissue from COX-2-deficient mice. These findings indicate that the attenuated adiposity that occurs in COX-2-deficient mice is associated with reduced macrophage recruitment and infiltration. Furthermore, the reduced adiposity that we observed in mice with attenuated production of 15d-PGJ2 suggests that any potential anti-inflammatory effects of this prostanoid do not significantly influence adipose inflammation.
15d-PGJ2 has been shown to be synthesized in mice following induction of an inflammatory response (43). Similar production of 15d-PGJ2 by inflammatory cells within inflamed adipose tissue would have the potential to enhance adipocyte differentiation. Further studies are required to determine the importance of the influence on adipose inflammation by COX-2 as a potential mechanism in the regulation of adipocyte differentiation.
Chronic inhibition of COX-2 has recently been shown to reduce macrophage-dependent adipose tissue inflammation in a rat model of obesity (44). COX-2 inhibition also increases adipose tissue expression of the anti-inflammatory adipokine adiponectin in a rat model of chronic inflammation (45). Our current findings show that as compared with wild-type controls, there is a significant increase in adiponectin mRNA expression in adipose tissue of COX-2-deficient mice. Adiponectin expression is known to be suppressed by pro-inflammatory cytokines such as TNFα (30). In the current report, TNFα mRNA expression was significantly attenuated in adipose tissue from COX-2-deficient mice. Previous studies have shown that macrophages are responsible for the majority of TNFα produced by adipose tissue of mice (40). Therefore, the enhanced gene expression of adiponectin that occurred in COX-2-deficient adipose tissue may result from reduced macrophage-dependent inflammation and attenuated production of TNFα.
Aging is associated with an increase in systemic levels of inflammatory mediators, and in mice, aging has been shown to significantly increase the level of adipose tissue inflammation (46, 47). As compared with young mice, the adipose tissue of aged mice expresses a significantly greater level of COX-2 (47). Our current studies did not identify a significant effect of COX-2 deficiency on body weight of mice at 2, 4, or 6 months of age. However, at 9 months of age, COX-2-deficient mice showed a reduction in total body weight and percent body fat, as compared with the wild-type controls. Furthermore, this effect on adiposity was similarly observed in mice that were either 11 or 12 months of age. Therefore, the contribution of COX-2 to increasing adiposity may primarily occur during the enhanced adipose tissue inflammation that is associated with aging.
There are numerous reports of conflicting findings that describe the roles of the individual COX isoforms in regulating adipogenesis. In cultured adipocytes, nonselective COX inhibitors and COX-2-selective inhibitors are reported to decrease adipocyte differentiation, whereas COX-1-selective inhibition has been shown to not alter differentiation (12, 48). The inhibition of either COX isoform has also been reported to enhance adipocyte differentiation (49). Recently, studies using antisense to attenuate endogenous COX-2 expression in cultured adipocytes suggest that COX-2 activity produces anti-adipogenic prostanoids, whereas pro-adipogenic prostanoids are derived from the activity of COX-1 (50). Thus, these findings conflict with some of the previous adipocyte culture studies that used pharmacological COX inhibition and with our current findings of reduced adiposity in COX-2-deficient mice. The basis for these divergent findings is not clear but may include complications resulting from comparing effects obtained with adipocyte cultures to those obtained from the mixed cell type population found in adipose tissue, particularly during inflammatory states.
In contrast to some of the previous reports utilizing adipocyte cultures, our current in vivo findings suggest that COX-2 functions to increase adipose tissue differentiation. Our findings are supported by previous studies also suggesting that COX-2 contributes to increased adiposity in mice. In mice on a high fat and high sugar diet, the deficiency of COX-2 significantly reduces gonadal fat pad weight, although this same report did show increased weight in mice heterozygous for COX-2 (51). In a separate study of mice on a high-fat diet, chronic administration of the COX-2-selective inhibitor rofecoxib significantly reduces total body weight and adipose tissue weight, without affecting the amount of diet consumed (13). Thus, these two reports indicate that the effect of COX-2 deficiency or COX-2 inhibition on reducing adiposity is observed after diet-induced weight gain (13, 51). Our current findings did not identify an effect of COX-2 deficiency in mice at 6 months of age or younger, suggesting that the role of COX-2 in contributing to adiposity may occur primarily during the increase in body weight associated with aging.
Footnotes
- PPAR
- peroxisome proliferator-activated receptor
- 15d-PGJ(2)
- 15-deoxy-Δ(12,14)-prostaglandin (PG) J2
- PG
- prostaglandin
- COX
- cyclooxygenase-2
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- A-FABP
- adipocyte fatty acid-binding protein
- pref-1
- preadipocyte factor-1
- C/EBP
- CCAAT/enhancer-binding protein
- MCP-1
- monocyte chemoattractant protein-1
- LPL
- lipoprotein lipase.
REFERENCES
- 1. Janssen I., Katzmarzyk P. T., Ross R. (2002) Arch. Intern. Med. 162, 2074–2079 [DOI] [PubMed] [Google Scholar]
- 2. Kershaw E. E., Flier J. S. (2004) J. Clin. Endocrinol. Metab. 89, 2548–2556 [DOI] [PubMed] [Google Scholar]
- 3. Rosen E. D., Sarraf P., Troy A. E., Bradwin G., Moore K., Milstone D. S., Spiegelman B. M., Mortensen R. M. (1999) Mol. Cell. 4, 611–617 [DOI] [PubMed] [Google Scholar]
- 4. Barak Y., Nelson M. C., Ong E. S., Jones Y. Z., Ruiz-Lozano P., Chien K. R., Koder A., Evans R. M. (1999) Mol. Cell. 4, 585–595 [DOI] [PubMed] [Google Scholar]
- 5. Forman B. M., Tontonoz P., Chen J., Brun R. P., Spiegelman B. M., Evans R. M. (1995) Cell 83, 803–812 [DOI] [PubMed] [Google Scholar]
- 6. Kliewer S. A., Lenhard J. M., Willson T. M., Patel I., Morris D. C., Lehmann J. M. (1995) Cell 83, 813–819 [DOI] [PubMed] [Google Scholar]
- 7. Scher J. U., Pillinger M. H. (2005) Clin. Immunol. 114, 100–109 [DOI] [PubMed] [Google Scholar]
- 8. Shibata T., Kondo M., Osawa T., Shibata N., Kobayashi M., Uchida K. (2002) J. Biol. Chem. 277, 10459–10466 [DOI] [PubMed] [Google Scholar]
- 9. Smith W. L., Garavito R. M., DeWitt D. L. (1996) J. Biol. Chem. 271, 33157–33160 [DOI] [PubMed] [Google Scholar]
- 10. Fujimori K., Aritake K., Urade Y. (2007) J. Biol. Chem. 282, 18458–18466 [DOI] [PubMed] [Google Scholar]
- 11. Bell-Parikh L. C., Ide T., Lawson J. A., McNamara P., Reilly M., FitzGerald G. A. (2003) J. Clin. Invest. 112, 945–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mazid M. A., Chowdhury A. A., Nagao K., Nishimura K., Jisaka M., Nagaya T., Yokota K. (2006) FEBS Lett. 580, 6885–6890 [DOI] [PubMed] [Google Scholar]
- 13. Lijnen H. R., Van Hoef B., Lu H. R., Gallacher D. J. (2008) Thromb. Haemost. 100, 338–342 [PubMed] [Google Scholar]
- 14. Madsen L., Pedersen L. M., Lillefosse H. H., Fjaere E., Bronstad I., Hao Q., Petersen R. K., Hallenborg P., Ma T., De Matteis R., Araujo P., Mercader J., Bonet M. L., Hansen J. B., Cannon B., Nedergaard J., Wang J., Cinti S., Voshol P., Døskeland S. O., Kristiansen K. (2010) PLoS One 5, e11391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vegiopoulos A., Müller-Decker K., Strzoda D., Schmitt I., Chichelnitskiy E., Ostertag A., Berriel Diaz M., Rozman J., Hrabe de Angelis M., Nüsing R. M., Meyer C. W., Wahli W., Klingenspor M., Herzig S. (2010) Science 328, 1158–1161 [DOI] [PubMed] [Google Scholar]
- 16. Morham S. G., Langenbach R., Loftin C. D., Tiano H. F., Vouloumanos N., Jennette J. C., Mahler J. F., Kluckman K. D., Ledford A., Lee C. A., Smithies O. (1995) Cell 83, 473–482 [DOI] [PubMed] [Google Scholar]
- 17. Sonoda E., Aoki S., Uchihashi K., Soejima H., Kanaji S., Izuhara K., Satoh S., Fujitani N., Sugihara H., Toda S. (2008) Endocrinology 149, 4794–4798 [DOI] [PubMed] [Google Scholar]
- 18. Sugihara H., Yonemitsu N., Toda S., Miyabara S., Funatsumaru S., Matsumoto T. (1988) J. Lipid Res. 29, 691–697 [PubMed] [Google Scholar]
- 19. Even P. C., Mokhtarian A., Pele A. (1994) Neurosci. Biobehav. Rev. 18, 435–447 [DOI] [PubMed] [Google Scholar]
- 20. Wang H., Ma W. G., Tejada L., Zhang H., Morrow J. D., Das S. K., Dey S. K. (2004) J. Biol. Chem. 279, 10649–10658 [DOI] [PubMed] [Google Scholar]
- 21. Bosetti F., Langenbach R., Weerasinghe G. R. (2004) J. Neurochem. 91, 1389–1397 [DOI] [PubMed] [Google Scholar]
- 22. Kirtikara K., Morham S. G., Raghow R., Laulederkind S. J., Kanekura T., Goorha S., Ballou L. R. (1998) J. Exp. Med. 187, 517–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xu A., Wang Y., Xu J. Y., Stejskal D., Tam S., Zhang J., Wat N. M., Wong W. K., Lam K. S. (2006) Clin. Chem. 52, 405–413 [DOI] [PubMed] [Google Scholar]
- 24. Liu L., Clipstone N. A. (2007) J. Cell. Biochem. 100, 161–173 [DOI] [PubMed] [Google Scholar]
- 25. Kageyama H., Hirano T., Okada K., Ebara T., Kageyama A., Murakami T., Shioda S., Adachi M. (2003) Biochem. Biophys. Res. Commun. 305, 22–27 [DOI] [PubMed] [Google Scholar]
- 26. Smas C. M., Sul H. S. (1993) Cell 73, 725–734 [DOI] [PubMed] [Google Scholar]
- 27. Hartman H. B., Hu X., Tyler K. X., Dalal C. K., Lazar M. A. (2002) J. Biol. Chem. 277, 19754–19761 [DOI] [PubMed] [Google Scholar]
- 28. Négrel R. (1999) Prostaglandins Leukot. Essent. Fatty Acids 60, 383–386 [DOI] [PubMed] [Google Scholar]
- 29. Aubert J., Saint-Marc P., Belmonte N., Dani C., Négrel R., Ailhaud G. (2000) Mol. Cell. Endocrinol. 160, 149–156 [DOI] [PubMed] [Google Scholar]
- 30. Maeda N., Takahashi M., Funahashi T., Kihara S., Nishizawa H., Kishida K., Nagaretani H., Matsuda M., Komuro R., Ouchi N., Kuriyama H., Hotta K., Nakamura T., Shimomura I., Matsuzawa Y. (2001) Diabetes 50, 2094–2099 [DOI] [PubMed] [Google Scholar]
- 31. Fitzpatrick F. A., Wynalda M. A. (1983) J. Biol. Chem. 258, 11713–11718 [PubMed] [Google Scholar]
- 32. Casimir D. A., Miller C. W., Ntambi J. M. (1996) Differentiation 60, 203–210 [DOI] [PubMed] [Google Scholar]
- 33. Rosen E. D., MacDougald O. A. (2006) Nat. Rev. Mol. Cell Biol. 7, 885–896 [DOI] [PubMed] [Google Scholar]
- 34. Tontonoz P., Hu E., Spiegelman B. M. (1994) Cell 79, 1147–1156 [DOI] [PubMed] [Google Scholar]
- 35. Tamori Y., Masugi J., Nishino N., Kasuga M. (2002) Diabetes 51, 2045–2055 [DOI] [PubMed] [Google Scholar]
- 36. Vassaux G., Gaillard D., Ailhaud G., Négrel R. (1992) J. Biol. Chem. 267, 11092–11097 [PubMed] [Google Scholar]
- 37. Darlington G. J., Ross S. E., MacDougald O. A. (1998) J. Biol. Chem. 273, 30057–30060 [DOI] [PubMed] [Google Scholar]
- 38. Tsuboi H., Sugimoto Y., Kainoh T., Ichikawa A. (2004) Biochem. Biophys. Res. Commun. 322, 1066–1072 [DOI] [PubMed] [Google Scholar]
- 39. Bosetti F. (2007) J. Neurochem. 102, 577–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Weisberg S. P., McCann D., Desai M., Rosenbaum M., Leibel R. L., Ferrante A. W., Jr. (2003) J. Clin. Invest. 112, 1796–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nishimura S., Manabe I., Nagasaki M., Seo K., Yamashita H., Hosoya Y., Ohsugi M., Tobe K., Kadowaki T., Nagai R., Sugiura S. (2008) J. Clin. Invest. 118, 710–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cuzzocrea S., Wayman N. S., Mazzon E., Dugo L., Di Paola R., Serraino I., Britti D., Chatterjee P. K., Caputi A. P., Thiemermann C. (2002) Mol. Pharmacol. 61, 997–1007 [DOI] [PubMed] [Google Scholar]
- 43. Rajakariar R., Hilliard M., Lawrence T., Trivedi S., Colville-Nash P., Bellingan G., Fitzgerald D., Yaqoob M. M., Gilroy D. W. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 20979–20984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hsieh P. S., Lu K. C., Chiang C. F., Chen C. H. (2010) Eur. J. Clin. Invest. 40, 164–171 [DOI] [PubMed] [Google Scholar]
- 45. Granado M., Martín A. I., Castillero E., López-Calderón A., Villanúa M. A. (2009) Eur. J. Pharmacol. 608, 97–103 [DOI] [PubMed] [Google Scholar]
- 46. Brüünsgaard H., Pedersen B. K. (2003) Immunol. Allergy Clin. North Am. 23, 15–39 [DOI] [PubMed] [Google Scholar]
- 47. Wu D., Ren Z., Pae M., Guo W., Cui X., Merrill A. H., Meydani S. N. (2007) J. Immunol. 179, 4829–4839 [DOI] [PubMed] [Google Scholar]
- 48. Fajas L., Miard S., Briggs M. R., Auwerx J. (2003) J. Lipid Res. 44, 1652–1659 [DOI] [PubMed] [Google Scholar]
- 49. Yan H., Kermouni A., Abdel-Hafez M., Lau D. C. (2003) J. Lipid Res. 44, 424–429 [DOI] [PubMed] [Google Scholar]
- 50. Chu X., Nishimura K., Jisaka M., Nagaya T., Shono F., Yokota K. (2010) Prostaglandins Other Lipid Mediat. 91, 1–9 [DOI] [PubMed] [Google Scholar]
- 51. Fain J. N., Ballou L. R., Bahouth S. W. (2001) Prostaglandins Other Lipid Mediat. 65, 199–209 [DOI] [PubMed] [Google Scholar]