

Abstract
Geosmin (1) is responsible for the characteristic odor of moist soil, as well as off-flavors in drinking water and foodstuffs (ref. 1 and 2). Geosmin is generated from farnesyl diphosphate (FPP, 2) by an enzyme that in the soil organism Streptomyces coelicolor A3(2) is encoded by the SCO6073 gene (ref. 3) We have now shown that the recombinant N-terminal half of this protein catalyzes the Mg2+-dependent cyclization of FPP to germacradienol (3) and germacrene D (4), while the highly homologous C-terminal domain, previously thought to be catalytically silent, catalyzes the Mg2+-dependent conversion of germacradienol to geosmin (1). Site-directed mutagenesis confirmed that the N- and C-terminal domain each harbors a distinct, independently functioning active site. A mutation in the N-terminal domain of germacradienol–geosmin synthase of a catalytically essential serine to alanine results in the conversion of FPP (2) to a mixture of sesquiterpenes that includes an aberrant product identified as isolepidozene (6), previously suggested to be an enzyme-bound intermediate in the cyclization of FPP to germacradienol.
Geosmin (1), whose name means “earth odor”, is a volatile microbial metabolite that is responsible for the characteristic smell of moist soil or freshly plowed earth1,2. Geosmin is produced by a number of microorganisms, including most Streptomyces and several species of cyanobacteria, myxobacteria, and fungi4–10. The detection and elimination of 1, which has an exceptionally low threshold for human detection of less than 10 parts per trillion, is of considerable economic importance due to its association with undesirable musty or off-flavors in drinking water, wine, fish and other foodstuffs, and its resistance to removal by conventional water treatment10,11.
The chemical origin of the characteristic odor of soil was first investigated in 1891 by Berthelot12, but not until 1965 was the responsible agent, (−)-geosmin (1), first isolated in pure form from the neutral extract of the fermentation broth of Streptomyces griseus and the structure assigned4,5. In 1981, incorporation of labeled acetate into geosmin using strains of S. antibioticus suggested that this bicyclic C12 metabolite might be a degraded sesquiterpene13. Although there have been more 800 papers dealing with the production, detection, and remediation of geosmin and other volatile metabolites in water supplies, aquaculture products, and wine, there were no further reports on the mechanism of microbial geosmin biosynthesis until five years ago14.
Expression in Escherichia coli of a 2181-bp gene from S. coelicolor A3(2) (SCO6073) gives a 726-amino acid protein with significant similarity in both the N-terminal and C-terminal halves to the well-characterized sesquiterpene synthase, pentalenene synthase15. The full length recombinant protein catalyzes the Mg2+-dependent conversion of farnesyl diphosphate (FPP, 2) to a mixture of germacradienol (3), germacrene D (4), octalin 5, and geosmin (1), without involvement of any cosubstrates or redox cofactors (Scheme 1)3,15,16. Incubation of FPP (2) with the closely related recombinant germacradienol–geosmin synthase from S. avermitilis (SAV2163, GeoA) yields an essentially identical mixture of 3, 4, 5, and geosmin (1)17. Deletion of the S. avermitilis geoA gene abolished both germacradienol and geosmin production, which could be restored by reintroduction of a copy of wild-type geoA17. Independently, researchers at the John Innes Institute have demonstrated that deletion of the S. coelicolor SCO6073 gene abolishes geosmin formation18.
Scheme 1.

Mechanism and stereochemistry of the cyclization of FPP (2) to germacradienol (3), germacrene D (4), octalin 5, and geosmin (1), catalyzed by S. coelicolor germacradienol–geosmin synthase (GS), showing branchpoint germacradienyl cation A and proposed intermediacy of isolepidozene (6).
Generation of germacradienol (3) and germacrene D (4) results from partitioning of a common intermediate, proposed to be carbocation A, the initial product of ionization and cyclization of FPP (2) (Scheme 1)16. Formation of germacrene D (4) involves a 1,3-hydride shift of H-1si of FPP16. The alternative formation of germacradienol (3) involves competing loss of the original H-1si proton of FPP (2) and cyclization to a proposed enzyme-bound, trans-fused bicyclic intermediate, isolepidozene (6), a known compound previously isolated from liverworts19 that would be converted to germacradienol by proton-initiated ring opening and capture of the resulting homoallyl cation by water. Further conversion of germacradienol to geosmin is proposed to involve protonation–cyclization, a novel retro-Prins type fragmentation resulting in loss of the 2-propanol side chain as acetone, and generation of the octalin intermediate 5. Reprotonation of 5 followed by a 1,2-hydride shift and quenching of the bridgehead cation by water will generate geosmin (1)20. The results of incubation reactions carried out in D2O are also consistent with this mechanistic proposal3.
Increasing either the concentration of germacradienol–geosmin synthase or the time of incubation enhances the relative proportion of geosmin to germacradienol as well as the absolute yield of 1.3,17 These results are inconsistent with the formation of germacradienol and geosmin by partitioning of a series of exclusively enzyme-bound intermediates. A substantial fraction of the initially generated germacradienol must be released from the enzyme before rebinding to the protein and cyclization–fragmentation to generate geosmin, without distinguishing whether germacradienol and geosmin formation take place at the same or at distinct active sites, nor whether transient release of germacradienol is a mandatory event in geosmin biosynthesis. We now report conclusive evidence that germacradienol–geosmin synthase is a bifunctional enzyme possessing two independent active sites with distinct catalytic functions.
Two strictly conserved motifs found in all sesquiterpene and monoterpene synthases are essential to binding of the Mg2+ cofactor: an aspartate-rich sequence DDXX(D/E), that is found at amino acid (aa) residues 80–120 in microbial synthases and at aa 290–310 in plant synthases, and an NSE triad of residues, (N/D)DXX(S/T)XXXE, generally found 140±5 aa downstream of the aspartate-rich motif21–25. The vast majority of bacterial and fungal terpene synthases are 330–400 aa in length, corresponding to a subunit Mr 35–45 kDa. The 726-aa germacradienol–geosmin synthase of S. coelicolor is unusual in that it is about twice the size of a typical terpene synthase. Notably, both the N- and C-terminal halves of the SCO6073 protein show significant sequence similarity to the well-characterized sesquiterpene synthase, pentalenene synthase (28% identity, 41% similarity over the aa 1–319; 29% identity, 46% similarity over aa 407–726)15. Both the N-and C-terminal regions of the protein harbor variants of the canonical aspartate rich domain, with a DDHFLE motif in the N-terminal half and an unusual DDYYP motif in the C-terminal half (Fig. 1). Both halves also display typical NSE motifs at NDLFSYQRE and NDVFSYQKE. Curiously, the N-terminal half also appears to harbor an unusual repeat of the upstream NSE motif, NDVLTSRLHQFE, located 38 aa downstream of the first. A 41-kDa truncated N-terminal mutant of germacradienol–geosmin synthase corresponding to aa 1–366 converts FPP to germacradienol, while a C-terminal truncation mutant (aa 383–726) catalyzes only slow solvolysis of FPP, with no detectable formation of cyclic products15. Deletion of the N-terminal domain of SCO6073 also abolishes geosmin production by S. coelicolor18.
Figure 1.
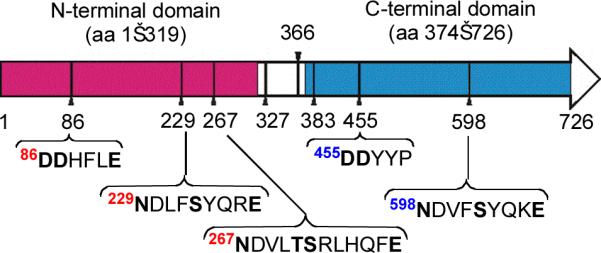
Organization of protein domains and conserved Mg2+-binding motifs in S. coelicolor germacradienol–geosmin synthase. The N-terminal domain, corresponding to aa 1–319, based on sequence alignments with known Streptomyces terpene synthases is shown in red, while the C-terminal domain, aa 374–726, is highlighted in blue. T366 and M327 are the N- and C-termini, respectively, of the recombinant N-terminal and C-terminal truncation proteins encoded by pRW22 and pJJ3.
Based on the discovery that full-length recombinant SCO6073 protein can convert FPP to germacradienol and geosmin3, we have sought to localize the geosmin synthase activity by examining the behavior of both N- and C-terminal truncation mutants. To this end, we generated a 56 aa-longer C-terminal construct using the NcoI-XhoI fragment of plasmid pRW31 encoding full length SCO6073 as template for PCR amplificaton of the region corresponding to aa M327–H726 (Fig. 1) and purified the derived pJJ3 protein (Mr 46,000), carrying a C-terminal His6-tag, to >95% homogeneity by Ni2+-NTA chromatography.
Incubation of the N-terminal truncated mutant (pRW22p)15 with 100 μM FPP (2) generated a mixture of germacradienol (3) and germacrene D (4) accompanied by small quantities of octalin 5, but no detectable geosmin (1), as determined by capillary gas chromatography–mass spectrometry (GC–MS) (Scheme 2a, Table 1, Supplementary Fig. 1a). Increasing the concentration of the N-terminal domain from 2.4 to 9.0 μM, conditions under which wild-type protein yields a mixture containing up to 15% geosmin, had minimal effect on the relative proportion of the three products. Significantly, addition of 2.4 μM recombinant C-terminal protein (pJJ3p) to the incubation mixture containing 5.4 μM truncated N-terminal protein and 100 μM FPP resulted in formation of geosmin (1), with a concomitant decrease in the proportion of germacradienol (3) (Scheme 2b, Table 1). Using 5.5 μM C-terminal protein increased the proportion of geosmin (1) to 17% while the fraction of germacradienol dropped to 66%, with insignificant variation in the proportion of germacrene D (4) in both cases and a doubling of the relative proportion of octalin 5 (Supplementary Fig. 1b). Control experiments reconfirmed that incubation of FPP (2) with the C-terminal protein alone generated only minor amounts of the acyclic solvolysis products (E)-α-farnesene (7) and nerolidol (8) along with a trace of (Z)-α-bisabolene (9) (Scheme 2c)15.
Scheme 2.

Role of the N-terminal and C-terminal domains of germacradienol–geosmin synthase. a. Incubation of FPP with recombinant N-terminal domain (pRW22p). b. Incubation of FPP with a mixture of recombinant N-terminal domain (pRW22p) and C-terminal domain (pJJ3p). c. Incubation of FPP with recombinant C-terminal domain (pJJ3p). d. Incubation of germacradienol (3) with recombinant C-terminal domain (pJJ3p).
Table 1.
Incubation of FPP with the N-terminal and C-terminal domains of S. coelicolor germacradienol-geosmin synthase.
| Protein (μM) | Product Relative Intensity (%) | |||||
|---|---|---|---|---|---|---|
| N-term | C-term | geosmin (1) | germacradienol (3) | germacrene D (4) | octalin (5) | α-humulene (16)a |
| 2.4 | --- | NDb | 3.80 (82%) | 0.71 (15%) | 0.11 (3%) | 1.00 |
| 5.3 | --- | ND | 5.10 (84%) | 0.87 (15%) | 0.05 (1%) | 1.00 |
| 9.0 | --- | ND | 4.72 (85%) | 0.73 (13%) | 0.08 (2%) | 1.00 |
| 5.4 | 2.4 | 0.25 (5%) | 3.89 (78%) | 0.71 (14%) | 0.16(3%) | 1.00 |
| 5.4 | 5.5 | 0.90 (17%) | 3.52 (66%) | 0.66 (12%) | 0.26 (5%) | 1.00 |
| 5.4 | 8.3 | 1.05 (15%) | 4.72 (68%) | 0.94 (13%) | 0.24 (4%) | 1.00 |
| --- | 8.3 | Trace products include (E)-α-farnesene (7), nerolidol (8), and (Z)-α-bisabolene (9). | ||||
α-Humulene (16) was added as internal standard.
ND, not detected.
These results established either that the C-terminal protein directly catalyzes the formation of geosmin from germacradienol produced by the N-terminal domain or, less likely, that the C-terminal protein in some way modulates the catalytic activity of the N-terminal domain. To distinguish these two possibilities, we incubated an 85:15 mixture of germacradienol (3) and germacrene D (4), containing ~1% octalin 5, obtained from incubation of FPP with N-terminal protein, with 1.6 μM C-terminal protein (pJJ3p) in the presence of 4 mM MgCl2 for 6 h at 30 °C. GC–MS analysis indicated a 2 % conversion to geosmin, which increased to 6% when the concentration of C-terminal mutant protein was augmented to 3.1 μM (Scheme 2d; Supplementary Fig. 2). Omission of Mg2+ from the incubation mixtures completely abolished geosmin formation.
To explore further the role of the C-terminal domain in the formation of geosmin from FPP and germacradienol (3), we used site-directed mutagenesis to target the conserved downstream aspartate-rich DDYYP and the NDVFSYQKE motifs of the full-length germacradienol–geosmin synthase (Supplementary Table 1). When incubated for 11 h with 49 μM FPP, the D455N/D456N double mutant (19.5 μM) generated a 87:11 mixture of germacradienol (3) and germacrene D (4), accompanied by a trace of octalin 5, without any detectable geosmin. The N598L, D599L, S602A, and E606Q mutants gave similar product mixtures, again with complete suppression of geosmin formation. Importantly, geosmin production could be restored by pre-incubation of 16.1 μM of the D455N/D456N double mutant with 43 μM FPP for 5.5 h at 30 °C, followed by addition of 2.7 μM of the C-terminal truncation protein and incubation for a further 5.5 h, yielding a mixture 89:8:1:2 of 3, 4, 5, and geosmin (1).
Site-directed mutagenesis of the presumptive Mg2+-binding domains21–25 in the N-terminal half of the germacradienol–germacrene D synthase resulted in decreases in catalytic efficiency as well as the generation of a variety of abnormal products that were characterized by GC-MS (Table 2). Thus the D86E mutant generated several aberrant products including the acyclic elimination products (E)-α-farnesene (7) and β-farnesene (10) and a monocyclic sesquiterpene tentatively identified by retention index (RI) and mass spectrum as β-elemene (11) (Scheme 3). Interestingly, the L90D mutant, with the canonical DDHFD sequence, was not only severely impaired catalytically, with a greater than 1,000-fold reduction in kcat/Km, but produced primarily a 1:6 mixture of β-elemene (11) and γ-elemene (12). Notably both the S272A and T271A mutants produced normal mixtures of 3, 4, 5, and geosmin (1), with only relatively minor reductions in kcat/Km, suggesting that the unusual downstream NSE triad found in the N-terminal half of the protein plays no significant role in catalysis. Most interestingly, the S233A mutant, which was severely impaired catalytically (2500-fold reduction in kcat/Km), produced, in addition to the normal products 1 (11%), 3 (57%), 4 (18%), 5 (3%), a new sesquiterpene hydrocarbon, identified as isolepidozene (6) (11%, m/z 204, RI 1483), identified by comparison of the mass spectrum and GC retention index with the data for authentic isolepidozene (Supplementary Fig. 3).
Table 2.
Steady-state kinetic parameters and products for wild-type S. coelicolor (SCO6073) germacradienol-geosmin synthase and mutants.
| protein | Steady-state kinetic parametersa.b |
products | ||
|---|---|---|---|---|
| kcat (s−1) | Km (μM) | kcat/Km (s−1M−1) | ||
| SCO6073 (wt)c | (6.2±0.5) × 10−3 | 0.062±0.008 | 1 × 105 | 1, 3, 4, 5 |
| D86E | (1.3±0.1) × 10−4 | 0.19±0.04 | 6.8 × 102 | 7, 10, 11 d |
| L90D | (5.5±0.3) × 10−4 | 5.8±0.6 | 95 | 11,12(1:6) |
| S233A | (8.8±0.3) × 10−5 | 2.2±0.2 | 40 | 1, 3, 4, 5, 6 |
| T271A | (3.3±0.2) × 10−4 | 0.092±0.015 | 3.6×103 | 1, 3, 4, 5 |
| S272A | (5.8±0.3) × 10−4 | 0.050±0.008 | 1.2×104 | 1, 3, 4, 5 |
Steady-state parameters are for total product formation.
Standard deviations for kcat and Km are the calculated statistical errors for the non-linear leastsquares regression fit of the data to the Michael-Menten expression, with R values typically >0.98
Steady-state parameters of wild-type SCO6073 protein are from Ref 15.
Additional unidentified minor products, m/z 204, were also detected.
Scheme 3.
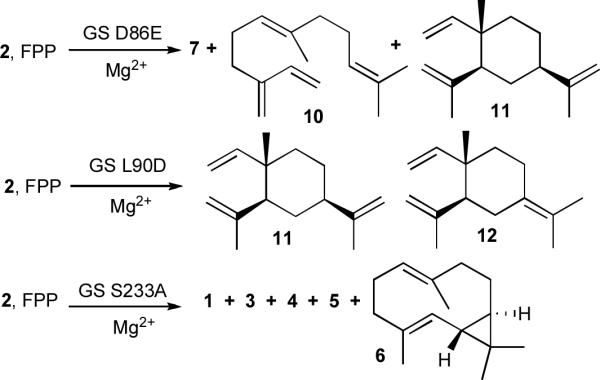
Formation of aberrant sesquiterpene products by incubation of FPP with germacradienol–geosmin synthase (GS) mutants D86E, L90D, and S233A.
The experiments described above demonstrate unambiguously that the SCO6073 germacradienol–geosmin synthase of S. coelicolor is a bifunctional enzyme in which the N-terminal half of the protein converts FPP (2), the universal acyclic sesquiterpene precursor, to an 85:15 mixture of germacradienol (3) and germacrene D (4). The C-terminal half of the protein then rebinds the germacradienol and catalyzes proton-intiated cyclization–fragmentation of 3 to give geosmin (1). Interestingly, the N-terminal domain also produces small amounts of octalin 5, thought to be an intermediate in the conversion of 3 to 1. Both the N- and C-terminal-catalyzed reactions have an absolute dependence on the divalent cation Mg2+. Site-directed mutagenesis clearly implicates the Mg2+-binding regions of each half of the protein in the conversion of FPP to germacradienol and thence to geosmin. While the role of Mg2+ in terpene synthase catalyzed cyclization of allylic diphosphate substrates is well understood21, the role of the divalent cation in the electrophilic polyene-type cyclization is unclear, since the germacradienol substrate does not have a pyrophosphate moiety that might interact with the bound Mg2+.
Although bifunctional sesquiterpene and monoterpene synthases have not previously been described, fungal ent-kaurene synthases have two distinct active sites that catalyze successive electrophilic cyclization reactions26,27. The first reaction in the sequence, known as a Type B reaction, is a proton-initiated polyene cyclization of geranylgeranyl diphosphate (GGPP, 13), catalyzed by the N-terminal domain of the protein, to generate the bicyclic intermediate copalyl diphosphate (CPP, 14) (Supplementary Scheme 1). CPP is then rebound by the C-terminal domain of the diterpene synthase which catalyzes an ionization–cyclization–rearrangement to yield the tetracyclic diterpene ent-kaurene (15) (Type A reaction). Interestingly, the order of the Type A and Type B electrophilic cyclization reactions is reversed in these fungal diterpene synthases compared to S. coelicolor germacradienol–geosmin synthase26. A DVDD motif in the N-terminal domain of ent-kaurene synthase is responsible for the proton-initiated polyene cyclization of the GGPP substrate, similar to the aspartate-rich DVDDTA motif of squalene-hopene cyclase, in which the D376 residue (bold) initiates the Type B polyene cyclization, rather than acting as a Mg2+-binding domain28. One or both aspartate residues in the DDYYP motif of SCO6073 may therefore play a similar role in the conversion of germacradienol to geosmin.
The genome sequences of a variety of geosmin-producing actinomycetes and cyanobacteria reveal at least ten additional presumptive geosmin synthases with a high level of sequence homology to SCO6073 (45–78% identity, 57–85% similarity over >700 aa) (Supplementary Table 2). The active site DDHFLE, NDLFSYQRE, DDYYP, and NDVFSYQKE motifs are especially highly conserved in all of these presumed geosmin synthases (Supplementary Fig. 4), as is the unusual, duplicated downstream NSE triad in the N-terminal domain that plays no apparent functional role in FPP binding or catalysis.
The detection of free germacradienol, as well as the demonstration that the C-terminal domain of SCO6073 converts germacradienol, produced by the N-terminal domain, to geosmin establishes that the germacradienol intermediate does not channel directly between the two active sites of the bifunctional protein. Instead substrate transfer involves release of the initially generated intermediate 3 into the medium and diffusive rebinding to the active site of the C-terminal domain.
Previous in vitro biochemical and in vivo molecular genetic studies are in agreement that the N-terminal domain of S. coelicolor SCO6073 is essential for the formation of geosmin15,18. The definitive demonstration that the N-terminal domain converts FPP to germacradienol but not to geosmin, and that the C-terminal domain catalyzes the cyclization–fragmentation of germacradienol to geosmin is at odds, however, with the earlier report that S. coelicolor mutants carrying an in-frame deletion of aa 380–721 of the C-terminal domain of SCO6073 apparently could still produce geosmin18.
The significant decreases in kcat and increases in Km as well as the formation of aberrant products as a consequence of mutations in the universally conserved Mg2+-binding domains of terpene synthases is well-precedented24,25. Frequently the aberrant products result from premature quenching or derailment of the normal reaction cascade of enzyme-bound carbocationic intermediates. The trans-fused bicyclic sesquiterpene isolepidozene (6) derailment product generated by the S233A mutant is in fact a postulated intermediate in the conversion of the germacradienyl cation A to germacradienol 3 (Scheme 1)3,17. The structure and stereochemistry of 6 are fully compatible with the observed regiochemistry and stereochemistry of labeling by chirally deuterated FPP of both germacradienol (3) and geosmin (1)3,16. Further experiments to establish the mechanistic details of the conversion of germacradienol to geosmin and the role of the protein in mediating this fascinating transformation are in progress.
METHODS
Materials and Instrumentation
General analytical, biochemical, and molecular biological procedures were as previously described3,15,29. Full-length recombinant S. coelicolor A3(2) germacradienol-geosmin synthase, corresponding to the SCO6073 gene, and the N-terminal truncation mutant, corresponding to a a 1–366, were expressed from E. coli BL21(DE3)pLys/pRW31 and E. coli BL21(DE3)pLys/pRW22, respectively, then resolublilized and refolded from protein inclusion bodies, purified to homogeneity and assayed as previously described3,15,16. Farnesyl diphosphate was synthesized and purified as previously described30. [1-3H]FPP [20 Ci/mmol (1 Ci = 37 GBq)] was purchased from American Radiolabeled Chemicals, Inc., diluted with synthetic FPP to a specific activity of 120 mCi/mmol, and pre-extracted with HPLC-grade pentane to remove any contaminating alcohols. GC–MS analysis was performed on a Hewlett-Packard Series 2 GC-MSD, at 70 eV electron impact (EI), operating in positive ion mode. DNA sequencing was carried out on an ABI 3730 DNA sequencer using Big Dye Terminator V3.0 sequencing chemistry at the U. C. Davis Automated DNA Sequencing Facility.
Other methods
See Supplementary Methods online for details of mutant construction and protein expression and purification.
Steady-state kinetic assay of germacradienol–geosmin synthase mutants
Purified germacradienol–geosmin synthase mutants were assayed as previously described for the wild-type S. coelicolor germacradienol–geosmin synthase15. To determine the steady-state kinetic parameters, we performed a series of assays in 10-ml glass tubes containing 1 ml of assay buffer (50 mM Tris-HCl, 20% glycerol, 10 mM MgCl2, pH 8.2) using a range of [1-3H]FPP concentrations (20, 40, 70, 100, 200, 300, 500 and 800 nM, 120 mCi/mmol). The reactions, which were carried out in parallel, were initiated by the addition of 10 μl of 50-fold diluted mutant protein solution (2.4 pmol), then immediately overlaid with 1.2 ml of hexane. After 30 min incubation at 30 °C (less than 10% conversion in all cases), the reaction was quenched by the addition of 75 μl of 0.5 M EDTA (pH 8.1). Control incubations utilized 70 nM and 300 nM [1-3H]FPP in the absence of enzyme. Ether (100 μl) was added and the mixture was vortexed for 30 s. The organic layer was passed through a 1-cm SiO2 column in a Pasteur pipette directly into a scintillation vial containing 7 ml of Opti-Fluor. The aqueous layer was extracted with an additional 1.2 ml of 11:1 hexane:ether which was added to the scintillation vial after passage through the SiO2 column which was then washed with 2 × 0.75 ml of ether. The steady-state kinetic parameters, kcat and Km for the conversion of FPP to combined hydrocarbons and alcohols were calculated by direct fitting of the data to the Michaelis-Menten equation by non-linear least squares regression using Kaleidagraph V3.6 (Adelbeck Software, Reading, PA, USA).
GC-MS analysis of products of germacradienol–geosmin synthase mutants
For GC-MS analysis, purified mutant proteins were incubated with 500 nmol of FPP in 5.0 ml of 50 mM Tris-HCl, pH 8.2, containing 20% glycerol, 4 mM MgCl2, overlaid with 3 ml of HPLC-grade pentane in 18 × 150 mm glass tubes fitted with a rubber septum for 6 h at 30 °C. The reaction mixture was extracted with 3×3 ml pentane/CH2Cl2 (5:1), and the combined organic layers were dried by passage through a Pasteur pipette containing 1 g of MgSO4 before concentration under reduced pressure at 0 °C to 200 μl. A 1-μl portion of the concentrated extract was analyzed by GC–MS (30 m × 0.25 mm HP5MS capillary column, using a temperature program of 50–280 °C, 20 °C min−1). Each compound was identified by direct comparison with authentic standards or with the spectra of the corresponding reference compounds in the MassFinder 3.0 Library (http://www.massfinder.com).
Incubation of N- and C-terminal truncated mutants
Purified N- and C-terminal truncated mutants were added to 5.0 ml of 50 mM Tris-HCl, pH 8.2, containing 20% glycerol, 4 mM MgCl2 and 100 μM FPP, overlaid with 3 ml of HPLC-grade pentane in 18 × 150 mm glass tubes fitted with a rubber septum and incubated for 6 h at 30 °C. The products were extracted and analyzed by GC–MS as described for the wild-type and full-length mutant proteins.
Incubation of germacradienol (3) with the C-terminal truncated mutant
A ~6:1 mixture of germacradienol (3) and germacrene D (4) containing a trace of octalin 5, extracted from the incubation of N-terminal truncated protein (pRW22) with FPP, was redissolved in 50 μl of MeOH and added to 5 ml of assay buffer containing 3.1 μM C-terminal protein (pJJ3) and 4 mM Mg2+. The mixture was incubated at 30 °C for 6 h without a pentane overlay, in order to avoid transfer of the germacradienol substrate out of the aqueous solution. A 1-μl portion of the concentrated pentane extract was analyzed by GC–MS (Supplementary Fig. 2). No geosmin could be detected when Mg2+ was omitted from the incubation mixture. A control incubation with a mixture of 3.3 μM N-terminal mutant and 3.1 μM C-terminal mutant for 6 h at 30 °C established that addition of up to 4% MeOH had minimal effect on the activity of the enzyme and the relative yield of geosmin.
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Schulz of the Technische Universität Braunschweig for a gift of synthetic octalin and T.-L. Shen for assistance with the mass spectrometric analysis. This research was supported by National Institutes of Health Grant GM30301 to D.E.C.
Footnotes
COMPETING FINANCIAL INTERESTS STATEMENT The authors declare that they have no competing financial interests.
Note: Supplementary information is available on the Nature Chemical Biology website.
References
- 1.Gerber NN. Volatile substances from actinomycetes: their role in the odor pollution of water. CRC Crit. Rev. Microbiol. 1979;7:191–214. doi: 10.3109/10408417909082014. [DOI] [PubMed] [Google Scholar]
- 2.Buttery RG, Garibaldi JA. Geosmin and methylisoborneol in garden soil. J. Agric. Food Chem. 1976;24:1246–1247. [Google Scholar]
- 3.Jiang J, He X, Cane DE. Geosmin biosynthesis. Streptomyces coelicolor germacradienol/germacrene D synthase converts farnesyl diphosphate to geosmin. J. Am. Chem. Soc. 2006;128:8128–8129. doi: 10.1021/ja062669x. [DOI] [PubMed] [Google Scholar]
- 4.Gerber NN, Lechevalier HA. Geosmin, an earthy-smelling substance isolated from actinomycetes. Appl. Microbiol. 1965;13:935–938. doi: 10.1128/am.13.6.935-938.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerber NN. Geosmin, from microorganisms, is trans-1,10-Dimethyl-trans-9-decalol. Tetrahedron Lett. 1968:2971–2974. [Google Scholar]
- 6.Pollak FC, Berger RG. Geosmin and related volatiles in bioreactor-cultured Streptomyces citreus CBS 109.60. Appl. Environ. Microbiol. 1996;62:1295–1299. doi: 10.1128/aem.62.4.1295-1299.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dickschat JS, Wenzel SC, Bode HB, Muller R, Schulz S. Biosynthesis of volatiles by the myxobacterium Myxococcus xanthus. Chembiochem. 2004;5:778–787. doi: 10.1002/cbic.200300813. [DOI] [PubMed] [Google Scholar]
- 8.Dickschat JS, Bode HB, Wenzel SC, Muller R, Schulz S. Biosynthesis and identification of volatiles released by the myxobacterium Stigmatella aurantiaca. Chembiochem. 2005;6:2023–2033. doi: 10.1002/cbic.200500174. [DOI] [PubMed] [Google Scholar]
- 9.Scholler CE, Gurtler H, Pedersen R, Molin S, Wilkins K. Volatile metabolites from actinomycetes. J. Agric. Food Chem. 2002;50:2615–2621. doi: 10.1021/jf0116754. [DOI] [PubMed] [Google Scholar]
- 10.La Guerche S, Chamont S, Blancard D, Dubourdieu D, Darriet P. Origin of (−)-geosmin on grapes: on the complementary action of two fungi, Botrytis cinerea and Penicillium expansum. Antonie Van Leeuwenhoek. 2005;88:131–139. doi: 10.1007/s10482-005-3872-4. [DOI] [PubMed] [Google Scholar]
- 11.Heil TP, Lindsay RC. Volatile compounds in flavor-tainted fish from the Upper Wisconsin River. J. Environ. Sci. Health B. 1988;23:489–512. doi: 10.1080/03601238809372621. [DOI] [PubMed] [Google Scholar]
- 12.Berthelot M, André G. Sur l'odeur propre de la terre. Compt. Rend. 1891;112:598–599. [Google Scholar]
- 13.Bentley R, Meganathan R. Geosmin and methylisoborneol biosynthesis in streptomycetes. Evidence for an isoprenoid pathway and its absence in non-differentiating isolates. FEBS Lett. 1981;125:220–222. doi: 10.1016/0014-5793(81)80723-5. [DOI] [PubMed] [Google Scholar]
- 14.Spiteller D, Jux A, Piel J, Boland W. Feeding of [5,5-2H2]-1-desoxy-D-xylulose and [4,4,6,6,6-2H5]-mevalolactone to a geosmin-producing Streptomyces sp. and Fossombronia pusilla. Phytochemistry. 2002;61:827–834. doi: 10.1016/s0031-9422(02)00282-0. [DOI] [PubMed] [Google Scholar]
- 15.Cane DE, Watt RM. Expression and mechanistic analysis of a germacradienol synthase from Streptomyces coelicolor implicated in geosmin biosynthesis. Proc. Natl. Acad. Sci. U S A. 2003;100:1547–1551. doi: 10.1073/pnas.0337625100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He X, Cane DE. Mechanism and stereochemistry of the germacradienol/germacrene D synthase of Streptomyces coelicolor A3(2) J. Am. Chem. Soc. 2004;126:2678–2679. doi: 10.1021/ja039929k. [DOI] [PubMed] [Google Scholar]
- 17.Cane DE, He X, Kobayashi S, Omura S, Ikeda H. Geosmin biosynthesis in Streptomyces avermitilis. Molecular cloning, expression, and mechanistic study of the germacradienol–geosmin synthase. J. Antibiot. (Tokyo) 2006;59:471–479. doi: 10.1038/ja.2006.66. [DOI] [PubMed] [Google Scholar]
- 18.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. U S A. 2003;100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hardt IH, Rieck A, König WA, Muhle H. Isolepidozene, a diastereomer of bicyclogermacrene, in some liverworts. Phytochem. 1995;40:605–606. [Google Scholar]
- 20.Dickschat JS, Bode HB, Mahmud T, Muller R, Schulz S. A novel type of geosmin biosynthesis in myxobacteria. J. Org. Chem. 2005;70:5174–5182. doi: 10.1021/jo050449g. [DOI] [PubMed] [Google Scholar]
- 21.Christianson DW. Structural biology and chemistry of the terpenoid cyclases. Chem. Rev. 2006;106:3412–3442. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]
- 22.Rynkiewicz MJ, Cane DE, Christianson DW. X-ray crystal structures of D100E trichodiene synthase and its pyrophosphate complex reveal the basis for terpene product diversity. Biochemistry. 2002;41:1732–1741. doi: 10.1021/bi011960g. [DOI] [PubMed] [Google Scholar]
- 23.Starks CM, Back K, Chappell J, Noel JP. Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-eristolochene synthase. Science. 1997;277:1815–1820. doi: 10.1126/science.277.5333.1815. [DOI] [PubMed] [Google Scholar]
- 24.Felicetti B, Cane DE. Aristolochene synthase: mechanistic analysis of active site residues by site-directed mutagenesis. J. Am. Chem. Soc. 2004;126:7212–7221. doi: 10.1021/ja0499593. [DOI] [PubMed] [Google Scholar]
- 25.Little DB, Croteau RB. Alteration of product formation by directed mutagenesis and truncation of the multiple-product sesquiterpene synthases δ-selinene synthase and γ-humulene synthase. Arch. Biochem. Biophys. 2002;402:120–135. doi: 10.1016/S0003-9861(02)00068-1. [DOI] [PubMed] [Google Scholar]
- 26.Kawaide H, Sassa T, Kamiya Y. Functional analysis of the two interacting cyclase domains in ent-kaurene synthase from the fungus Phaeosphaeria sp. L487 and a comparison with cyclases from higher plants. J. Biol. Chem. 2000;275:2276–2280. doi: 10.1074/jbc.275.4.2276. [DOI] [PubMed] [Google Scholar]
- 27.Toyomasu T, et al. Cloning of a full-length cDNA encoding ent-kaurene synthase from Gibberella fujikuroi: functional analysis of a bifunctional diterpene cyclase. Biosci. Biotechnol. Biochem. 2000;64:660–664. doi: 10.1271/bbb.64.660. [DOI] [PubMed] [Google Scholar]
- 28.Feil C, Sussmuth R, Jung G, Poralla K. Site-directed mutagenesis of putative active-site residues in squalene-hopene cyclase. Eur. J. Biochem. 1996;242:51–55. doi: 10.1111/j.1432-1033.1996.0051r.x. [DOI] [PubMed] [Google Scholar]
- 29.Tetzlaff CN, et al. A gene cluster for biosynthesis of the sesquiterpenoid antibiotic pentalenolactone in Streptomyces avermitilis. Biochemistry. 2006;45:6179–6186. doi: 10.1021/bi060419n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cane DE, Chiu HT, Liang PH, Anderson KS. Pre-steady-state kinetic analysis of the trichodiene synthase reaction pathway. Biochemistry. 1997;36:8332–8339. doi: 10.1021/bi963018o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.