

Abstract
The chloroplast gene rbcL encodes the large subunit of the CO2-fixing enzyme ribulose-bisphosphate carboxylase. In previous work a target for photo-accelerated degradation of Chlamydomonas reinhardtii rbcL transcripts in vivo was found to lie within the first 63 nucleotides, and a sequence element required for increasing the longevity of transcripts of rbcL-reporter genes was found to occur between nucleotides 170 and 350. Photo-accelerated degradation of rbcL transcripts has been found to require nucleotides 21 to 41. Transcript nucleotides lying between 329 and 334 and between 14 and 27 are essential for stabilizing transcripts in vivo; mutations in either region reduce the longevity of transcripts. It is postulated that the effectiveness of photo-accelerated endonuclease attacks on the nucleotide 21 to 41 region is reduced by physical blockage or distortion of the target sequence by interacting proteins that associate with nucleotides in the 14 to 27 and 329 to 334 regions of the transcripts. Both the nucleotide +329 to +334 stabilizing sequence of rbcL and a transcription enhancing sequence that lies between +126 and +170 encode well conserved (cyanobacteria through angiosperms) amino acid sequences; the evolution of expression control elements within the protein coding sequence of rbcL is considered.
Control of mRNA stability can be important in posttranscriptional regulation of gene expression (1) but molecular mechanisms involved in the degradation of mRNAs are not very well understood. Sequences in the 5′ untranslated leader are sites of determinants of transcript longevity in mRNAs of Chlamydomonas reinhardtii chloroplast genes rbcL (2), psbD (3), petD (4), and psbB (5). Furthermore, instability of psbD transcripts is correlated with a mutation at the nuclear nac2 locus and the loss of binding of a 47-kDa peptide to the leader RNA (3)).
The chloroplast gene rbcL in C. reinhardtii is an especially interesting object for studying some aspects of transcript longevity. The basic promoter of this gene extends from approximately −22 (taking the transcription start site as +1) to +60 (6). Reporter transcripts containing the first 63 (or 97) nucleotides have half-lives of 5 h in unilluminated cells but only 0.3 h in illuminated cells (2). The translation start site for the protein that the gene encodes, the large subunit of Rubisco (ribulose-bisphosphate carboxylase), lies at +93. A target for mRNA degradation was found to lie within the transcribed portion of the promoter, i.e., between +1 and +63, and an antidegradation (or stabilizing) sequence is within the protein coding region between +297 and +350 (2). Also, an enhancer that increases expression from the basic promoter by 10- to 100-fold is within the protein coding region, between +126 and +170 (6).
In the present work we have identified a region within the +1 to +63 span of C. reinhardtii rcbL transcripts that is required for photo-accelerated degradation of rbcL-reporter gene transcripts. We have also identified sequences within the 5′ untranslated and 5′ protein coding regions of the transcript that are required, together, for greatly increasing the longevity of rbcL sequence-containing reporter mRNAs.
Materials and Methods
Chlamydomonas Strains and Culture.
C. reinhardtii nonphotosynthetic mutant strain CC-373 (ac-uc-2–21) and its photosynthetic transformants were grown on 12-h light/12-h dark cycles as described earlier (2, 7, 8).
Chloroplast Transformation.
Chimeric gene constructs (rbcL:GUS, rbcL:AADA, etc.) were cloned into a Chlamydomonas chloroplast transformation vector that promoted integration (by homologous recombination) of the foreign DNA into the chloroplast genome upstream of the 3′ end of the atpB gene (7, 8). DNA was introduced into Chlamydomonas CC373 cells (Chlamydomonas Genetics Center, Duke University, Durham, NC) by using a PDS-1000 Bio-Rad particle gun or a Finer et al. (9) gun. Transformants were selected and verified according to published protocols (10, 11).
Total Nucleic Acids and RNA Isolation.
Total nucleic acids and cellular RNAs were isolated as described earlier (2).
RNA Slot-Blot and Hybridization Analyses.
RNA slot blots were prepared by using GeneScreenPLUS membrane according to the manufacturer's instructions (DuPont/NEN). Briefly, the cellular RNA was denatured in denaturing solution [50% deionized formamide, 6% formaldehyde, 0.5× MOPS (pH 7.0); 1× MOPS is 0.02 M morpholinopropanesulfonic acid/0.005 M sodium acetate/0.5 mM EDTA] by incubating at 60°C for 15 min. The mixture was then placed on ice and an equal volume of TE buffer (10 mM Tris⋅HCl, pH 7.5/1 mM EDTA, pH 8.0) was added. The solution was mixed well and kept on ice until ready to apply to the membrane. GeneScreenPLUS membrane was soaked in distilled water for 15 min, placed on a slot manifold, and assembled. Each well of the manifold was prewashed with 100 μl of TE buffer. RNA sample solutions were drawn through the membrane by suction. RNA was then crosslinked to the membrane by using a Stratagene UV-linker. 32P-labeled DNA probes (GUS, aadA, and rbcL) were prepared (2) by using a random primer labeling kit (GIBCO/BRL) according to the protocol suggested by the vendor.
RNA Secondary Structure Prediction.
The predicted RNA secondary structure analyses were conducted by using the mfold program of the Wisconsin Package version 9.1 (Genetics Computer Group, Madison, WI) and other procedures (12, 13).
Construction of PatpB-rbcL-GUS Plasmids.
For experiments designed to better delimit an RNase target region in the rbcL +1 to +63 sequence, a set of constructs carrying rbcL fragments located between PatpB (atpB promoter) and the β-glucuronidase (GUS) reporter sequence was created. First, an NcoI restriction site was introduced into plasmid pCrc32 (6) immediately downstream of the SmaI site. For this purpose the entire GUS sequence was amplified from the pCrc32 template by PCR using the primer GUS-SN-U (5′ primer), which included SmaI and NcoI restriction sites, and the primer GUS-SN-L (3′ primer), which included an XbaI restriction site. The amplified GUS DNA with the newly created NcoI site was cut with SmaI and XbaI and inserted into pCrc32 DNA cut with the same enzymes. The resulting plasmid is designated pCrc32Nco. Next, the +1 to +62 rbcL sequence was inserted into the pCrc32Nco plasmid through the use of SmaI and NcoI sites. The synthetic +1 to +62 rbcL DNA had blunt 5′ GGG ends and sticky 3′ CATG ends complementary to the NcoI sequence. Ligation with pCrc32Nco DNA cut with SmaI and NcoI resulted in the plasmid pCrc32(1–62) with recovered SmaI and lost NcoI sites. The third step was to create PatpB-rbcL-GUS constructs carrying rbcL sequences +1 to +41, +1 to +20, and +1 to +10 located between PatpB and GUS sequences. All three constructs were created through the use of PCR and pCrc32(1–62) DNA as a template. The primer ATPB306 was used in all three PCRs as the 5′ primer, and RB41–2, RB20, and RB10 primers were used for the production of +1 to +41, +1 to +20, and +1 to +10 rbcL DNA fragments, respectively. The ATPB306 primer extended from position −114 in the atpB promoter region and included an XhoI site. The RB41–2, RB20, and RB10 primers started from the positions +41, +20, and +10, respectively, in the rbcL sequence and included both EcoRV and NcoI sites. PCR products were cut with XhoI and EcoRV and ligated into pCrc32 DNA cut with XhoI and SmaI; the resulting plasmids were designated sD41, sD20, and sD10, respectively. The NcoI site in the new plasmids facilitated screening of recombinant clones. The sequences of the primers used in this portion of the work were as follows:
 |
Conditions for all PCRs were 94°C, 1 min; 56°C, 1.5 min; 72°C, 30 sec. Total = 30 cycles.
Escherichia coli strain Stbl2 (GIBCO/BRL) was used as the host in cloning the PatpB-rbcL-GUS plasmids as well as the ST27, ST30, and ST27–30 plasmids described below.
ST27, ST30, and ST27–30 Mutations.
The +17 to +22 sequence of the rbcL gene was mutagenized by using MU15 (2) DNA and the complementary primers ST27U and ST27L. Wild-type sequences were replaced by SspI recognition sequences. With MU15 (2) DNA as the template in a PCR, primers ST27U and RBST350–3′ were used to produce a mutagenized −4 to +350 segment of rbcL. Primer RB1 and the mutagenized primer ST27L were used with MU15 DNA as a template to produce rbcL −82 to +47 DNA with mutations in the +17 to +22 region. The mutagenized −4 to +350 DNA produced in the former reaction and the −82 to +47 DNA product of the second reaction were mixed, denatured, and used in a final PCR employing the RB1 and RBST350–3′ primers. The product of the latter was digested with XhoI and EcoRV to produce a mutagenized −82 to +350 rbcL fragment that could be cloned into pCrc32 (6) DNA digested with XhoI and SmaI. The primers used were as follows:
 |
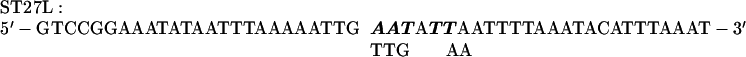 |
 |
 |
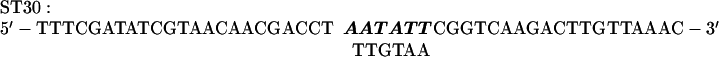 |
Mutations in the +329 to +334 region of the rbcL sequence were made by a PCR using MU15 (2) DNA as a template and the primers RB1 and ST30. ST30 introduces six mutations into the +329 to +334 sequence of rbcL:
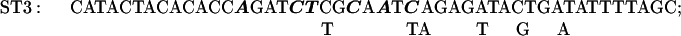 |
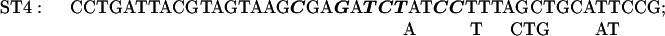 |
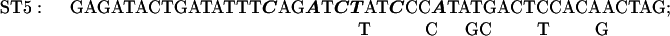 |
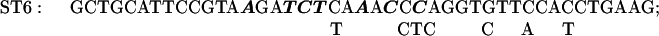 |
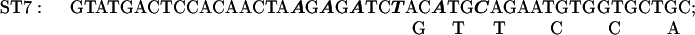 |
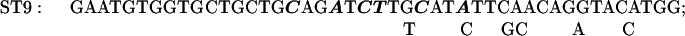 |
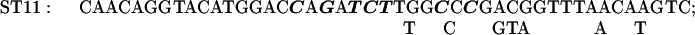 |
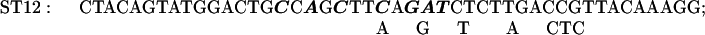 |
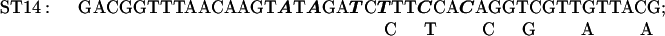 |
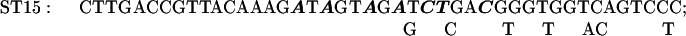 |
The mutagenized −82 to +350 fragment was digested with XhoI and EcoRV and cloned into pCrc32 digested with XhoI and SmaI to produce the plasmid pST30.
To produce a −82 to +350 rbcL fragment carrying both the ST27 and ST30 mutations, ST27 DNA was amplified by using RB1 and ST30 primers. The product was processed and cloned into pCrc32 as described above for ST27 and ST30 separately. The plasmid with the two sets of mutations produced in this way is designated ST27–30. The mutations in ST27 are complementary to those in ST30.
Other ST Mutations.
The Transformer site-directed mutagenesis kit (CLONTECH) was used to prepare the ST3–9, 11, 12, 14, and 15 mutations. The plasmid MU15 (2), containing the GUS reporter, was altered to construct mutants ST11, 14, and 15; the plasmid 15inUK19 (see below), containing the aadA reporter, was altered to construct mutants ST3, 4, 5, 7, 9, and 12.
The sequences of the mutagenic primers used are shown below; each changed/mutated nucleotide is shown in boldface type in the primer sequence in the upper line in each pair of lines and the nucleotide at that position in the original sequence is shown below each altered nucleotide.
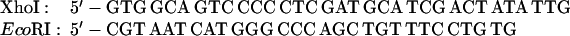 |
To facilitate the selection of mutagenized plasmids, XhoI and EcoRI sites were eliminated and new unique NSI and ApaI sites, respectively, were created through the use of synthetic oligonucleotide selection primers. [In the parent MU15 plasmid (2), the original XhoI site is upstream of the rbcL promoter. The modified EcoRI site was downstream of the reporter gene.] The sequences of the selection primers are as follows:
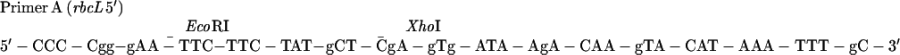 |
The selection primers are outside of the promoter or reporter gene sequences. Mutagenized MU15 DNAs were used to transform the nonphotosynthetic C. reinhardtii strain CC373. Transformants were selected for their ability to grow photoautotrophically (10, 11) and were checked for the presence of the GUS gene sequence.
As the first step in constructing aadA reporter-containing chimeric genes, a −709 to +350 rbcL sequence was amplified by PCR from MU15 (2) DNA by using the following synthetic primers, designed to create EcoRI or XhoI sites at the 5′ end and an NcoI site at the 3′ end. These restriction sites were designed to facilitate the cloning of the rbcL sequence in frame with the aadA reporter gene by using the atpX-aadA plasmid (14).
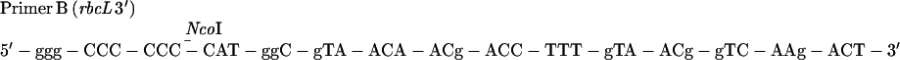 |
The PCR product was digested with EcoRI and NcoI and ligated with the atpX-aadA plasmid (12) DNA digested with EcoRI and NcoI. Then, the atpX-aadA plasmid containing the cloned rbcL sequence was digested with XhoI and SpeI. The XhoI–SpeI fragment, containing the rbcL sequence (−709 to +350 region) joined in-frame with the reporter aadA gene sequence, was ligated with the plasmid pMU19 (6) digested with XhoI and SpeI to obtain the plasmid 15inUK19 (also designated UK19–10). The plasmid 15inUK19 was used to generate all of the ST-series mutants containing the aadA reporter gene (i.e., ST3, 4, 5, 7, 9, and 12) by using the site-directed mutagenesis method and the various mutagenic primers/oligonucleotides and selection primers (i.e., XhoI and EcoRI primers) described above in this section. The Chlamydomonas transformation plasmid vector, 15inUK19, containing various mutations in the +170 to +350 region of the rbcL gene sequence (ST series), was used to transform the nonphotosynthetic C. reinhardtii strain Cc-373. Transformants were selected for their ability to grow photoautotrophically in the presence of spectinomycin and were checked for the presence of the aadA gene sequence.
Results
The present work stems from the observations that in C. reinhardtii chloroplasts: (i) the half-lives of rbcL transcripts are about 21 h during dark growth (in cells growing on cycles of 12 h light/12 h darkness) but only 3.5 h to 5 h in illuminated cells (2, 15); (ii) transcripts of chimeric genes containing the rbcL basic promoter sequence (≈−20 to ≈+60) and extending to +97 fused to the protein coding sequence of the bacterial uidA (GUS) have half-lives of 5 h in cells growing in darkness, but only 20 min in similar cells brought into the light [as a result of the latter, most of the transcripts that accumulate in darkness are degraded in cells that have been illuminated for 1 h (2)]; (iii) transcripts of a reporter gene composed of the rbcL promoter and sequences to +350 followed by the same GUS sequence as in the reporter gene described above accumulate in darkness but, unlike reporter transcripts with only 63 or 97 nucleotides transcribed from rbcL sequences, are almost completely stable during the first hour that cells are illuminated (2). These experiments showed that sequences that lie between +97 and +350 in the 5′ portion of the rbcL structural gene reverse the destabilizing effect of the presence of the +1 to +63 region in the transcript (2). The destabilizing and stabilizing effects of the rcbL sequences on the reporter transcripts were also exerted on chimeric genes containing bacterial aadA (14) or maize Lc protein coding regions (16) rather than GUS, showing that the survival behavior of the transcripts in these experiments was not a consequence of the presence of the GUS coding region.
Localization of the 5′ Degradation Target Region in Transcripts Containing rbcL Sequences.
The rbcL sequence between +1 and +63 (Fig. 1), which comprises the transcribed portion of the promoter extending from about −20 to +63 (6, 8), contains the target for degrading a reporter gene transcript rapidly after cells growing in darkness have been transferred into the light (2). About 70% of the transcripts of a GUS reporter gene containing rbcL nucleotides +1 to +63 in MU7 (2) that accumulate during 12 h of growth in darkness are destroyed during the first hour of illumination (Fig. 2). In contrast, only about 10% of the transcripts of MU15, a comparable reporter gene with rbcL nucleotides +1 to +350 (2), are destroyed.
Figure 1.

The sequence of C. reinhardtii rbcL from +1 (the transcription start site) through +350. Translation starts at +93. The basic promoter of this gene extends to +63 (2) and a transcriptional enhancer sequence lies between +63 and +170 (2). In some of the present experiments reporter genes were tested that contained the first 10 (+1 to +10), 20 (+1 to +20), 41 (+1 to +41), or 62 (+1 to +62) nucleotides of rbcL introduced downstream of the C. reinhardtii chloroplast atpB promoter (Figs. 3 and 4). In other experiments (Figs. 6 and 7), reporter genes were tested that contained the −709 to +350 segment of rbcL followed by a uidA (GUS) or aadA protein coding sequence and the 3′ terminal sequence of the psaB gene. Except for the control reporter, each reporter gene had a mutation at ST27, ST3, ST4, ST5, ST6, ST7, ST9, ST11, ST12, ST14, ST15, or ST30. A reporter gene with mutations at both ST27 and ST30 was also tested. Each mutagenized region is over- or underlined, and the nucleotides substituted within each mutated region are shown.
Figure 2.

The longevity of transcripts of MU15 and MU7 (2) reporter genes. Each contains a GUS coding sequence and a psaB 3′ terminus. MU15 contains rbcL sequences extending to +350 and MU7 rbcL to +97. Cells were grown on cycles of 12 h light/12 h darkness. D (dark) samples were taken just before illumination and L (light) samples were taken 1 h later. RNA was estimated by slot blotting using a GUS coding sequence probe. Representative blots are shown at right. The light/dark ratios (the fraction of GUS transcripts present in cells after 1 h of illumination compared with the amount of transcript present just before illumination) are calculated from values obtained by densitometry and shown at left.
We wanted to determine which part of the ribonucleotide 1 to 63 region is the target by testing transcripts of genes with portions of the +1 to +63 region deleted. However, previous work had shown that 3′ to 5′ deletions from +63 sharply reduce the activity of the rbcL promoter in vivo (6, 8). Consequently, as illustrated in Fig. 3, we introduced the +1 to +60 region and 3′ to 5′ deletions of this region of rbcL just downstream of a Chlamydomonas atpB promoter (11) and immediately upstream of the protein coding region of the E. coli uidA gene in pCrc32 (2). The cloned DNA was deposited on microprojectiles which were shot into cells of the C. reinhardtii strain ac-uc-2–21 (CC 373), which cannot carry on photosynthesis because part of the atpB gene is deleted. Transformants were selected by their capacity to grow photoautotrophically.
Figure 3.

A diagram showing the organization of reporter genes containing rbcL sequences +1 to +10, +1 to +20, +1 to +41, or +1 to +62 followed by a GUS protein coding sequence and the 3′ portion of psaB. The reporter genes are under the control of an atpB promoter (PatpB). The SmaI site into which the rbcL sequences were inserted is 111 nucleotides downstream of the transcription start site.
Transformed cells were grown on 12-h light/12-h dark cycles. Cells were sampled just before and 1 h after the beginning of the light period. RNA prepared from these cells was probed on slot blots with a GUS sequence as described in Materials and Methods. The ratio of GUS sequence-containing transcripts present in the cells after 1 h of illumination to such transcripts present at the end of the dark growth period was 1.1 in the control lacking the rcbL sequence (Fig. 4). Inclusion of ribonucleotides 1 to 20 transcribed from rbcL had no effect on the longevity of the reporter transcripts, but inclusion of 21 or 42 additional rbcL-encoded ribonucleotides (in Fig. 4, +1 to +41 and +1 to +62, respectively) reduced the lifetime of transcripts. Thus, the sequence transcribed from rbcL +20 to +41 is the target, or an essential part of the target, for an enzyme (probably an endonuclease) responsible for the commencement of photo-accelerated degradation.
Figure 4.

Longevity of transcripts of atpB promoter: GUS reporter genes containing rbcL sequences as shown in Fig. 3 (also see Materials and Methods). pCrc32 is the parent construct without any rbcL sequence inserted.
Identification of Stabilizing Sequences in rbcL.
The plasmid pMU15 (2) contains a chimeric gene composed of the −709 to +350 segment of rbcL followed by the GUS coding region and the psaB 3′ terminus. In the initial experiments, sets of 6 or 7 mutations were made in 12 to 14 nucleotide-long regions lying between +170 and +350. The substituted nucleotides are shown in Fig. 1 above or below the original sequence; the solid line above or below the sequence designates the extent of the mutagenized region. All but one of the chimeric genes tested in these experiments contained one of the sets of mutations shown in Fig. 1. The exception was a reporter gene with both ST27 and ST30 mutations, which is designated ST27–30. Each chimeric gene was cloned into a Chlamydomonas chloroplast transformation vector that promoted integration of the constructs by homologous recombination into C. reinhardtii chloroplast DNA adjacent to the 3′ end of the atpB gene (7, 11). As in the degradation target experiments described above, cells were grown on 12-h light/12-h dark cycles; cell samples were taken at the end of the dark period and after 1 h into the light period. Total cellular RNA from each transformant was analyzed by slot blotting. As indicated in Materials and Methods, the slot blots were probed with the 32P-labeled GUS or aadA DNA, and either rbcL DNA or 16S rDNA. (The latter served as an internal loading control.) The results of densitometric analyses of autoradiograms (corrected for loading) are presented in Fig. 5.
Figure 5.

The effects of each of a series of mutations in the +170 to +350 region of the C. reinhardtii rbcL gene sequence on the survival of rbcL: GUS or aadA reporter gene transcripts in cells illuminated for 1 h. See legends for Fig. 1 for the sites of the mutations and the substitutions made; see Figs. 2 and 4 (as well as the text and Materials and Methods) for experimental procedures. UK19–10 is a control wild-type construct.
The ratio of GUS transcripts present after 1 h in the light to those present at the end of the dark period was 0.93 for the control UK19–10. Mutation ST14 (+321 to +334) strikingly reduced the transcript-stabilizing effect of this region of the gene. Smaller effects were seen as the result of mutations ST15 (+337 to +350) and ST7 (+230 to +244).
A folding model (15, 16) for the nucleotide 1 to 350 portion of the transcript (Fig. 6) indicates that the mutations in ST14 could lie within a stem and loop (designated the +327 loop) structure predicted between nucleotides 323 and 340. The +327 loop would extend from nucleotide 327 to 336. The mutations present at the 5′ end of ST15 would lie in the stem of the same stem–loop structure; other mutations in ST15 would be beyond the predicted stem–loop. (The +327 loop and stem persist in folding models predicted under a number of different limiting parameters.) Another prominent (and larger) loop-and-stem structure in this model extends from nucleotides 1 to 41. The predicted loop itself, designated the +14 loop, would start at nucleotide 14 and extend to 27. Six of the eight nucleotides in the predicted +327 loop are complementary to nucleotides in the predicted +14 loop.
Figure 6.

A folding model for the RNA that would be transcribed from +1 to +350 of the C. reinhardtii rbcL gene (12, 13).
To investigate whether nucleotides in the predicted +14 loop play a role in the stabilization process, perhaps by interacting directly with nucleotides in a predicted +327 loop, mutations were made in the +17 to +22 (ST27) segment of the rbcL sequence. Transcripts of the reporter gene with the ST27 mutations show about the same reduced survival in illuminated cells as transcripts with mutations in ST14 (Fig. 7). These results indicate that nucleotides which were modified in ST27 are part of the rbcL transcript-stabilizing apparatus but, because the ST27 mutations do not abolish degradation of the transcripts (Fig. 4), they are not a part of the target for destruction of the transcripts.
Figure 7.

The effects of mutations ST27, ST30, and ST27–30 (Fig. 1) on the survival of rbcL-GUS reporter transcripts in illuminated cells. See legend for Fig. 5.
To investigate whether a direct interaction between the predicted +14 and +327 loops could have a role in stabilizing the transcript, mutations complementary to those in ST27 were made between +329 and +334 in the predicted +327 loop (ST30). Transcripts with ST30 mutations alone (i.e., modifications only in nucleotides of the predicted +327 loop) behaved similarly to transcripts with ST14 mutations with regard to survival after illumination of the cells. Thus, these mutations (in this predicted loop alone) are enough to eliminate the stabilizing effect of this region of the transcript. However, combining the ST30 and the complementary ST27 mutations in a single transcript did not restore the stabilizing capacity despite the complementarity of the ST30 and ST27 mutations. This observation suggests that direct interaction between these two sequences (which may lie in two loops) alone is probably not important for stabilization.
Discussion
We determined previously that the target for photo-accelerated degradation of transcripts of the Chlamydomonas chloroplast gene rbcL lies within the first 63 ribonucleotides (2). In the present work we have found that rbcL nucleotides 21 to 41 are required for the photo-accelerated degradation in vivo of the transcript of a reporter gene composed (from 5′ to 3′) of an atpB promoter, an embedded rbcL sequence, a GUS coding sequence, and a psaB 3′ termination sequence (2). A folding model for the rbcL transcript (Fig. 6) predicts the presence of a stem–loop structure extending from ribonucleotide 1 to 41. The stem is predicted to contain ribonucleotides 1 to 14 and 27 to 41. The loop itself is predicted to be composed of ribonucleotides 14 to 27. Elimination of the sequence from 20 to 41 abolishes the stem and half of the loop in models of transcripts of rbcL and of the chimeric genes containing rbcL sequences inserted behind atpB promoters constructed for these experiments. Transcripts with mutations in the loop region alone (ST27; Fig. 7) are degraded more rapidly than controls; therefore, this loop region does not appear to be part of the target for degradation. Consequently, the simplest interpretation of these data is that the target lies between ribonucleotides 27 and 41. However, the data do not exclude the possibility that the target is a region in the double-stranded RNA of which ribonucleotides 1 to 14 (as well as 27 to 41) are predicted to be parts.
In addition to providing more specific information regarding the target sequence than was available before, the data from experiments with transcripts of atpB-rbcL chimeras indicate that the initial attack on the rbcL transcript is most likely by an endonuclease. Based on the disappearance of the GUS transcript reporter, rather than production of discrete fragments detectable in Northern blots, we judge that subsequent destruction of the rbcL transcript could be by a 5′-to-3′ exonuclease and/or by one or more endonucleases that act only after the initial attack on the transcript. A 5′-to-3′ exo-RNase has recently been shown to be a component of the degradation pathway for petD transcripts in C. reinhardtii chloroplasts (17).
In our earlier work (2), we found that the longevity of rbcL sequence-containing transcripts is increased in the presence of gene sequences transcribed from between +170 and +350. The ST14 mutations, at +321 to +334 in a predicted stem and loop in the 5′ untranslated region, and the ST30 mutations, in a group of rbcL nucleotides in only the predicted loop region at +329 to +334, each one alone sharply reduces the effectiveness of this portion of the transcript for stabilization. The effects of these mutations are about the same as of the ST27 mutations in the predicted loop at the 5′ end of the transcript. We judge from these results that sequences in both the predicted +14 and +327 loops are involved in the stabilization process. Other sequences that we have identified (e.g., ST7) and sequences in regions we have not examined in detail (e.g., in the 5′ as well as in some 3′ portion of the transcripts) may also be involved. The possibility that other regions may be involved is suggested by the observations (i) that transcripts with mutations only at ST 15, ST14, or ST7 are not destroyed as rapidly as those lacking the entire ribonucleotide sequence transcribed from rbcL +98 to +350 (MU7 Fig. 2)—only about 30% of the transcripts survive after cells have been in the light for 1 h if only the first 97 nucleotides are included, but about 40% to 70% of the transcripts survive if any one of the other sets of downstream mutations is present in the first 350 nucleotides—and (ii) the half-lives of transcripts of the endogenous rbcL gene are substantially greater than those of reporter genes containing rbcL sequences only to +350 (2, 3).
We have not demonstrated that there is a direct or indirect (via proteins) physical interaction between the predicted +327 and +14 loops. Some nucleotides in the +329 region are complementary to nucleotides in the +17 region, but transcripts of constructs with compensatory changes in the two regions (i.e., in the ST27 and ST30 mutations) are not stabilized. This observation argues against the notion that direct interaction between nucleotides in the +327 and +14 regions (i.e., in the predicted 5′ and 3′ loops) is an important element of the stabilization process.
Another possibility is that in the presence of the rbcL sequence transcribed to ribonucleotide 350, one or more proteins associates with sequences in the 321 to 350 region (perhaps the ST7 and ST15 regions are also involved). Then, either this same protein (or set of proteins) or another protein (or set of proteins) associates with sequences in the 14 to 27 region. As a result of direct RNA–RNA interactions, which are argued against but not excluded by our experiments, or protein-mediated interactions with the nucleotide 14 to 27 region, the postulated endonuclease target site could be physically blocked or could become less accessible as a result of distortion (Fig. 8); the latter could obtain especially if the target is double-stranded RNA. The postulated endonuclease(s), 5′ to 3′ exonuclease(s), and proteins that bind to sequences required for stabilization, etc. all remain to be identified. Salvador and Klein (18) have determined that blocking photosynthetic electron transport prevents the previously observed (13) light-dependent breakdown of GUS transcripts in transgenic MU7 (2) C. reinhardtii cells. Furthermore, the presence of the oxidizing agent diamide in cultures of MU7 cells delayed the degradation of transcripts in the light, whereas addition of the photosynthetic electron transport blocker 3-(3,4-dichlorophenyl)-1,1-dimethylurea (DCMU) together with the reductant DTT resulted in induced degradation. This work shows that one or more of the postulated nucleases could be directly or indirectly activated by reduction.
Figure 8.

A diagrammatic representation. NS, the predicted nuclease site in the 21 to 40 region of the transcript; +14, the loop predicted to form in the 5′ region of the rbcL sequence of the transcript; +327, the loop predicted to occur downstream, within the protein coding sequence of rbcL. It is postulated that, for example, one protein associates with the +14 loop and another associates with the +327 loop. When the two proteins interact, the nuclease target is distorted and the effectiveness of endonuclease attacks is reduced. (See Discussion.)
The C. reinhardtii rbcL gene is also interesting from an evolutionary point of view. How did protein-encoding gene sequences become parts of the transcriptional and posttranscriptional expression-regulating apparatus of the gene? One can imagine that a sequence of events like the following could have occurred in the evolution of C. reinhardtii rbcL. Rubisco is a central enzyme in photosynthetic CO2 fixation and its abundance can limit plant growth (19). Consequently, evolutionary pressure would be toward increasing the abundance of the enzyme. The rbcL promoter, like promoters for other Chlamydomonas chloroplast genes that encode proteins, includes about 20 nucleotides upstream of the transcription start site and approximately 60 nucleotides that are transcribed (6, 8). Mutations in the basic ≈−20 to ≈+60 promoter that result in more active transcription would be favored. However, alterations in nucleotides in the transcribed portion of the promoter could inadvertently increase the susceptibility of the transcript to a ribonuclease. Such a negative effect on the abundance of the transcript would be ameliorated if another part of the transcript's sequence already existed or could evolve that could prolong the life of the transcript in some manner, e.g., by interfering with an initial step in degradation. In this case, there are severe constraints that limit mutations in the essential +329 to +334 stabilizer sequence. This sequence lies within the protein coding region of the gene. National Center for Biotechnology Information blast searches (20) reveal that the three encoded amino acids (Arg, Tyr, and two of the nucleotides in the Lys codon) are completely conserved in some angiosperms (e.g., Zea mays, Arabidopsis thaliana), ferns, and cyanobacteria (e.g., Synechocystis 6803, Prochloron, Anabaena nidulans, Prochlorothrix hollandica and some strains of Synechococcus and Prochlorococcus—in other strains of these species Phe replaces Arg) The encoding nucleotide sequence CGT TAC AAA in C. reinhardtii is present in rbcLs of Z. mays, ferns, and the cyanobacteria A. nidulans, S. 6803, and Prochloron. Consequently, the present situation in C. reinhardtii raises interesting evolutionary questions. The Arg-Tyr-Lys coding sequence was probably in place when the promoter was evolving. If so, the evolution of the promoter, in the presence of a potentially destructive endonuclease, was constrained by the existing sequences in the transcript.
Stabilization through direct physical interactions between the nucleotide 17 and the 329 regions of the transcript (regardless of whether or not stem–loop structures form) was postulated above because it seems to be a simpler way for the proposed stabilization process to have started. The interaction (if indeed that is how stabilization is effected) is likely to involve a protein or proteins that interact with the 327 and 14 sequences. Proteins that bind RNA or others present in the cell early could have been recruited and subsequently selected for increased effectiveness. The probable constraints on mutations in the +327 coding region of the gene and possibly counterproductive changes in the +14 transcribed but untranslated promoter region could all have influenced protein recruiting and fitting processes.
The rate of C. reinhardtii transcription is 10- to 100-fold greater from a reporter gene that includes rbcL DNA extending to +170 than that if only the basic promoter (≈−20 to +60) is present. The transcription enhancement depends on the presence of a sequence within the +126 to +170 region of the gene (6). The 15-aa residues encoded by this part of the gene are mostly conserved from some cyanobacteria through flowering plants (19). The exact extent of the essential enhancer sequence has not been determined but, as in the case of the +327 stabilizing region, the possibilities for nucleotide variation—including insertion of a protein binding sequence—are highly restricted. The problem of recruiting and fitting a DNA-binding enhancer protein to an existing DNA sequence is comparable to that discussed above with regard to the development of the transcript stabilizing system.
Regardless of the precise evolutionary history of the gene, at some point it most likely began to evolve not through the assembly of bits but as a unit including the transcribed portion of the promoter—located in the 5′ untranslated region—and the enhancer plus the 3′ portion of the stabilizing sequence, both of which are in the protein coding region.
Acknowledgments
We are grateful to Profs. Uwe Klein and Maria Louisa Salvador for helpful discussions and for the very useful suggestions they made in the course of this work and in the preparation of this manuscript. This work was supported by a research grant from the National Institute of General Medical Sciences. A. B. is on leave from the Institute of Basic Problems in Biology RAS, Puschino, Russia.
Abbreviation
- GUS
β-glucuronidase
References
- 1.Atwater J A, Wisdom R, Verma I M. Annu Rev Genet. 1990;24:519–541. doi: 10.1146/annurev.ge.24.120190.002511. [DOI] [PubMed] [Google Scholar]
- 2.Salvador M L, Klein U, Bogorad L. Proc Natl Acad Sci USA. 1993;90:1556–1560. doi: 10.1073/pnas.90.4.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nickelsen J van, Dillewijn J, Rahire M, Rochaix J D. EMBO J. 1994;13:3182–3191. doi: 10.1002/j.1460-2075.1994.tb06617.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Higgs D C, Shapiro R S, Kindle K L, Stern D B. Mol Cell Biol. 1999;12:8479–8491. doi: 10.1128/mcb.19.12.8479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Nickelsen J, Fleischmann M, Boudreau E, Rahire M, Rochaix J D. Plant Cell. 1999;11:957–970. doi: 10.1105/tpc.11.5.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein U, Salvador M L, Bogorad L. Proc Natl Acad Sci USA. 1994;91:10819–10823. doi: 10.1073/pnas.91.23.10819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blowers A D, Ellmore G J, Klein U, Bogorad L. Plant Cell. 1990;2:1059–1070. doi: 10.1105/tpc.2.11.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klein U, De Camp J D, Bogorad L. Proc Natl Acad Sci USA. 1992;89:3453–3457. doi: 10.1073/pnas.89.8.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finer J J, Vain P, Jones M W, McMullen M D. Plant Cell Rep. 1992;11:323–328. doi: 10.1007/BF00233358. [DOI] [PubMed] [Google Scholar]
- 10.Boynton J E, Gillham N W, Harris E H, Hosler J P, Johnson A M, Jones A R, Randolph-Anderson B L, Robertson D, Klein T M, Shark K B, et al. Science. 1988;240:1534–1538. doi: 10.1126/science.2897716. [DOI] [PubMed] [Google Scholar]
- 11.Blowers A D, Bogorad L, Shark K B, Sanford J C. Plant Cell. 1989;1:123–132. doi: 10.1105/tpc.1.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuker M. Science. 1989;244:48–52. doi: 10.1126/science.2468181. [DOI] [PubMed] [Google Scholar]
- 13.Jaeger J A, Turner D H, Zuker M. Proc Natl Acad Sci USA. 1989;86:7706–7710. doi: 10.1073/pnas.86.20.7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldschmidt-Clermont M. Nucleic Acids Res. 1991;19:4083–4089. doi: 10.1093/nar/19.15.4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salvador M L, Klein U, Bogorad L. Plant J. 1993;3:213–219. doi: 10.1046/j.1365-313x.1993.t01-13-00999.x. [DOI] [PubMed] [Google Scholar]
- 16.Ludwig S R, Habera L F, Delaporta S L, Wessler S R. Proc Natl Acad Sci USA. 1989;86:7092–7096. doi: 10.1073/pnas.86.18.7092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drager R G, Higgs D C, Kindle K L, Stern D B. Plant J. 1999;19:521–531. doi: 10.1046/j.1365-313x.1999.00546.x. [DOI] [PubMed] [Google Scholar]
- 18.Salvador M L, Klein U. Plant Physiol. 1999;121:1367–1374. doi: 10.1104/pp.121.4.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodermel S R, Abbott M S, Bogorad L. Cell. 1988;55:673–681. doi: 10.1016/0092-8674(88)90226-7. [DOI] [PubMed] [Google Scholar]
- 20.Altschul S F, Madden T L, Schaffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]