

Abstract
Background
Gene-environment interplay modulates Inflammatory Bowel Diseases [IBD]. Dioxin-like compounds can activate the Aryl Hydrocarbon Receptor [AhR] and alter macrophage function as well as T cell polarization. We hypothesized that attenuation of the AhR signaling pathway will ameliorate colitis in a murine model of IBD.
Design
DSS colitis was induced in C57BL/6 AhR null mice [AhR −/−], heterozygous mice [AhR−/+], and their wild type [WT] littermates. Clinical and morphopathological parameters were used to compare the groups. Patients: AhR pathway activation was analyzed in biopsy specimens from 25 IBD patients and 15 healthy controls.
Results
AhR −/− mice died before the end of the treatment. However, AhR −/+ mice exhibited decreased disease activity compared to WT mice. The AhR −/+ mice expressed less proinflammatory cytokines such as TNFα (6.1 versus 15.7 fold increase) and IL17 (23.7 versus 67.9 fold increase) and increased antiinflammatory IL-10 (2.3 fold increase) compared with the AhR+/+ mice in the colon. Colonic macrophage infiltration was attenuated in the AhR −/+ group. AhR and its downstream targets were significantly upregulated in IBD patients versus control (CYP1A1 – 19.9, and IL8-10 fold increase).
Conclusion
Attenuation of the AhR receptor expression resulted in a protective effect during DSS-induced colitis, while the absence of AhR exacerbated the disease. Abnormal AhR pathway activation in the intestinal mucosa of IBD patients may promote chronic inflammation. Modulation of AhR signaling pathway via the diet, cessation of smoking or administration of AhR antagonists could be viable strategies for the treatment of IBD.
Keywords: Inflammatory Bowel Diseases, Aryl hydrocarbon Receptor, Adiponectin, Angiotensin, ER stress, Th1, Th2, Th17
Introduction
Inflammatory Bowel Disease [IBD] is characterized by an inappropriate immune response to commensal flora. (1, 2) In Western countries, 1 in 200 patients are affected by Ulcerative Colitis [UC] or Crohn’s Disease [CD], the major forms of IBD and their incidence is steadily increasing. (3) The causes of IBD are unknown and the diagnosis is based on clinical, endoscopic, radiological and histological criteria. (4) Differences between familial and geographic clustering point toward environmental factors. In Caucasians, smoking has the strongest association with the severity of the gut inflammation. (5, 6)
Aryl Hydrocarbon receptor [AhR] is the only known receptor for dioxin, a potent immunomodulating environmental contaminant. Studies showed that cigarette smoke contains dioxins and dioxin-like chemicals. (7–9) The unbound AhR is present in the cytoplasm of all immune system cells. Furthermore, many genes involved in immune regulation possess multiple dioxin response elements [DREs] in their promoter region. (10, 11) Studies performed in AhR −/− mice have shown an enhanced inflammatory response to cigarette smoke or endotoxin, with elevated levels of tumor necrosis factor-α [TNFα] and interleukin-6 [IL-6]. (12, 13) AhR is essential in the regulation of cell cycle, lipid metabolism (14) circadian rhythm (15) and immune response. (16) Although AhR seems to be a crucial co-factor in regulation of both homeostasis and inflammation, its role in the gut autoimmune pathology is poorly described. Surprisingly, sustained activation of AhR by its high affinity, prototypical agonist, 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin [TCDD], resulted in altered bone marrow and thymic development, as well as susceptibility to infectious diseases and cancer in mice. (17, 18) Collectively, these observations suggest that a low to normal activation of AhR is beneficial for the protection against environmental insults, and represents an important link between environment and chronic diseases.
Our preliminary experiments indicated that AhR −/− mice are highly sensitive to DSS-induced colitis. In the current study, we examined the outcome of DSS-induced colitis in wild type and AhR heterozygous mice. Interestingly, moderation of the AhR signaling pathway in the AhR heterozygous mice was sufficient to ameliorate colitis. We also provide evidence that the level of AhR expression correlates with the profile of cytokines, adipokines and cellular stress response. These data indicate that attenuation of the AhR signaling pathway corresponds to an attenuated inflammatory response in the colon in disease-free conditions and during experimentally induced colitis. Furthermore, we extended our observations in IBD patients were AhR pathway was significantly upregulated when compared to healthy controls.
Materials and Methods
Subjects and sample collection
Colonic biopsies were obtained from 15 healthy controls and 25 IBD [Crohn’s Disease] patients. Control subjects were healthy individuals who underwent screening colonoscopy and had no inflammatory bowel conditions. IBD patients had an established diagnosis, based on standard endoscopic, radiologic, and histologic criteria. Informed consent was obtained before participation and the study protocol was approved by the Institutional Review Board of the University of Kentucky.
To examine the AhR activation in the lower digestive tract, biopsies were taken from the colon during standard colonoscopy. Two biopsies from each subject were fixed in formalin or RNA Later until analysis. The formalin fixed subset of biopsies was subject to standard histological staining and fluorescent immunohistochemistry. The RNA later subset of biopsies was used to assess the total mRNA using MagnaPure Compact RNA Isolation Kit [Roche]. cDNA was obtained using Transcription High Fidelity cDNA Synthesis Kit [Roche]. Specific mRNA levels were quantified by real time reverse transcription-polymerase chain reaction [RT-PCR], using the IQ iCycler [Bio-Rad], and SYBR Green qPCR Supermix [Bio-Rad]. Primers were obtained from SABiosciences, Frederick, MD. The mRNA levels for test genes were normalized to reference gene according to the comparative CT method also referred to as the 2−ΔΔCT method. The formula used: [2− [CT test − CT reference]] × 100%.
Animal treatments and sample collection
Male, 3 months old C57BL/6 and AhR −/− mice [Jackson Laboratory], or AhR −/+ mice bred in-house to C57BL/6 mice [n = 10 mice/group] were housed in a pathogen-free environment with free access to food and water. Mice were administered 3.5% [wt/vol] DSS [ICN Biochemical] in water for a week, followed by 3 days of water only. Body weight, stool consistency and rectal bleeding were monitored daily. On day ten, mice were euthanized with ketamine/xylazine [100/10 mg/kg ip] for blood and tissue harvest. The colons were removed and perfused with phosphate-buffered saline [PBS - pH 7.4], and measured. Half of each colon was fixed in RNAlater [Qiagen], and stored at −20°C. The other half was made in a “swiss roll”, cut and fixed in 10% buffered formalin [Sigma Chemical]. The Animal Care and Use Committee at the University of Kentucky approved all procedures.
Analysis of mRNA gene expression levels in mouse colonic tissue
On day 10, total RNA was purified using MagnaPure Compact RNA Isolation Kit [Roche] from whole colon tissue and cDNA was obtained using Transcription High Fidelity cDNA Synthesis Kit [Roche]. Specific mRNA levels were quantified by real time reverse transcription-polymerase chain reaction [RT-PCR], using the IQ iCycler [Bio-Rad], and SYBR Green qPCR Supermix [Bio-Rad]. Primers were designed using the Primer 3 software [SourceForge] and the sequences are shown in Table 1 [supplemental material].
The mRNA levels for test genes were normalized to reference gene according to the comparative CT method also referred to as the 2−ΔΔCT method. The formula used: [2− [CT test − CT reference]] × 100%.
Measurement of cytokines in the colonic tissue
At day 10 we measured cytokines in the colonic tissue homogenate using a bead based immunoassay [Lincoplex] multianalyte detection platform [Luminex - Mouse Cytokine Panel]. Values are expressed in picograms per milliliter and as mean ± SE, n=10/group.
Histology
Serial sections [5–7 μm] of paraffin embedded colons [swiss roles] were stained with hematoxylin and eosin. A pathologist blinded to the group allocation assessed the severity of colitis. The scoring system evaluated the following characteristics: [1] percentage of area involved, [2] number of follicle aggregates, [3] edema, [4] erosion/ulceration, [5] crypt loss, and [6] infiltration of mononuclear and polymorphonuclear cells. The total score ranges from 0 to 26. (19)
Proliferating Cell Nuclear Antigen [PCNA] assay
The sections were deparaffinized and treated with Antigen Retrieval Solution [DAKO, Carpinteria, CA] followed by incubation in 0.3% H2O2-methanol for 10 min, and wash. Sections were incubated in normal blocking serum for 90 min followed by overnight incubation in primary antibody, NCL-PCNA (1:200) [Novocastra, Leica Microsystems] at 4°C. The biotinilated secondary antibody [Elite ABC kit, Vector] was then applied for 2 hours. The slides were counterstained with Methyl Green [DAKO]. Images were taken with an Olympus BX51 microscope [Olympus America Inc.].
Fluorescent Immunohistochemistry
Formalin-fixed, paraffin embedded colon sections (5–7 μm) placed on coated slides were sequentially deparaffinized and rehydrated using xylene and ethanol. Slides were then treated for 10 min with a citric acid-based antigen-unmasking solution (Vector Laboratories, Burlingame, CA). Next, slides were incubated for 1 hour at room temperature in normal blocking serum (1% in PBS) and incubated overnight at 4°C with the respective primary antibody (dilution 1:50): Angiotensin II (BGN/0856/21) - a mouse anti-human monoclonal antibody, AT1 (306: sc-579) - a rabbit anti-human polyclonal antibody or ACE (H-170: sc-20791) - a rabbit anti-human polyclonal antibody, (Santa Cruz Biotechnology, INC, CA). On the second day, after washing, sections were incubated for 1 h with a mixture of Cy2-conjugated goat anti-mouse IgG and Cy3-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, catalog numbers 115-225-146 and 111-165-144). Sections were counterstained with 4′, 6-diamidino-2-phenylindole dihydrochloride (DAPI) (Molecular Probes Invitrogen, Eugene, OR) to visualize nuclei, and mounted with VECTASHIELD® medium (Vector Laboratories, Burlingame, CA). Images were taken with an Olympus BX51 microscope, using a 20x objective. We generated the composite images using Image-ProPlus 5.0 (Media Cybernetics, Inc.) software.
Statistical analysis
Data are expressed as Mean ± SE. Data were analyzed using unpaired t-test and one-way analysis of variance (ANOVA) [GraphPad Prism 5] followed by Tukey’s test with significance accepted at p < 0.05. Body weight data were analyzed by one-way ANOVA with repeated measures on time. Survival percentage has been calculated using Kaplan-Meier method. Significance was accepted at p < 0.05.
Results
The severity of DSS- induced colitis is decreased in AhR −/+ mice
Preliminary experiments showed that AhR KO mice do not survive beyond day 7 to DSS induced colitis [Fig 1A]. Therefore, in the current study, AhR −/+ and WT mice were analyzed. There was circa 50% reduction in AhR mRNA expression in colonic tissue of AhR −/+ mice that was maintained during colitis [Fig 1B]. Before DSS administration, there were no differences in body weight among groups. On day ten, WT mice lost significantly more weight [Fig 1C. p=0.001], had a significant drop in the hematocrit [Fig 1D, p=0.002], and the postmortem exam revealed swollen and shortened colons compared to AhR −/+ mice exposed to DSS [Fig 1E, p=0.01]. Microscopically, colitis was mild in the AhR −/+ group as compared with significant mucosal ulceration, crypt abscesses and altered architecture present in WT mice and reflected in the histology score [Fig 1F, p=0.001], and H&E stained colon sections [Fig 2A]. Furthermore, Proliferating cell nuclear antigen [PCNA] staining revealed a significant decrease in crypt proliferative activity in WT group as compared to the AhR −/+ group [Fig 2B], consistent with defective epithelial turnover. Importantly, there were no significant differences between AhR −/+ with and without colitis and the control water fed WT mice.
Figure 1. The severity of DSS-induced colitis is attenuated in AhR −/+ mice.

Mice were administered 3.5% DSS dissolved in water for 7 days followed by 3 days of water. Control mice received water alone for 10 days. [A] Kaplan-Meier plot showing premature lethality of male AhR −/− when compared with WT and AhR −/+ littermates [B] AhR mRNA gene expression in the colonic tissue [C] Body weight [D] Hematocrit [E] Colon length [F] Histopathological grading of colonic inflammation at the end of the experiment. Data are expressed as mean ± SE, n=10/group.
Figure 2. Decreased histological severity in AhR −/+ mice during DSS-induced colitis.
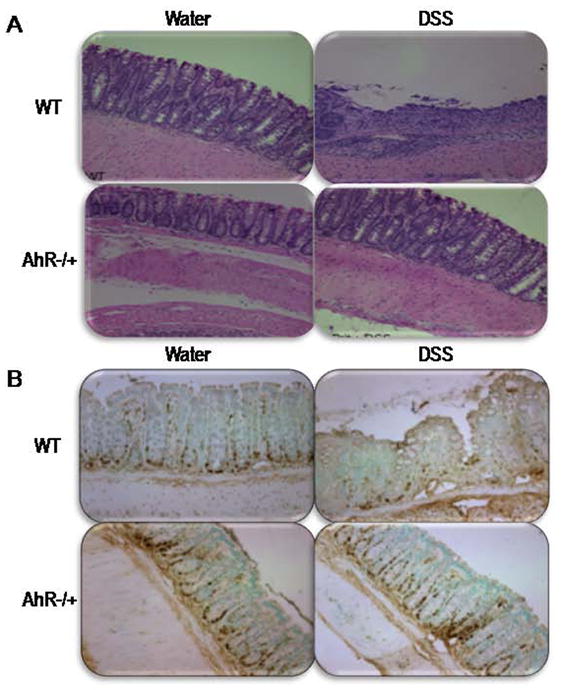
[A] H&E-staining of colons from WT [upper panel] and AhR −/+ mice [lower panel]. [B] Immunohistochemistry for epithelial cell proliferative index [PCNA] in sections of the colonic tissue of WT [upper panel] and AhR −/+ mice [lower panel].
AhR −/+ genotype favors Th2 over Th1 type cytokine expression
The cytokine milieu within the gut defines the outcome of both innate and acquired immune cells activation. We investigated the expression of classical Th1/Th2 cytokines previously documented in mice with DSS induced colitis and IBD patients [27]. Wild type mice exposed to DSS developed colitis that was characterized by increased expression of TNFα gene and protein level [Fig 3A, 3B, p=0.001]. Importantly, AhR −/+ mice had significantly lower baseline TNFα gene [Fig 3A] and protein level [Fig 3B] expression as compared to that in wild type mice and did not change during colitis. Intestinal macrophages play a central role in the immune response to commensal flora and become a source of Th1 cytokines in IBD. [28] The mRNA gene expression of the macrophage marker F4/80 [Fig 3C, p=0.018] was downregulated in AhR −/+ mice treated with water or DSS. The immunostaining of gut sections from AhR −/+ mice with colitis showed a significant decrease in macrophage infiltration [Macrophage-restricted protein-F4/80 -green, Fig 7], consistent with a dampened innate immune response as compared with WT mice with colitis. Although the monocyte chemoattractant protein [MCP1] expression increased during colitis in AhR −/+ mice, this was significantly lower compared to wild type mice [Fig 3D, p=0.005]. Baseline expression of IL10 within the colon was similar in control AhR −/+ and WT mice. Nevertheless, during DSS colitis there was significant upregulation of this anti - inflammatory cytokine only in AhR −/+ mice [Fig 3E, 3F, p=0.005]. In addition, we observed a more prominent induction of anti-inflammatory Secretory leukocyte protease inhibitor [SLPi] in AhR −/+ mice [suppl. Fig 10, p=0.0001] as compared to that in the WT mice during DSS induced colitis. SLPI is mainly expressed in the colon by the epithelial cells and is a potent inhibitor of Th1 like cytokines. (20)
Figure 3. The expression of pro-inflammatory cytokines and macrophage marker are reduced inAhR −/+ mice during colitis.

[A] TNFα gene expression, [B] TNFα protein level, [C] F4/80 gene expression [D] MCP1 gene expression, [E] IL10 gene expression and [F] IL10 protein level in colonic tissue. Data are expressed as mean ± SE, n=10/group
Figure 7. Reduced macrophage recruitment during DSS colitis in the AhR −/+ mice.

Fluorescent immunohistochemistry showing macrophage-restricted protein - F4/80 expression in colonic tissue of AhR−/+ mice with colitis. [A] Macrophage-restricted protein-F4/80 [green] in control groups, [B] Macrophage-restricted protein-F4/80 [green] in DSS-treated mice groups.
Th17 to Treg shift occurs during DSS-induced colitis in AhR −/+ mice
Aside from the classical Th1/Th2 paradigm, we further investigated the expression of master regulators for Th17 and Treg cells during colitis. Under basal conditions there were no differences between the AhR −/+ mice and WT [Fig 4A–D]. During DSS colitis there was a significant up regulation of Treg specific transcription factor FOXp3 gene expression in AhR −/+ mice as compared to that in the WT mice [Fig 4A, p=0.004]. In contrast, the mRNA expression of Th17 specific transcription factor, RORγ was significantly downregulated in AhR −/+ mice relative to the WT mice [Fig 4B, p=0.021]. Consistent with the latter finding we observed a significant attenuation of IL17 gene expression and protein level in AhR −/+ mice [Fig 4C–D].
Figure 4. Differential expression of master regulators for Treg and Th17 cells in the colon of WT and AhR −/+ mice.

Increased expression of [A] FoxP3 mRNA gene expression and attenuated gene expression of [B] RORγ and [C] IL17 with a low level of [D] IL17 protein expression in AhR −/+ mice treated with DSS. Data are expressed as mean ± SE, n=10/group.
Pro-inflammatory adipokines are decreased in AhR −/+ mice during DSS colitis
Mesenteric adipose tissue can become a source of inflammatory adipokines like angiotensin and osteopontin. Thus, during postmortem collection of the colons, we carefully removed the adjacent fat tissue. Nevertheless, these mediators can also be expressed in gut epithelia and lamina propria immune cells. [29–31] We confirmed that both AhR −/+ and WT C57BL/6 mice express components of the angiotensin system [Fig 5A–E] as well as osteopontin [ Fig 5D] within the colon. Pharmacological blockade of renin-angiotensin system has been shown to decrease the severity of colitis in several mouse models. [30, 32] We show that AhR −/+ mice treated with DSS presented a significant downregulation of the only known precursor of angiotensin I/II - angiotensinogen [Fig 5A, p=0.0001], as well as the angiotensin converting enzyme ACE [Fig 5B, p=0.0048], and angiotensin receptor AT1 [Fig 5C, p=0.003] gene expression compared to the WT group. Importantly, the AhR −/+ mice presented significant less AT1 mRNA gene expression under basal conditions [Fig 5C, p=0.023]. Compared to WT mice with colitis, the immunohistochemistry of colon sections of AhR −/+ mice with colitis revealed decreased angiotensin II, AT1 receptor [Fig 5E] and ACE expression [suppl. Fig 11].
Figure 5. Proinflammatory adipokines are decreased in AhR −/+ mice treated with DSS.

The pro-inflammatory adipokines [A] Angiotensinogen [B] ACE, [C] AT1a receptor, [D] Osteopontin gene expression were significantly lower in AhR heterozygous mice during colitis. Data are expressed as mean ± SE, n=10/group. [E] Fluorescence immunohistochemistry for angiotensin II [green] and AT1aR [red] in AhR −/+ and WT mice treated with DSS. [a], [b] show dual staining.
Adiponectin, the anti-inflammatory adipokine is increased in AhR −/+ mice during DSS induced colitis
Adiponectin, the only known anti-inflammatory adipokine, is considered an exclusive product of adipose tissue. Here, we show that adiponectin gene is also expressed in the colon [Fig 6A]. Next, we asked whether induction of colitis impairs adiponectin expression, and/or promotes a state of adiponectin resistance by down regulating its receptors. All the AhR −/+ mice presented a higher basal adiponectin mRNA gene expression that did not change during treatment. Importantly, adiponectin mRNA gene expression dropped significantly in the wild type mice during colitis [Fig 6A, p=0.0006]. Consequently, adiponectin receptors [AdipoQ R1 and R2] were downregulated during DSS colitis in all groups [Fig 6C, D]. Interestingly, T-Cadherin mRNA gene expression, a novel adiponectin receptor that serves to anchor adiponectin to cell surface (21), was upregulated only in the AhR −/+ mice and not in the WT mice with colitis [Fig 5B, p=0.037]. Moreover, AhR −/+ mice exposed to DSS had a lower expression of Protein of 44 kDa [Erp44] that inhibits the secretion of adiponectin oligomers from the endoplasmic reticulum [ER]. In addition, the ER oxidoreductase 1-Lα [Ero1-Lα], an ER chaperone that releases adiponectin trapped by Erp44 had similar expression in all groups. [Fig 6D–E, p=0.002]. Therefore, by modulating the ratio of these ER chaperones the attenuated AhR signaling pathway favors the secretion of the anti-inflammatory adiponectin during experimentally induced colitis.
Figure 6. Adiponectin is negatively regulated during colitis only in WT mice and not in the AhR −/+ mice.

[A] Adiponectin gene expression in WT mice treated with DSS was significantly lower compared to AhR −/+ mice. [B] Decreased T- Cadherin receptor expression in WT mice with colitis. [C] and [D] Decreased adiponectin receptors in the context of low adiponectin expression may further impair this adipokine signaling pathway. [E] Increased gene expression of chaperone protein ERP44 relative to its partner, endoplasmic reticulum oxidoreductin [ERO] can block the secretion of adiponectin in WT mice with colitis. Data are expressed as mean ± SE, n=10/group.
The AhR+/− mice with DSS induced colitis have less endoplasmic reticulum [ER] stress response
The development of colitis during DSS administration is associated with high cellular stress due to accumulation of misfolded proteins within the colonic epithelium. ER binding protein [BIP/Grp78] acts as an intracellular sensor and is considered a marker of ER stress (22). X-Box Binding Protein 1 [XBP1] is a transcription factor induced by ER stress that exerts a protective role and thus allows cells to recover. (23) In our study we observed a significant downregulation of ER stress markers BIP [p=0.018] and XBP1 [p=0.002] in the AhR −/+ mice exposed to DSS as compared to the WT mice with colitis [Suppl. Fig 9]. Since ER stress response is coupled to the cell death program, we investigated the mRNA expression of two pro-apoptotic molecules: C/EBP Homologous Protein [CHOP] and Caspase 12 [Casp12]. There was a significant downregulation of the expression of both pro-apoptotic genes only in the AhR −/+ mice exposed to DSS [Suppl. Fig 9].
AhR pathway is activated in patients with Inflammatory Bowe Diseases
In this study we have shown that AhR pathway can modulate the inflammatory response during experimental colitis. Then, using immunohistochemistry we investigated the pattern of AhR expression in healthy controls and patients with Crohn’s Disease. In human subjects without IBD, AhR expression was confined to the epithelial layer [Fig 8C], whereas in Crohn’s Disease there was a significant influx of AhR+ cells in the lamina propria compartment [Fig 8D] corresponding to pro-inflammatory hematopoietic cells. AhR pathway activation was assessed by measuring the expression of its downstream target, CYP1A1. There was negligible expression in healthy controls while a 19.9 fold upregulation was noted in IBD patients [Fig 8A]. IL-8 is a chemokine, which promotes neutrophil recruitment in patients with IBD. Human IL-8 promoter contains xenobiotic responsive elements [XRE] and thus can be upregulated by AhR signaling. (24) Similar to the prototypical AhR target CYP1A1, IL-8 expression was upregulated 10 fold in patients with Crohn’s Disease [Fig 8B].
Figure 8. AhR activation in patients with IBD.

(A) CYP1A1 mRNA gene expression level is increased in IBD patients compared with control. (B) IL8 mRNA gene expression increases during the inflammatory process in IBD patients. Control n=15, IBD n=25. Data are expressed as median. (C, D) Fluorescent Immunohistochemistry staining of the AhR in a control patient (C) versus a patient with Crohn’s disease (D). Representative images are shown of biopsies obtained from a control subject (C) and from a patient with Crohn’s disease (D). Red staining indicates binding of the antibody to human AhR. All samples were counterstained with DAPI (blue) to visualize nuclei. Magnification is 20X. E - Enterocytes and LP- Lamina Propria.
Discussion
Although the precise pathogenesis of IBD still needs to be unraveled, recent studies have reinforced the strong association between smoking, disease severity, complications and resistance to treatment. (6, 25) Mahid et al. found a greater prevalence of IBD patients in Kentucky, a state that ranks second in the nation in tobacco production and first in its use. (26) Mainstream cigarette smoke contains high level of dioxins and dioxin-like chemicals that are known to be strong inducers of AhR. (7, 8) In addition to their prolonged half life, these ubiquitous contaminants are highly lipophilic and accumulate in the adipose tissue (27), possibly leading to a prolonged activation of the AhR signaling pathway. (28)
Persistent activation of the AhR has been extensively researched using TCDD, a high affinity AhR agonist. (29) Administration of TCDD to laboratory animals induces inflammation by promoting tissue migration of immune cells (30, 31), and an increase in pro-inflammatory cytokine expression. (32, 33) Surprisingly, macrophages isolated from mice lacking the AhR produce higher amounts of the inflammatory cytokines IL-1, IL-6 and TNFα in response to LPS. (34, 35) Earlier studies indicated that AhR null mice develop colitis and rectal prolapse and have a propensity to develop colon cancer. (36) The inferred conclusion from the data obtained from either the ligand-activated AhR cell lineages or AhR null mice clearly states the physiological importance of this receptor in cell growth (37, 38), cell apoptosis (39), elaborate cross-talk with NF-kB (40, 41), and ER stress response. (42)
To explore the association between the AhR pathway and IBD pathogenesis, we induced colitis in AhR −/−, AhR −/+, and AhR +/+ mice. We found that mice lacking the AhR succumbed early to inflammation while the WT developed severe colitis. In comparison, the AhR heterozygous mice had a good clinical outcome during DSS administration and presented little structural changes in intestinal mucosa architecture.
Macrophages play an important role in the innate immune response to bacteria. In patients with Crohn’s disease, there is an influx of CD14+ macrophages within the gut, which become a rich source of TNFα. (43) Blockade of this proinflammatory cytokine induces disease remission in patients with IBD and in experimental models of colitis. (44, 45) We found that AhR −/+ mice express significantly lower levels of TNFα as compared to that observed in the WT groups under both, disease-free conditions and during experimentally induced colitis. Moreover, the expression of the Macrophage-restricted protein F4/80 was decreased in the colon of AhR −/+ mice during colitis. It is increasingly acknowledged that the AhR pathway modulates a number of immune responses.(46) While the specific AhR-induced mechanisms that underlie its effects on the immune system are poorly understood, numerous genes activated during the immune response have been found to contain DNA recognition sites for the AhR/ARNT heterodimers. [47, 48] Furthermore, the AhR pathway has been shown to play a significant role in the development of both Th17, and Treg cells. (47) The pathogenic role of Th17 cells as well as defective function of T-regulatory cells has been found in patients with Crohn’s disease and animal models of IBD. (48, 49) In the current study, we show that there is significant downregulation of the Th17 lineage master regulator, RORγ, with a corresponding upregulation of Treg transcription factor Foxp3 in the colon of AhR−/+ mice during colitis. Consistent with these changes, we have also noted a significant decrease in the IL-17 expression and a corresponding increase in IL-10 expression. Thus, the attenuation of the AhR signaling pathway correlates with the outcome of experimental colitis and the Th17/Treg balance in the colon.
The adipokines and cytokines secreted by adipose tissue have been increasingly recognized as bona-fide immune regulators. Angiotensin, a proinflammatory adipokine is generated from angiotensinogen through the proteolytic activity of ACE and tissue chymases. AT1a is the main receptor that mediates the pro-inflammatory actions of angiotensin. (50) Intestinal mucosa is a rich source of ACE, while macrophages express the full renin-angiotensin system. Increased ACE was reported in Crohn’s disease patients, while Ace/AT1 blockade protects mice from experimental colitis. (51, 52) In our study, AhR wild type mice that developed colitis had high expression of the angiotensin system components. Furthermore, there was a significant increase in lamina propria cells expressing both angiotensin and AT1a. We have previously shown that AhR agonists induce expression of angiotensinogen and proinflammatory cytokines in cultured adipocytes. (27) Our current finding that AhR −/+ mice fail to upregulate angiotensin system during DSS colitis further implicate this system in AhR- mediated inflammation.
Adiponectin, the primary adipokine with anti-inflammatory activity was downregulated in the wild type AhR mice that developed severe colitis, while the opposite was found in AhR −/+ group. We have previously demonstrated that AhR activation downregulates the expression of this adipokine in fat cells. (27) In the current study, we report that attenuated AhR expression/activity in the AhR −/+ mice, correlates with increased colonic adiponectin and its T-Cadherin receptor expression during DSS treatment. Moreover, our study indicates that AhR modulates the expression of ER chaperone proteins that regulate adiponectin secretion. These correlations might be very important since adiponectin has a protective role in experimental colitis as recently shown by the Nishihara group. (53) Furthermore, it was demonstrated that adiponectin treatment protects adiponectin KO mice from developing DSS-induced colitis. In addition, adiponectin has been shown to induce IL-10 (54), and thus, promote alternative activation of macrophages and resolution of chronic inflammation. In our study, the increased adiponectin in AhR −/+ mice may have had an important role in maintaining tissue homeostasis during DSS induced colitis.
Recent studies have shown that an increased endoplasmic reticulum stress response in epithelial cells promotes colitis. Importantly, mutations in XBP1, a key component of this response, have been associated with Crohn’s disease. (55) In the current study, DSS-induced colitis resulted in an increase expression of ER stress markers in wild-type mice, while they were downregulated in the AhR −/+ mice. Furthermore, increased IL-10 expression in the latter group may have also reduced the stress response associated with colitis. (56) It is also possible that the ER stress response during colitis facilitates AhR signaling and their reciprocal induction leads to the inflammatory response associated with DSS induced colitis. (57)
The effect of AhR activation on T cells is ligand dependent. TCDD induces persistent activation of AhR in Treg cells. (11) On the other hand dietary derived, short acting ligands, such as FICZ [6-formylindolo, 3, 2-b-carbazole] induce Th17 differentiation. (58) The relative abundance of the different ligands, along with the AhR system polymorphisms may further modulate the response. (59, 60) In our DSS model, AhR activation most likely occurred through dietary [i.e. FICZ] and endogenous ligands with similar kinetics, thus explaining the upregulation of IL-17 and RORγ. The increased expression of colonic macrophages in wild type mice compared to AhR −/+ further supports the role of this receptor in colitis. Recent in-vitro studies in macrophages described an ARNT independent, non-genomic pathway downstream of AhR that induces an inflammatory response. (61) We hypothesize that low AhR expression may be coupled to the classical signal that downregulates macrophage function whereas increased receptor expression preferentially activate the non-genomic pathway and hence promotes inflammation.
Our studies in patients without IBD indicate that AhR is mainly expressed in the epithelial layer. Nevertheless, this translated in minimal activation of this pathway and likely fulfills a physiologic role such as cell cycle regulation and metabolism of diet derived xenobiotics. AhR is important for the development of Th17 cells and can upregulate human macrophage expression of IL-8.(24) These activities are highly relevant for the ongoing intestinal inflammation of IBD patients. The robust AhR activation shown in our Crohn’s Disease patients was secondary to the influx of lamina propria AhR+ mononuclear cells, like macrophages and T cells. Consistent with this observation, we found high IL-8 expression that mirrored the AhR classical target, CYP1A1.
In summary, we provide novel evidence that dysregulated expression of the AhR alters the outcome of colitis. The extreme phenotypes of AhR null and wild types groups indicate that AhR pathway fulfills both tissue homeostatic and inflammatory roles. Furthermore, we show that AhR pathway activation distinguishes Crohn’s Disease patients from healthy controls. Modulation of this pathway through diet, cigarette smoking cessation, as well as pharmacological antagonism of the AhR could be viable strategies for the treatment of IBD.
Supplementary Material
mRNA gene expression in colonic tissue. Data are expressed as mean ± SE, n=10/group.
Fluorescent immunohistochemistry shows expression of ACE [red – upper panels], Angiotensin [green – lower panels] and their co-localization [middle panels] in the colonic tissues of mice treated with water [left panels] compared with DSS treatment [right panels].
Decreased mRNA gene expression of endoplasmic reticulum stress response proteins [BIP, XBP1] and related pro-apoptotic factors [CHOP, Casp12]. Data are expressed as mean ± SE, n=10/group
Acknowledgments
This work was supported by grants from NIH - DK07778-07 [VA], from BROAD MEDICAL FOUNDATION [RA] and University of Kentucky Physician-Scientist Award [RA]. There are no competing financial interests to declare.
Abbreviations
- Ao
Angiotensinogen
- ACE
Angiotensin Converting Enzyme
- AdipoQ
Adiponectin
- AdipoQ R1
2, Adiponectin Receptor 1, 2
- AhR
Aryl Hydrocarbon receptor
- ARNT
Aryl Hydrocarbon Receptor Nuclear Translocator
- AT1a
Angiotensin Receptor 1a
- BIP
Endoplasmic reticulum-binding protein [Hsp70]
- CASP12
Caspase 12
- CHOP
C/EBP homologous protein
- CYP1A1
Cytochrome P450, family 1, subfamily A
- ER
Endoplasmic Reticulum
- ERO
Endoplasmic reticulum oxidoreductin
- F4/80
Macrophage-restricted protein
- FOXp3
Forkhead box P3
- ERP44
ER protein of 44 kDa
- IBD
Inflammatory Bowel Disease
- MCP1
Monocyte chemotactic protein-1
- PCNA
Proliferating cell nuclear antigen
- SLPi
Secretory leukoprotease inhibitor
- RORγ
Retinoid-related orphan receptor gamma
- TCDD
dioxin
- TNFα
Tumor Necrosis Factor
- XBP1
X-box binding protein 1
References
- 1.Marquez A, Orozco G, Martinez A, et al. Novel association of the interleukin 2-interleukin 21 region with inflammatory bowel disease. Am J Gastroenterol. 2009;104(8):1968–75. doi: 10.1038/ajg.2009.224. [DOI] [PubMed] [Google Scholar]
- 2.Arsenescu R, Bruno ME, Kaetzel CS, et al. Signature biomarkers in Crohn’s disease: toward a molecular classification. Mucosal Immunol. 2008;1(5):399–411. doi: 10.1038/mi.2008.32. [DOI] [PubMed] [Google Scholar]
- 3.Bjorksten B. Disease outcomes as a consequence of environmental influences on the development of the immune system. Curr Opin Allergy Clin Immunol. 2009;9(3):185–9. doi: 10.1097/ACI.0b013e32832abfc2. [DOI] [PubMed] [Google Scholar]
- 4.Longstreth GF, Thompson WG, Spiller RC, et al. Functional bowel disorders. Gastroenterology. 2006;130(5):1480–91. doi: 10.1053/j.gastro.2005.11.061. [DOI] [PubMed] [Google Scholar]
- 5.Bhat M, Nguyen GC, Pare P, et al. Phenotypic and genotypic characteristics of inflammatory bowel disease in French Canadians: comparison with a large North American repository. Am J Gastroenterol. 2009;104(9):2233–40. doi: 10.1038/ajg.2009.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Calkins BM. A meta-analysis of the role of smoking in inflammatory bowel disease. Dig Dis Sci. 1989;34(12):1841–54. doi: 10.1007/BF01536701. [DOI] [PubMed] [Google Scholar]
- 7.Kitamura M, Kasai A. Cigarette smoke as a trigger for the dioxin receptor-mediated signaling pathway. Cancer Lett. 2007;252(2):184–94. doi: 10.1016/j.canlet.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 8.Kasai A, Hiramatsu N, Kitamura M, et al. High levels of dioxin-like potential in cigarette smoke evidenced by in vitro and in vivo biosensing. Cancer Res. 2006;66(14):7143–50. doi: 10.1158/0008-5472.CAN-05-4541. [DOI] [PubMed] [Google Scholar]
- 9.Stevens EA, Mezrich JD, Bradfield CA. The aryl hydrocarbon receptor: a perspective on potential roles in the immune system. Immunology. 2009;127(3):299–311. doi: 10.1111/j.1365-2567.2009.03054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quintana FJ, Cohen IR. Regulatory T cells and immune computation. Eur J Immunol. 2008;38(4):903–7. doi: 10.1002/eji.200838143. [DOI] [PubMed] [Google Scholar]
- 11.Ho PP, Steinman L. The aryl hydrocarbon receptor: a regulator of Th17 and Treg cell development in disease. Cell Res. 2008;18(6):605–8. doi: 10.1038/cr.2008.63. [DOI] [PubMed] [Google Scholar]
- 12.Thatcher TH, Maggirwar SB, Baglole CJ, et al. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB. Am J Pathol. 2007;170(3):855–64. doi: 10.2353/ajpath.2007.060391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swanson HI, Bradfield CA. The AH-receptor: genetics, structure and function. Pharmacogenetics. 1993;3(5):213–30. doi: 10.1097/00008571-199310000-00001. [DOI] [PubMed] [Google Scholar]
- 14.Minami K, Nakajima M, Yokoi T, et al. Regulation of insulin-like growth factor binding protein-1 and lipoprotein lipase by the aryl hydrocarbon receptor. J Toxicol Sci. 2008;33(4):405–13. doi: 10.2131/jts.33.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shimba S, Watabe Y. Crosstalk between the AHR signaling pathway and circadian rhythm. Biochem Pharmacol. 2009;77(4):560–5. doi: 10.1016/j.bcp.2008.09.040. [DOI] [PubMed] [Google Scholar]
- 16.Marshall NB, Kerkvliet NI. Dioxin and immune regulation: emerging role of aryl hydrocarbon receptor in the generation of regulatory T cells. Ann N Y Acad Sci. 1183:25–37. doi: 10.1111/j.1749-6632.2009.05125.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Currier N, Solomon SE, Demicco EG, et al. Oncogenic signaling pathways activated in DMBA-induced mouse mammary tumors. Toxicol Pathol. 2005;33(6):726–37. doi: 10.1080/01926230500352226. [DOI] [PubMed] [Google Scholar]
- 18.Andersson P, McGuire J, Rubio C, et al. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc Natl Acad Sci U S A. 2002;99(15):9990–5. doi: 10.1073/pnas.152706299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ten Hove T, Corbaz A, Amitai H, et al. Blockade of endogenous IL-18 ameliorates TNBS-induced colitis by decreasing local TNF-alpha production in mice. Gastroenterology. 2001;121(6):1372–9. doi: 10.1053/gast.2001.29579. [DOI] [PubMed] [Google Scholar]
- 20.Mizoguchi E, Podolsky DK, Mizoguchi A, et al. Colonic epithelial functional phenotype varies with type and phase of experimental colitis. Gastroenterology. 2003;125(1):148–61. doi: 10.1016/s0016-5085(03)00665-6. [DOI] [PubMed] [Google Scholar]
- 21.Hug C, Tsao TS, Lodish HF, et al. T-cadherin is a receptor for hexameric and high-molecular-weight forms of Acrp30/adiponectin. Proc Natl Acad Sci U S A. 2004;101(28):10308–13. doi: 10.1073/pnas.0403382101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee AS. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods. 2005;35(4):373–81. doi: 10.1016/j.ymeth.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 23.Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23(21):7448–59. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Podechard N, Lecureur V, Fardel O, et al. Interleukin-8 induction by the environmental contaminant benzo(a)pyrene is aryl hydrocarbon receptor-dependent and leads to lung inflammation. Toxicol Lett. 2008;177(2):130–7. doi: 10.1016/j.toxlet.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Bagaitkar J, Demuth DR, Scott DA. Tobacco use increases susceptibility to bacterial infection. Tob Induc Dis. 2008;4(1):12. doi: 10.1186/1617-9625-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahid SS, Stromberg AJ, Galandiuk S, et al. Active and passive smoking in childhood is related to the development of inflammatory bowel disease. Inflamm Bowel Dis. 2007;13(4):431–8. doi: 10.1002/ibd.20070. [DOI] [PubMed] [Google Scholar]
- 27.Arsenescu V, Arsenescu RI, Cassis LA, et al. Polychlorinated biphenyl-77 induces adipocyte differentiation and proinflammatory adipokines and promotes obesity and atherosclerosis. Environ Health Perspect. 2008;116(6):761–8. doi: 10.1289/ehp.10554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bohonowych JE, Denison MS. Persistent binding of ligands to the aryl hydrocarbon receptor. Toxicol Sci. 2007;98(1):99–109. doi: 10.1093/toxsci/kfm085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mandal PK. Dioxin: a review of its environmental effects and its aryl hydrocarbon receptor biology. J Comp Physiol B. 2005;175(4):221–30. doi: 10.1007/s00360-005-0483-3. [DOI] [PubMed] [Google Scholar]
- 30.Kerkvliet NI, Oughton JA. Acute inflammatory response to sheep red blood cell challenge in mice treated with 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD): phenotypic and functional analysis of peritoneal exudate cells. Toxicol Appl Pharmacol. 1993;119(2):248–57. doi: 10.1006/taap.1993.1066. [DOI] [PubMed] [Google Scholar]
- 31.Pande K, Moran SM, Bradfield CA. Aspects of dioxin toxicity are mediated by interleukin 1-like cytokines. Mol Pharmacol. 2005;67(5):1393–8. doi: 10.1124/mol.105.010983. [DOI] [PubMed] [Google Scholar]
- 32.Moos AB, Oughton JA, Kerkvliet NI. The effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on tumor necrosis factor (TNF) production by peritoneal cells. Toxicol Lett. 1997;90(2–3):145–53. doi: 10.1016/s0378-4274(96)03838-6. [DOI] [PubMed] [Google Scholar]
- 33.Vogel CF, Nishimura N, Matsumura F, et al. Modulation of the chemokines KC and MCP-1 by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in mice. Arch Biochem Biophys. 2007;461(2):169–75. doi: 10.1016/j.abb.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 34.Kimura A, Naka T, Nakahama T, et al. Aryl hydrocarbon receptor in combination with Stat1 regulates LPS-induced inflammatory responses. J Exp Med. 2009;206(9):2027–35. doi: 10.1084/jem.20090560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sekine H, Mimura J, Oshima M, et al. Hypersensitivity of AhR-deficient mice to LPS-induced septic shock. Mol Cell Biol. 2009 doi: 10.1128/MCB.00337-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawajiri K, Kobayashi Y, Ohtake F, et al. Aryl hydrocarbon receptor suppresses intestinal carcinogenesis in ApcMin/+ mice with natural ligands. Proc Natl Acad Sci U S A. 2009;106(32):13481–6. doi: 10.1073/pnas.0902132106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ray SS, Swanson HI. Dioxin-induced immortalization of normal human keratinocytes and silencing of p53 and p16INK4a. J Biol Chem. 2004;279(26):27187–93. doi: 10.1074/jbc.M402771200. [DOI] [PubMed] [Google Scholar]
- 38.Puga A, Marlowe J, Barnes S, et al. Role of the aryl hydrocarbon receptor in cell cycle regulation. Toxicology. 2002;181–182:171–7. doi: 10.1016/s0300-483x(02)00276-7. [DOI] [PubMed] [Google Scholar]
- 39.Marlowe JL, Fan Y, Chang X, et al. The aryl hydrocarbon receptor binds to E2F1 and inhibits E2F1-induced apoptosis. Mol Biol Cell. 2008;19(8):3263–71. doi: 10.1091/mbc.E08-04-0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vogel CF, Sciullo E, Matsumura F. Involvement of RelB in aryl hydrocarbon receptor-mediated induction of chemokines. Biochem Biophys Res Commun. 2007;363(3):722–6. doi: 10.1016/j.bbrc.2007.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vogel CF, Matsumura F. A new cross-talk between the aryl hydrocarbon receptor and RelB, a member of the NF-kappaB family. Biochem Pharmacol. 2009;77(4):734–45. doi: 10.1016/j.bcp.2008.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shimada T, Hiramatsu N, Hayakawa K, et al. Dual suppression of adipogenesis by cigarette smoke through activation of the aryl hydrocarbon receptor and induction of endoplasmic reticulum stress. Am J Physiol Endocrinol Metab. 2009;296(4):E721–30. doi: 10.1152/ajpendo.90829.2008. [DOI] [PubMed] [Google Scholar]
- 43.Kamada N, Hisamatsu T, Okamoto S, et al. Unique CD14 intestinal macrophages contribute to the pathogenesis of Crohn disease via IL-23/IFN-gamma axis. J Clin Invest. 2008;118(6):2269–80. doi: 10.1172/JCI34610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pache I, Rogler G, Felley C. TNF-alpha blockers in inflammatory bowel diseases: practical consensus recommendations and a user’s guide. Swiss Med Wkly. 2009;139(19–20):278–87. doi: 10.4414/smw.2009.12549. [DOI] [PubMed] [Google Scholar]
- 45.Tilg H, Moschen A, Kaser A. Mode of function of biological anti-TNF agents in the treatment of inflammatory bowel diseases. Expert Opin Biol Ther. 2007;7(7):1051–9. doi: 10.1517/14712598.7.7.1051. [DOI] [PubMed] [Google Scholar]
- 46.Esser C. The immune phenotype of AhR null mouse mutants: not a simple mirror of xenobiotic receptor over-activation. Biochem Pharmacol. 2009;77(4):597–607. doi: 10.1016/j.bcp.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 47.Quintana FJ, Basso AS, Iglesias AH, et al. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453(7191):65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 48.Brand S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut. 2009;58(8):1152–67. doi: 10.1136/gut.2008.163667. [DOI] [PubMed] [Google Scholar]
- 49.Bai A, Chen J, Peng Z, et al. All-trans retinoic acid down-regulates inflammatory responses by shifting the Treg/Th17 profile in human ulcerative and murine colitis. J Leukoc Biol. 2009;86(4):959–69. doi: 10.1189/jlb.0109006. [DOI] [PubMed] [Google Scholar]
- 50.Cassis LA, Lu H, Daugherty A, et al. Bone marrow transplantation reveals that recipient AT1a receptors are required to initiate angiotensin II-induced atherosclerosis and aneurysms. Arterioscler Thromb Vasc Biol. 2007;27(2):380–6. doi: 10.1161/01.ATV.0000254680.71485.92. [DOI] [PubMed] [Google Scholar]
- 51.Jaszewski R, Tolia V, Ehrinpreis MN, et al. Increased colonic mucosal angiotensin I and II concentrations in Crohn’s colitis. Gastroenterology. 1990;98(6):1543–8. doi: 10.1016/0016-5085(90)91088-n. [DOI] [PubMed] [Google Scholar]
- 52.Santiago OI, Rivera E, Appleyard CB, et al. An angiotensin II receptor antagonist reduces inflammatory parameters in two models of colitis. Regul Pept. 2008;146(1–3):250–9. doi: 10.1016/j.regpep.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nishihara T, Matsuda M, Araki H, et al. Effect of adiponectin on murine colitis induced by dextran sulfate sodium. Gastroenterology. 2006;131(3):853–61. doi: 10.1053/j.gastro.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 54.Wolf AM, Enrich B, Tilg H, et al. Adiponectin induces the anti-inflammatory cytokines IL-10 and IL-1RA in human leukocytes. Biochem Biophys Res Commun. 2004;323(2):630–5. doi: 10.1016/j.bbrc.2004.08.145. [DOI] [PubMed] [Google Scholar]
- 55.Kaser A, Lee AH, Franke A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134(5):743–56. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shkoda A, Ruiz PA, Daniel H, et al. Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: impact on chronic inflammation. Gastroenterology. 2007;132(1):190–207. doi: 10.1053/j.gastro.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 57.Horikawa K, Oishi N, Nakagawa J, et al. Novel potential of tunicamycin as an activator of the aryl hydrocarbon receptor -- dioxin responsive element signaling pathway. FEBS Lett. 2006;580(15):3721–5. doi: 10.1016/j.febslet.2006.05.061. [DOI] [PubMed] [Google Scholar]
- 58.Veldhoen M, Hirota K, Westendorf AM, et al. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature. 2008;453(7191):106–9. doi: 10.1038/nature06881. [DOI] [PubMed] [Google Scholar]
- 59.Esser C, Rannug A, Stockinger B. The aryl hydrocarbon receptor in immunity. Trends Immunol. 2009;30(9):447–54. doi: 10.1016/j.it.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 60.Stockinger B, Veldhoen M, Hirota K. Modulation of Th17 development and function by activation of the aryl hydrocarbon receptor--the role of endogenous ligands. Eur J Immunol. 2009;39(3):652–4. doi: 10.1002/eji.200839134. [DOI] [PubMed] [Google Scholar]
- 61.Sciullo E, Vogel C, Li W, Matsumura F, et al. Initial and extended inflammatory messages of the nongenomic signaling pathway of the TCDD-activated Ah receptor in U937 macrophages. Arch Biochem Biophys. 2008;480(2):143–55. doi: 10.1016/j.abb.2008.09.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
mRNA gene expression in colonic tissue. Data are expressed as mean ± SE, n=10/group.
Fluorescent immunohistochemistry shows expression of ACE [red – upper panels], Angiotensin [green – lower panels] and their co-localization [middle panels] in the colonic tissues of mice treated with water [left panels] compared with DSS treatment [right panels].
Decreased mRNA gene expression of endoplasmic reticulum stress response proteins [BIP, XBP1] and related pro-apoptotic factors [CHOP, Casp12]. Data are expressed as mean ± SE, n=10/group