

Abstract
G-protein coupled receptor kinase 2 (GRK2) is a member of a kinase family originally discovered for its role in the phosphorylation and desensitization of G-protein coupled receptors. It is expressed in high levels in myeloid cells and its levels are altered in many inflammatory disorders including sepsis. To address the physiological role of myeloid cell-specific GRK2 in inflammation, we generated mice bearing GRK2 deletion in myeloid cells (GRK2Δmye). GRK2Δmye mice exhibited exaggerated inflammatory cytokine/chemokine production, and organ injury in response to lipopolysaccharide (LPS, a TLR4 ligand) when compared to wild type littermates (GRK2fl/fl). Consistent with this, peritoneal macrophages from GRK2Δmye mice showed enhanced inflammatory cytokine levels when stimulated with LPS. Our results further identify TLR4-induced NFκB1p105-ERK pathway to be selectively regulated by GRK2. LPS-induced activation of NFκB1p105-MEK-ERK pathway is significantly enhanced in the GRK2Δmye macrophages compared to GRK2fl/fl cells and importantly, inhibition of the p105 and ERK pathways in the GRK2Δmye macrophages, limits the enhanced production of LPS-induced cytokines/chemokines. Taken together, our studies reveal previously undescribed negative regulatory role for GRK2 in TLR4-induced p105-ERK pathway as well as in the consequent inflammatory cytokine/chemokine production and endotoxemia in mice.
Introduction
G-protein coupled receptor kinases (GRKs) are enzymes that phosphorylate activated G-protein coupled receptors (GPCRs) and cause desensitization of G-protein-dependent signaling. GRK2 is one of seven members of GRKs and is widely expressed (De Blasi et al., 1995; Loudon et al., 1996). GRK2 levels are altered in immune cells from human patients with a variety of inflammatory disorders, as well as, in a number of animal disease models (Giorelli et al., 2004; Lombardi et al., 2001; Lombardi et al., 1999; Vroon et al., 2005; Vroon et al., 2003). In particular GRK2 levels are markedly increased in neutrophils from septic patients (Arraes et al., 2006). Treatment of neutrophils and macrophages (Mϕ) with TLR ligands upregulates GRK2 levels significantly (Alves-Filho et al., 2009; Loniewski et al., 2008). This increase in GRK2 levels has been postulated to be important in limiting chemokine receptor (a GPCR)-induced chemotaxis of immune cells. In fact, neutrophils from human septic patients show significantly attenuated chemotaxis (Arraes et al., 2006). Several other studies have also determined the role of GRK2 in immune cell chemotaxis owing to the fact that chemokine receptors belong to the GPCR family and that the observations are remarkably in-tune with the known classic role for GRK2, i.e. GPCR desensitization. In spite of these seminal advances in GRK2 biology, role of GRK2 in Mϕ, particularly in response to non-GPCRs, is not well understood. More importantly, the role of myeloid cell-specific GRK2 in lipopolysaccharide-induced inflammation and endotoxemia in vivo is not known.
Lipopolysaccharides (LPS) activate a class of innate immune receptors called the Toll-like receptors (TLRs) which act as the first line of host defense against bacterial infections (Beutler, 2009). Among the TLRs, TLR4 is activated by LPS from gram-negative bacteria that triggers an inflammatory response (Beutler, 2009). Under endotoxemic conditions, however, this system is over-stimulated and the exaggerated cytokine response elicited by the host turns harmful and leads to endotoxic shock and eventual death (Salomao et al., 2008). In addition to endotoxic shock and sepsis, TLR4 is now proposed to be an important player in a number of human and animal inflammatory diseases (Beutler, 2009). Activation of TLR4 by LPS triggers the recruitment of adapter proteins such as TRIF and Myd88 as well as other TIR domain containing proteins that eventually activates the inhibitor of κB kinase (IKK) complex (O'Neill and Bowie, 2007). The activated IKK complex then phosphorylates IκBα (an inhibitor of NF-κB) thereby targeting it for ubiquitination and proteasomal degradation. IκBα degradation enables the release and nuclear translocation of NF-κB, which then regulates the expression of genes involved in inflammation and innate and adaptive immune responses. In macrophages, activation of IKK complex also phosphorylates NFκB1 p105 (another IκB protein), which normally is stoichiometrically bound to a MAP3K called TPL2. LPS stimulation and phosphorylation of p105 leads to partial degradation and subsequent release of TPL2. P105-free TPL2 activates MEK1/2, and ultimately the ERK1/2 pathway (Beinke et al., 2004; Waterfield et al., 2004). In addition to these pathways, LPS also mediates the activation of p38, JNK, and Akt signaling pathways (Symons et al., 2006). TLR4-induced activation of these signaling pathways and the subsequent activation of transcription factors, such as NFκB, AP-1 and EGR-1, mediate the pathogenesis of inflammation and endotoxemia and shock (Salomao et al., 2008; Symons et al., 2006; Wong and Tergaonkar, 2009).
Even though one consequence of TLR4-mediated regulation of GRK2 levels in Mϕ and neutrophils is modulation of GPCR activity, we postulated that this regulation might have a feed back role in TLR4 signaling. To test this hypothesis, we generated myeloid cell-specific knockout of GRK2 and determined the role and mechanisms by which GRK2 regulates TLR4 signaling in vivo and in primary Mϕ. We demonstrate here that myeloid cell-specific GRK2 negatively regulates TLR4-induced inflammatory cytokines/chemokines and limits the pathogenesis of endotoxemic shock in mice. Furthermore, we also provide evidence that the pro-inflammatory phenotype of GRK2-deficient Mϕ is in part due to enhanced NFκB1p105-ERK pathway.
Materials and methods
Materials
Antibodies were from Santacruz Biotech (ERK, p50 and actin-HRP) or from Cell Signaling Technology (most antibodies). E. coli LPS (0111:B4) was from Sigma and ultrapure LPS was from Invivogen.
Animals
Animals were housed four to five mice per cage at 22–24°C in rooms with 50% humidity and a 12-h light–dark cycle. All animals were given mouse chow and water ad libitum. All animal procedures were approved by the Michigan State University Institutional Animal Care and Use Committee and conformed to NIH guidelines.
Generation of myeloid cell specific GRK2 deficient mice
GRK2fl/fl mice in which exons 3-6 of GRK2 are flanked by LoxP sites were crossed with LysMCre mice to generate GRK2fl/fl LysMCre mice (Matkovich et al., 2006). A breeding colony was maintained by mating GRK2fl/fl with GRK2fl/fl+LysMCre. The mice were generated on a mixed C57BL6/129sv background. GRK2fl/fl+LysMCre were used in experiments and compared to littermate GRK2fl/fl controls. LysMCre and GRK2lox/lox mice were genotyped as described previously (Clausen et al., 1999; Matkovich et al., 2006). Myeloid cell GRK2 deleted mice will be referred to as GRK2Δmye and littermate controls as GRK2 fl/fl mice. Male mice 6-8 weeks of age were used for the experiments.
Peritoneal Macrophage and Neutrophil isolation
To isolate peritoneal cells, mice were injected with 1 ml of 4% thioglycollate (i.p.) for 3 hours (for Neutrophils) or 4 days (for macrophages). Cells were collected as described before (Greten et al., 2007). Cells were plated in 12 well plates and serum starved for 3-4 hours before stimulation.
Cytokine analysis
A mouse 23-plex assay was used to determine the cytokine/chemokine concentrations according to manufacturer's instructions via Luminex 100 technology as described previously (Appledorn et al., 2008). Plasma cytokine levels are expressed as pg/ml of plasma. Cytokine levels in the cell culture supernatants were normalized to cellular protein levels and expressed as pg/μg of total cellular protein.
Bronchioalveolar lavage fluid (BALF)
Bronchioalveolar lavage fluid was collected at different intervals following LPS injection (using 2 ml of 0.9% Normal saline). In each mouse, around 90% of the total injected volume was consistently recovered. The BALF was centrifuged at 450 × g for 10 min and the supernatants were used to determine the concentration of total proteins using Bradford assay with bovine serum albumin (BSA) as a standard.
Western blot analysis
Cells were lysed in lysis buffer (20 mM Tris-HCl (pH 7.4), 1 mM EDTA, 150 mM NaCl) containing 1% Triton X-100 and protease inhibitors (Protease inhibitor cocktail, Roche Diagnostics). Insoluble material was removed by centrifugation (13,000 × g, 10 min., 4°C) and protein concentration of the supernatants determined by Bradford assay. Western blotting was performed as described previously (Loniewski et al., 2007; Patial et al., 2009).
Morphological assessment of liver injury
Livers from GRK2fl/fl and GRK2Δmye mice were fixed in 10% buffered formalin solution for histological examination. Tissue sectioning (5 micron) and staining with Hematoxylin and Eosin were performed at MSU Investigative Histolopathology Lab. For glycogen staining, tissue sections were stained to demonstrate the periodic acid-schiff (PAS) reaction in which an intense red dye reacts with aldehyde groups on carbohydrate-rich macromolecules like glycogen, mucus, and basement membrane material. Subsequent treatment with diastase (an amylase) enzymatically dissolves glycogen, leaving a clear or non-staining area where the previously red PAS-positive glycogen had been detected in the cell cytoplasm. Mucus and other non-glycogen continue to stain intensely red after diastase treatment. All sections were blinded with regard to the genotype before microscopic analysis.
Survival study
Six to eight week old mice were injected with LPS (20 μg/gm body weight, i.p.) from Escherichia Coli (serotype 0111:B4; Sigma-Aldrich, St. Louis, MO). The mice were monitored for LPS induced lethality every 6 hours for a period of 48 hours. Differences in survival were analyzed using a Kaplan-Meier test (Prism 5 software, Graph Pad Software, La Jolla, CA).
Statistical analysis
All values are represented as mean ± SEM. Each “N” represents individual mouse. Data were analyzed and statistics performed using GRAPHPAD PRISM software (La Jolla, California). The Student's t-test was used to compare mean values between two experimental groups and Analysis of Variance (ANOVA) with Bonferroni post-test was used to compare more than two groups. P value of less than 0.05 was considered significant.
Results
Generation of myeloid cell specific GRK2 deficient mice was accomplished by breeding mice expressing Cre recombinase under the control of the Lysozyme-M (LysM) promoter [LysM-Cre mice express Cre-recombinase specifically in myeloid cells (Clausen et al., 1999)] with mice homozygous for Lox-P flanked GRK2 alleles (GRK2fl/fl) (Matkovich et al., 2006). Double heterozygous mice obtained from this breeding (GRK2fl/- LysMCre+/-) were further intercrossed to obtain GRK2fl/fl LysMCre+/- mice (myeloid -specific GRK2 deficient: GRK2Δmye) and GRK2 fl/fl (control mice). A breeding colony was then maintained to generate both GRK2fl/fl LysMCre+/- as well as GRK2fl/fl mice littermates needed for these studies. Using this strategy, peritoneal macrophages (Mϕ), neutrophils and bone marrow derived macrophages (but not lungs and spleen homogenates) showed marked decrease in GRK2 protein (~95-99%) in GRK2Δmye mice compared to GRK2fl/fl mice (Fig 1).
Figure 1. Generation and characterization of myeloid-cell specific GRK2 knockout.

A. Schematic representation of the presence of loxP sites surrounding exons 3-6 that were targeted for deletion with Cre recombinase. Cre mediated recombination leads to the deletion of exons 3-6 of GRK2 gene.
B. Western blot showing the GRK2 protein levels in thioglycollate elicited macrophages, neutrophils and bone-marrow derived macrophages, lungs and spleen from GRK2Δmye and littermate GRK2fl/fl mice. Tubulin is shown as a loading control.
LPS-induced MIP-1α, IL-12p40 and IL-10 are enhanced in GRK2Δmye mice in vivo
To determine if GRK2 regulates TLR4 signaling in vivo, GRK2Δmye and GRK2fl/fl mice were injected with LPS (30 μg/g body weight, i.p., (Greten et al., 2007)) and levels of various cytokines/chemokines determined in the plasma using 23-plex cytokine assay. Of the various cytokines, plasma levels of MIP1α, IL-12(p40) and IL-10 were significantly enhanced in the GRK2Δmye mice compared to GRK2fl/fl. In particular, levels of MIP1α were markedly enhanced in the GRK2Δmye mice at 1 and 3 hours post-LPS injection, whereas IL-12p40 and IL-10 levels were significantly elevated in the GRK2Δmye mice at 12-hours post-LPS injection (Fig 2). Although levels of MIP1β, IL-6 and IL-17 were also enhanced in GRK2Δmye mice, it did not reach statistical significance. A similar trend was also observed for IL-1α, IL-1β, eotaxin, RANTES, MCP1 and IL-9 (data not shown). In contrast, plasma levels of IL-2, IL-3, IL-4, IL-5, IL-12(p70), IL-13, TNFα, IFNγ, GCSF, GMCSF and KC were not different between the two genotypes (data not shown). Taken together, these results suggest that myeloid cell GRK2 negatively regulates a group of inflammatory cytokines/chemokines in vivo.
Figure 2. Enhanced plasma inflammatory cytokines in GRK2Δmye mice.

Mice were injected with LPS and blood collected at the indicated time points. Levels of cytokines and chemokines were assessed using a Biorad 23-plex. N=11 mice per genotype each for 1, 3 and 12 hour time point; N=6 mice per genotype each for 0, 6 and 18 hour time points. *p<0.05; **p<0.01.
Enhanced tissue injury and mortality in GRK2Δmye mice
The profound increase in inflammatory factors elicited by LPS in mice is in part responsible for organ injury observed in this model (Rittirsch et al., 2007). To determine if the enhanced cytokine levels observed in the GRK2Δmye mice also leads to exaggerated tissue injury, we focused on liver and lung after LPS injection.
Liver injury
To assess liver injury, we performed histopathological analysis of the liver samples from LPS-injected, GRK2fl/fl and GRK2Δmye mice. As predicted, the inflammatory response to LPS in the GRK2Δmye mice was considerably enhanced as evidenced by a much more intense infiltration of inflammatory cells within the parenchyma and a prominence of portal areas due to focally marked perivascular edema with accumulation of fibrin strands. (Fig 3A-D). Furthermore, we also observed that LPS-induced glycogen depletion in the liver was markedly attenuated in the GRK2Δmye mice compared to the GRK2fl/fl mice (Fig 3E-J), suggesting that myeloid cell-specific GRK2Δmye mice also have dysregulated glycogen metabolism.
Figure 3. Enhanced tissue injury and mortality in GRK2Δmye mice.

A-D. Morphological changes in liver of mice (GRK2fl/fl and GRK2Δmye) injected with LPS (12 hours). A. GRK2fl/fl mice. B. GRK2Δmye mice. Arrows show examples of increased parenchymal immune cells, arrowheads show fibrin in venous spaces. (H&E, original magnification × 10). C. GRK2fl/fl mice liver. Immune cells and minimal edema present in periportal areas. D. GRK2Δmye mice liver. Numerous immune cells; star shows edema; arrowhead shows fibrin strands in prominent periportal areas (H&E, original magnification, × 20).
E-J. PAS reaction staining (for glycogen) of mouse liver section from PBS injected GRK2fl/fl (E), PBS injected GRK2Δmye (F), LPS injected GRK2fl/fl (G), LPS-injected GRK2Δmye mice (H). Specificity of cytoplasmic glycogen staining (after diastase treatment) is demonstrated in I and J, by loss of intense red staining in cell cytoplasm due to dissolution of glycogen by enzyme. Focal clusters of glycogen-rich cells show cytoplasmic clearing after diastase treatment in J but rare individual cells show clearing in I.
K. Mice were injected with LPS and broncho-alveolar lavage fluid collected after 12 hours. The cells were pelleted and the total protein content of the supernatant measured using Bradford assay (N=6 mice per genotype)
L. Survival of GRK2Δmye and GRK2fl/fl mice after LPS injection. N=10 mice per genotype.
Lung Injury
Acute lung injury in sepsis is associated with lung edema with extravasation of plasma proteins due to increased vascular permeability. Thus, detection of proteins in bronchoalveolar lavage (BAL) fluid serves as one useful indicator of lung injury (Duniho et al., 2002). GRK2fl/fl and GRK2Δmye mice were injected with LPS for 12 hours and total protein in bronchoalveolar lavage (BAL) fluid was determined. As shown in Fig. 3K there was a significant increase in protein content in GRK2Δmye compared GRK2fl/fl mice suggesting that the lung injury in response to LPS is worse in the GRK2Δmye mice. Taken together, these results suggest that the inflammatory response and tissue injury to LPS develops much more excessively in myeloid cell deficient GRK2 mice.
Endotoxic mortality
Organ failure in response to endotoxemia eventually leads to mortality in mice. Because lung and liver injury were exaggerated in the GRK2Δmye mice, we hypothesized that the mortality of GRK2Δmye mice would be higher than the GRK2fl/fl mice. As predicted, 50% of the GRK2Δmye mice died within the first 24 hours of LPS injection compared to only 20% in the GRK2fl/fl group. By 42 hours after LPS injection, 100% of GRK2Δmye mice died whereas 30% of GRK2fl/fl mice were alive by 48 hours (Fig. 3L). Thus, although not statistically significant, there was a trend for higher mortality of GRK2Δmye mice compared to GRK2fl/fl mice after LPS injection.
Enhanced LPS-induced cytokine/chemokine response in GRK2Δmye macrophages and neutrophils
Results so far indicate that myeloid cell GRK2 limits LPS-induced cytokine levels as well as organ injury in mice. To further understand the cellular mechanisms of our in vivo findings, we isolated thioglycollate-elicited peritoneal Mϕ from GRK2fl/fl and GRK2Δmye mice and stimulated with LPS (1 μg/ml) for various time points. As observed in vivo, LPS-induced cytokine/chemokine responses were significantly enhanced in primary Mϕ in vitro in the GRK2Δmye mice. There was a markedly enhanced secretion of many inflammatory cytokines including IL-1β, IL-1α, IL-6, IL-9, IL-5, MIP1α, KC, RANTES, GCSF and MCP-1 in the GRK2Δmye mice Mϕ compared to GRK2fl/fl cells (Fig 4A). LPS-induced TNFα showed a biphasic response and was enhanced in GRK2Δmye only at the 6-hour time point (data not shown). In contrast to these cytokines, IL-2, IL-12p40, IFNγ and IL-17 did not differ between GRK2fl/fl and GRK2Δmye Mϕ (data not shown). Interestingly, neutrophils from GRK2Δmye mice showed a significantly enhanced secretion of only G-CSF and KC (Fig. 4B). Other cytokines, such as IL-1α, IL-1β, IL-6, and MIP1β showed a similar trend although not statistically significant. Taken together, these results suggest that GRK2 regulates TLR4-induced inflammatory cytokine production both in vivo and in vitro in a similar manner.
Figure 4. LPS-induced inflammatory cytokine is enhanced in GRK2Δmye macrophages and neutrophils.

A. Thioglycollate-elicited peritoneal Mϕ were stimulated with LPS (1 μg/ml) for the indicated time points. Levels of inflammatory mediators in culture supernatants were determined using Biorad-23 plex assay. Levels of cytokines and chemokines were normalized to the total cellular protein content and expressed as pg/μg of total cellular protein. *p<0.05; **p<0.01; ***p<0.001. N=5 mice per genotype.
B. Thioglycollate-elicited neutrophils (collected and purified using MACS column as described before (Greten et al., 2007) were stimulated with LPS (1μg/ml) in 12-well cell culture plates and cell culture supernatants collected at the indicated time points. Levels of inflammatory mediators were determined using Biorad-23 plex assay and normalized to the total cellular protein and expressed as pg/μg of total cellular protein. **p<0.01. N=5 mice per genotype.
GRK2 negatively regulates NF-κB1-p105-TPL2-MEK-ERK pathway in primary macrophages
To understand the mechanisms of the results described above, we tested the effect of LPS in GRK2Δmye and GRK2fl/fl Mϕ on various signaling pathways including pERK1/2, pJNK1/2, pP38, pIκBα, pAkt and pGSK3 [pathways/proteins known to be regulated by GRK2, see Ref (Patial et al., 2009; Peregrin et al., 2006; Ribas et al., 2007)]. Interestingly, LPS-induced ERK1/2 phosphorylation (but not JNK, p38, IκBα, Akt or GSK3) was significantly enhanced in the GRK2Δmye Mϕ (Fig 5A-C).
Figure 5. LPS-induced phosphorylation of ERK1/2 is selectively enhanced in GRK2Δmye macrophages.

GRK2Δmye and GRK2fl/fl Mϕ were stimulated with LPS (1 μg/ml) for the indicated time points and phosphorylation of ERK1/2, JNK, p38, IκBα, Akt and GSK3 determined by western blotting. Representative blots and quantitation for p-ERK and ERK are shown in (A). Representative blots for p-p38, p-JNK, and p-IκBα, are shown in (B) along with JNK/actin for loading control. Representative blots for pAkt and p-GSK3 along with loading controls (Akt and tubulin) are shown in (C). *p<0.05 and ***p<0.001 compared to GRK2fl/fl. N=6-7 mice for each genotype.
To further elucidate the biochemical mechanisms by which GRK2 regulates the ERK pathway, we examined the upstream regulators of ERK phosphorylation in primary Mϕ. Previous studies have demonstrated that ERK activation in Mϕ is regulated via LPS-stimulated IKKβ-NFκB1 p105 pathway (Beinke et al., 2004; Cho et al., 2005; Loniewski et al., 2007; Waterfield et al., 2004). LPS stimulation of p105 phosphorylation (by IKKβ) results in the ubiquitination and partial degradation of p105 that then releases TPL2 (a MAP3K, which is stoichiometrically bound to p105 under unstimulated conditions). Free TPL2 phosphorylates MEK1/2, which then activates ERK1/2. Note that since p105 is also bound to NFκBp50 under unstimulated conditions, partial degradation of p105 also releases p50 that then evokes changes in NFκB-dependent gene transcription. We first confirmed the existence of IKKβ-mediated ERK signaling pathway in peritoneal Mϕ. As shown in Fig 6, we observed that the pharmacological inhibition of IKKβ [with BMS345541 @ 5 μM, Ref (Burke et al., 2003)] inhibited ERK phosphorylation confirming the existence of IKKβ-NFκB1p105-ERK pathway. To investigate at what level GRK2 regulates this pathway, we examined the phosphorylation of MEK1/2 and p105 after LPS stimulation in GRK2fl/fl and GRK2Δmye Mϕ. As shown in Fig 7, LPS-induced MEK1/2 and p105 phosphorylation were significantly enhanced in GRK2Δmye Mϕ compared to GRK2fl/fl cells. These results suggest that GRK2 negatively regulates ERK pathway possibly at the level of p105.
Figure 6. Existence of IKKβ-ERK pathway in peritoneal macrophages.
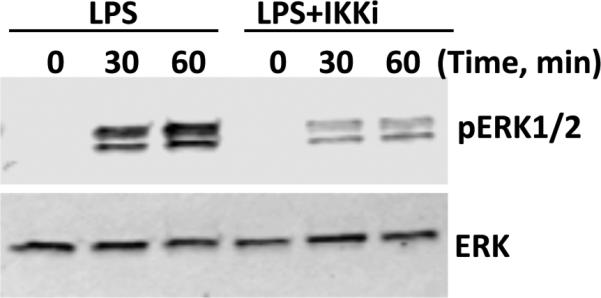
Peritoneal macrophages from GRK2fl/fl mice were stimulated with either LPS alone or pretreated with BMS345541 (5 μM, 30 minutes prior to stimulation with LPS (1μg/ml) for various time points as shown and immunoblotting performed as described in the methods. A representative blot from 4 such experiments is shown.
Figure 7. LPS-induced NFκB1p105-MEK pathway is enhanced in GRK2Δmye macrophages.

GRK2Δmye and GRK2fl/fl Mϕ were stimulated with LPS (1 μg/ml) for the indicated time points and phosphorylation of NFκB1p105 and MEK1/2 determined by western blotting. Representative blots (for pMEK and pP105) are shown in the top and quantitation for pP105 is shown in the bottom (N=7 mice for each genotype) *P<0.05 compared to GRK2fl/fl.
Inhibition of p105-ERK pathway limits the exaggerated inflammatory response in GRK2Δmye macrophages
Knockout of GRK2 in primary macrophages results in the enhanced activation of p105-ERK pathway, which is associated with an enhanced inflammatory cytokine response observed in Mϕ as well as in vivo in mice. As noted before, activation of p105 not only stimulates the ERK pathway, it also activates p50-mediated NFκB pathway. Thus to differentiate the downstream effects of IKKβ-p105-NFκBp50 and IKKβ-p105-ERK pathways and further to demonstrate that the enhanced cytokine responses observed in the GRK2Δmye cells are mediated via these pathways, we examined LPS-induced cytokine responses using pharmacological inhibitors of MEK-ERK (PD98059) (Davies et al., 2000) and IKKβ (BMS345541) (Burke et al., 2003). We treated peritoneal Mϕ from GRK2fl/fl and GRK2Δmye mice with LPS in the presence or absence of these two inhibitors. Inhibition of ERK significantly blocked the enhanced levels of (LPS-induced) IL-1α, MCP1 and GCSF in the GRK2Δmye Mϕ and importantly, the levels of these factors returned to the levels observed in GRK2fl/fl cells (treated with LPS) (Fig 8A). In addition, these cytokines were only modestly affected in the GRK2fl/fl Mϕ by PD98059. These results demonstrate that the enhanced secretion of IL-1α, MCP1 and GCSF observed in GRK2Δmye cells is due to enhanced ERK activation. Furthermore, inhibition of IKKβ (by BMS345541) significantly blocked LPS-induced IL-1α and GCSF levels in the GRK2Δmye cells. Taken together these results suggest that these factors are likely regulated by GRK2 via the IKKβ-p105-ERK pathway (Fig 8A).
Figure 8. Inhibition of ERK and IKKβ blocks the enhanced cytokine levels in GRK2Δmye macrophages.

Peritoneal Mϕ were stimulated or not with LPS along with PD98059 (10 μM) or BMS345541 (5 μM) and the cell culture supernatants collected 24 hours later. Levels of cytokines and chemokines were assessed using Biorad 23-plex assay. Cytokines blocked by PD98059 are shown in (A), cytokines blocked primarily by BMS345541 are shown in (B), and IL-9 is shown in (C). ERKi=PD98059; IKKi=BMS345541; N=5 mice per genotype each *p<0.05; **p<0.01, compared to corresponding GRK2fl/fl. PD98059 or BMS345541 alone did not affect secretion of any of the cytokine/chemokine tested (data not shown).
Interestingly, inhibition of IKKβ but not ERK blocked the enhanced levels of IL-1β, KC, MIP1α, RANTES, IL-5 and IL-6 observed in LPS treated GRK2Δmye cells (Fig. 8B). In the presence of the IKKβ inhibitor, levels of these cytokines in the GRK2Δmye cells returned to levels observed in GRK2fl/fl Mϕ, suggesting that this subset of cytokines is likely regulated by GRK2 exclusively via the IKKβ-p105-p50 pathway. In contrast to these results, neither BMS345541 nor PD98059 inhibited IL-9 secretion in cells from both genotypes (Fig 8C) suggesting neither pathways being involved. Taken together, these data suggest that GRK2 regulation of the p105-ERK pathway negatively regulates IL-1α, and GCSF. Furthermore, these results also suggest that regulation of the p105 (ERK-independent) pathway by GRK2 negatively modulates IL-1β, KC, MIP1α, RANTES, IL-5, and IL-6 in response to LPS stimulation (Fig 9).
Figure 9. Proposed model of GRK2 regulation of TLR4-induced inflammatory response.

TLR4 activates ERK pathway via IKKβ-mediated phosphorylation of p105. Based on the results presented here, we propose that GRK2 negatively regulates IKKβ-induced p105 phosphorylation and therefore the ERK pathway. Because p105 phosphorylation is also essential for p50 mediated NFκB activation, negative regulation of p105 by GRK2 also modulates a subset of cytokines that are regulated by this pathway. Note that there are two exceptions: 1. Because MCP-1 was inhibited by ERK inhibitor but not by the IKKβ inhibitor, it is possible that MCP-1 is being regulated by GRK2 in an ERK-dependent but p105-independent mechanism. 2. Also, because MIP1α was modestly inhibited by PD98059, it is possible that a small component of the ERK pathway may also regulate MIP1α. Previous studies in primary macrophages and neutrophils have demonstrated that TLR4 activation increases the expression of GRK2 significantly and thus GRK2 might act as a negative feedback regulator for TLR4 signaling.
DISCUSSION
It has been well documented that the expression levels of GRK2 are altered in specific cell types in human diseases including active relapsing-remitting multiple sclerosis (MS) or with secondary progressive MS (Vroon et al., 2005), Alzheimer's disease (Leosco et al., 2007), and rheumatoid arthiritis (Lombardi et al., 1999). Related to these changes in GRK2 levels in humans, rodent models with decreased GRK2 levels exhibit altered disease pathogenesis ((Tarrant et al., 2008) (Vroon et al., 2005) (Nijboer et al., 2008). In addition to the human diseases outlined above, GRK2 levels are increased in immune cells from patients with sepsis. Consistent with this, TLR ligands enhance GRK2 levels in primary macrophages and neutrophils in culture (Alves-Filho et al., 2009; Loniewski et al., 2008). Therefore, we reasoned that GRK2 levels in myeloid cells might regulate the pathogenesis of endotoxemia and sepsis. Based on this reasoning, we generated myeloid cell-specific GRK2 knockout to determine the pathogenesis of endotoxemia in mice. Our results clearly suggest that GRK2 deficiency in the myeloid cells enhances liver and lung injury in response to high dose LPS injections.
In addition to liver injury, we also observed that glycogen metabolism is altered in the GRK2Δmye mice compared to the GRK2fl/fl mice. Although the significance of this to liver injury is not clear, dysregulated glycogen and lipid metabolism are observed in human septic patients as well as in animal models of sepsis (Ali et al., 2008; Frazier et al., 2009; Hirshberg et al., 2008; Van den Berghe et al., 2006). Attenuated glycogen depletion suggests inadequate glycogenolysis in the GRK2Δmye mice during endotoxemia. Whether this is directly related to liver injury or the cytokine levels is not clear. However, sympathetic activation in endotoxemia plays a major role in liver carbohydrate metabolism and therefore it is possible that the adrenergic signals that regulate glycogenolysis might be dysregulated in the GRK2Δmye mice (Goldstein and Elwyn, 1989).
Migration of immune cells to the inflammatory sites although important for host defense, is also in part responsible for tissue damage observed in sepsis. MIP1α, MIP1β, MCP1, Rantes and eotaxin are members of CC chemokine family, which serve as major chemoattractants for neutrophils, mononuclear cells as well as eosinophils and therefore contribute to tissue injury observed in sepsis (Baggiolini, 1998; Jose et al., 1994; Kapp et al., 1994; Standiford et al., 1995). In this study the levels of MIP1α and MCP1 were significantly increased in the Mϕ culture supernatants of GRK2Δmye mice, and interestingly MIP1α was also elevated in vivo (in response to LPS) in the GRK2Δmye mice. MIP1α is produced by various immune cells including Mϕ and plays a crucial role in activation and chemotaxis of several populations of Mϕ. It also stimulates the proliferation of mature tissue Mϕ and has been shown to induce the secretion of TNFα, IL-6 and IL-1α from peritoneal Mϕ (Fahey et al., 1992). Consistent with the previously reported role for MIP1α in lung injury (Standiford et al., 1995; Standiford et al., 1993), plasma MIP1α level was enhanced early on in the GRK2Δmye mice and was associated with enhanced lung capillary damage in the GRK2Δmye mice. Together these results suggest a possible role for MIP1α in causing exaggerated tissue injury in the GRK2Δmye mice. In other studies Fan and Malik (Fan and Malik, 2003) have proposed that MIP-2-CXCR2-mediated neutrophil migration is negatively regulated by GRK2, such that decrease in GRK2 levels enhances neutrophil migration. In addition, TLR4 activation was shown to enhance CXCR2-induced neutrophil migration by downregulating GRK2 levels. In the in vivo context, these results would suggest that reduced GRK2 levels may result in enhanced neutrophil infiltration into the organs, and therefore cause exaggerated organ damage, consistent with the results presented here.
Our studies show that in addition to MIP1α LPS-induced IL-12p40 and IL10 are enhanced in vivo in the GRK2Δmye mice. IL12p40, through its role in forming functional IL12, is an important mediator of cell-mediated immunity and plays an important role in inflammatory diseases including experimental autoimmune encephalitis and sepsis (Adorini, 1999; Becher et al., 2002). IL-10 is an important anti-inflammatory cytokine that prevents an over-exuberant immune response, leading to immune-suppression shifting the balance toward lethal consequences. Based on the present data, it is not clear which of the specific cytokines/chemokines regulated in the plasma or in the macrophages/neutrophils specifically contribute to the exaggerated tissue injury observed in GRK2Δmye mice.
Results from signaling experiments suggest that GRK2 acts at the level of NFκB1p105. Previous studies have shown that GRK2 and NFκB1 p105 interact in yeast two-hybrid assays (http://www.signaling-gateway.org/data/Y2H/cgi-bin/y2h.cgi). Furthermore, we have shown that the RH domain of GRK2 directly interacts with the carboxy terminus of p105 in direct interaction assays, although we did not find any functional significance of that interaction in Raw264.7 macrophage cell line using RNAi (Parameswaran et al., 2006). Previous studies also demonstrated that NF-κB1 p105 is a poor substrate for GRK2, suggesting that the regulation might be phosphorylation-independent (Parameswaran et al., 2006). This is not entirely surprising given the role of the RH domain of GRKs in mediating phosphorylation-independent cell signaling, potentially as a scaffolding protein (Ribas et al., 2007). Future studies will determine whether direct interaction of GRK2 with p105 might block IKKβ phosphorylation of p105 thus preventing p105 degradation and TPL2 release (Beinke et al., 2004; Waterfield et al., 2004). Together, results from primary Mϕ demonstrate that GRK2 is a negative regulator of IKKβ-induced p105-ERK pathway.
Other studies have shown that GRK2 can regulate a number of different signaling pathways depending on the cellular context and receptor involved. Studies on the role of GRK2 in the regulation of ERK pathway however, have shown conflicting results (Jimenez-Sainz et al., 2006) (Kleibeuker et al., 2008). While these studies have focused on chemokine receptor (a GPCR)-induced ERK activation, our studies address the role of GRK2 in TLR4 signaling. Thus although GRK2 has very important roles in GPCR signaling, we demonstrate that GRK2 is equally important in regulating TLR4-induced p105 and ERK activation and the consequent cytokine/chemokine production. In contrast to the results presented here on p38, Peregrin et al showed that GRK2 can interact with and phosphorylate p38 MAPK and this can lead to the inactivation of p38 MAPK (Peregrin et al., 2006). In addition, Nijboer et al (Nijboer et al., 2009), demonstrated that ischemic injury in the brain is regulated by GRK2 via p38 MAPK. Although the reason for the differences in our results compared to the other studies is not clear, one possible reason may be due to the different mouse models and cell types used.
In summary, mice with a deficiency of GRK2 in myeloid cells are more susceptible to LPS-mediated endotoxemia and this is associated with enhanced secretion of several cytokines/chemokines. We further demonstrate that the increased cytokine/chemokine production in the GRK2Δmye Mϕ is related to enhanced activation of the NFκB1p105-ERK pathways. We propose that the increase in GRK2 levels observed in immune cells under septic conditions might function as a “negative feedback regulator” of TLR4 signaling and therefore function to limit the progression of sepsis (Fig 9).
Acknowledgments
We gratefully acknowledge the support from NIH (grants HL095637, AR055726 and AR056680 to N.P.) and American Heart Association (Mid West affiliate) grant-in-aid (to N.P.). We thank the university lab animal resources for taking excellent care of our animals, the MSU histopathology and DCPAH laboratories for their excellent service. We are also thankful to Megan Hull for her excellent assistance.
References
- Adorini L. Interleukin-12, a key cytokine in Th1-mediated autoimmune diseases. Cell Mol Life Sci. 1999;55(12):1610–1625. doi: 10.1007/s000180050400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali NA, O'Brien JM, Jr., Dungan K, Phillips G, Marsh CB, Lemeshow S, Connors AF, Jr., Preiser JC. Glucose variability and mortality in patients with sepsis. Crit Care Med. 2008;36(8):2316–2321. doi: 10.1097/CCM.0b013e3181810378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alves-Filho JC, Freitas A, Souto FO, Spiller F, Paula-Neto H, Silva JS, Gazzinelli RT, Teixeira MM, Ferreira SH, Cunha FQ. Regulation of chemokine receptor by Toll-like receptor 2 is critical to neutrophil migration and resistance to polymicrobial sepsis. Proc Natl Acad Sci U S A. 2009;106(10):4018–4023. doi: 10.1073/pnas.0900196106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appledorn DM, Patial S, McBride A, Godbehere S, Van Rooijen N, Parameswaran N, Amalfitano A. Adenovirus vector-induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo. J Immunol. 2008;181(3):2134–2144. doi: 10.4049/jimmunol.181.3.2134. [DOI] [PubMed] [Google Scholar]
- Arraes SM, Freitas MS, da Silva SV, de Paula Neto HA, Alves-Filho JC, Auxiliadora Martins M, Basile-Filho A, Tavares-Murta BM, Barja-Fidalgo C, Cunha FQ. Impaired neutrophil chemotaxis in sepsis associates with GRK expression and inhibition of actin assembly and tyrosine phosphorylation. Blood. 2006;108(9):2906–2913. doi: 10.1182/blood-2006-05-024638. [DOI] [PubMed] [Google Scholar]
- Baggiolini M. Chemokines and leukocyte traffic. Nature. 1998;392(6676):565–568. doi: 10.1038/33340. [DOI] [PubMed] [Google Scholar]
- Becher B, Durell BG, Noelle RJ. Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J Clin Invest. 2002;110(4):493–497. doi: 10.1172/JCI15751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beinke S, Robinson MJ, Hugunin M, Ley SC. Lipopolysaccharide activation of the TPL-2/MEK/extracellular signal-regulated kinase mitogen-activated protein kinase cascade is regulated by IkappaB kinase-induced proteolysis of NF-kappaB1 p105. Mol Cell Biol. 2004;24(21):9658–9667. doi: 10.1128/MCB.24.21.9658-9667.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler B. Microbe sensing, positive feedback loops, and the pathogenesis of inflammatory diseases. Immunol Rev. 2009;227(1):248–263. doi: 10.1111/j.1600-065X.2008.00733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JR, Pattoli MA, Gregor KR, Brassil PJ, MacMaster JF, McIntyre KW, Yang X, Iotzova VS, Clarke W, Strnad J, Qiu Y, Zusi FC. BMS-345541 is a highly selective inhibitor of I kappa B kinase that binds at an allosteric site of the enzyme and blocks NF-kappa B-dependent transcription in mice. J Biol Chem. 2003;278(3):1450–1456. doi: 10.1074/jbc.M209677200. [DOI] [PubMed] [Google Scholar]
- Cho J, Melnick M, Solidakis GP, Tsichlis PN. Tpl2 (tumor progression locus 2) phosphorylation at Thr290 is induced by lipopolysaccharide via an Ikappa-B Kinase-beta-dependent pathway and is required for Tpl2 activation by external signals. J Biol Chem. 2005;280(21):20442–20448. doi: 10.1074/jbc.M413554200. [DOI] [PubMed] [Google Scholar]
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8(4):265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351(Pt 1):95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Blasi A, Parruti G, Sallese M. Regulation of G protein-coupled receptor kinase subtypes in activated T lymphocytes. Selective increase of beta-adrenergic receptor kinase 1 and 2. J Clin Invest. 1995;95(1):203–210. doi: 10.1172/JCI117641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duniho SM, Martin J, Forster JS, Cascio MB, Moran TS, Carpin LB, Sciuto AM. Acute changes in lung histopathology and bronchoalveolar lavage parameters in mice exposed to the choking agent gas phosgene. Toxicol Pathol. 2002;30(3):339–349. doi: 10.1080/01926230252929918. [DOI] [PubMed] [Google Scholar]
- Fahey TJ, 3rd, Tracey KJ, Tekamp-Olson P, Cousens LS, Jones WG, Shires GT, Cerami A, Sherry B. Macrophage inflammatory protein 1 modulates macrophage function. J Immunol. 1992;148(9):2764–2769. [PubMed] [Google Scholar]
- Fan J, Malik AB. Toll-like receptor-4 (TLR4) signaling augments chemokine-induced neutrophil migration by modulating cell surface expression of chemokine receptors. Nat Med. 2003;9(3):315–321. doi: 10.1038/nm832. [DOI] [PubMed] [Google Scholar]
- Frazier WJ, Wang X, Wancket LM, Li XA, Meng X, Nelin LD, Cato AC, Liu Y. Increased inflammation, impaired bacterial clearance, and metabolic disruption after gram-negative sepsis in Mkp-1-deficient mice. J Immunol. 2009;183(11):7411–7419. doi: 10.4049/jimmunol.0804343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorelli M, Livrea P, Trojano M. Post-receptorial mechanisms underlie functional disregulation of beta2-adrenergic receptors in lymphocytes from Multiple Sclerosis patients. J Neuroimmunol. 2004;155(1-2):143–149. doi: 10.1016/j.jneuroim.2004.05.013. [DOI] [PubMed] [Google Scholar]
- Goldstein SA, Elwyn DH. The effects of injury and sepsis on fuel utilization. Annu Rev Nutr. 1989;9:445–473. doi: 10.1146/annurev.nu.09.070189.002305. [DOI] [PubMed] [Google Scholar]
- Greten FR, Arkan MC, Bollrath J, Hsu LC, Goode J, Miething C, Goktuna SI, Neuenhahn M, Fierer J, Paxian S, Van Rooijen N, Xu Y, O'Cain T, Jaffee BB, Busch DH, Duyster J, Schmid RM, Eckmann L, Karin M. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007;130(5):918–931. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirshberg E, Larsen G, Van Duker H. Alterations in glucose homeostasis in the pediatric intensive care unit: Hyperglycemia and glucose variability are associated with increased mortality and morbidity. Pediatr Crit Care Med. 2008;9(4):361–366. doi: 10.1097/PCC.0b013e318172d401. [DOI] [PubMed] [Google Scholar]
- Jimenez-Sainz MC, Murga C, Kavelaars A, Jurado-Pueyo M, Krakstad BF, Heijnen CJ, Mayor F, Jr., Aragay AM. G protein-coupled receptor kinase 2 negatively regulates chemokine signaling at a level downstream from G protein subunits. Mol Biol Cell. 2006;17(1):25–31. doi: 10.1091/mbc.E05-05-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose PJ, Griffiths-Johnson DA, Collins PD, Walsh DT, Moqbel R, Totty NF, Truong O, Hsuan JJ, Williams TJ. Eotaxin: a potent eosinophil chemoattractant cytokine detected in a guinea pig model of allergic airways inflammation. J Exp Med. 1994;179(3):881–887. doi: 10.1084/jem.179.3.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapp A, Zeck-Kapp G, Czech W, Schopf E. The chemokine RANTES is more than a chemoattractant: characterization of its effect on human eosinophil oxidative metabolism and morphology in comparison with IL-5 and GM-CSF. J Invest Dermatol. 1994;102(6):906–914. doi: 10.1111/1523-1747.ep12383399. [DOI] [PubMed] [Google Scholar]
- Kleibeuker W, Jurado-Pueyo M, Murga C, Eijkelkamp N, Mayor F, Jr., Heijnen CJ, Kavelaars A. Physiological changes in GRK2 regulate CCL2-induced signaling to ERK1/2 and Akt but not to MEK1/2 and calcium. J Neurochem. 2008;104(4):979–992. doi: 10.1111/j.1471-4159.2007.05023.x. [DOI] [PubMed] [Google Scholar]
- Leosco D, Fortunato F, Rengo G, Iaccarino G, Sanzari E, Golino L, Zincarelli C, Canonico V, Marchese M, Koch WJ, Rengo F. Lymphocyte G-protein-coupled receptor kinase-2 is upregulated in patients with Alzheimer's disease. Neurosci Lett. 2007;415(3):279–282. doi: 10.1016/j.neulet.2007.01.034. [DOI] [PubMed] [Google Scholar]
- Lombardi MS, Kavelaars A, Cobelens PM, Schmidt RE, Schedlowski M, Heijnen CJ. Adjuvant arthritis induces down-regulation of G protein-coupled receptor kinases in the immune system. J Immunol. 2001;166(3):1635–1640. doi: 10.4049/jimmunol.166.3.1635. [DOI] [PubMed] [Google Scholar]
- Lombardi MS, Kavelaars A, Schedlowski M, Bijlsma JW, Okihara KL, Van de Pol M, Ochsmann S, Pawlak C, Schmidt RE, Heijnen CJ. Decreased expression and activity of G-protein-coupled receptor kinases in peripheral blood mononuclear cells of patients with rheumatoid arthritis. Faseb J. 1999;13(6):715–725. doi: 10.1096/fasebj.13.6.715. [DOI] [PubMed] [Google Scholar]
- Loniewski K, Shi Y, Pestka J, Parameswaran N. Toll-like receptors differentially regulate GPCR kinases and arrestins in primary macrophages. Mol Immunol. 2008;45(8):2312–2322. doi: 10.1016/j.molimm.2007.11.012. [DOI] [PubMed] [Google Scholar]
- Loniewski KJ, Patial S, Parameswaran N. Sensitivity of TLR4- and -7-induced NF kappa B1 p105-TPL2-ERK pathway to TNF-receptor-associated-factor-6 revealed by RNAi in mouse macrophages. Mol Immunol. 2007;44(15):3715–3723. doi: 10.1016/j.molimm.2007.04.002. [DOI] [PubMed] [Google Scholar]
- Loudon RP, Perussia B, Benovic JL. Differentially regulated expression of the G-protein-coupled receptor kinases, betaARK and GRK6, during myelomonocytic cell development in vitro. Blood. 1996;88(12):4547–4557. [PubMed] [Google Scholar]
- Matkovich SJ, Diwan A, Klanke JL, Hammer DJ, Marreez Y, Odley AM, Brunskill EW, Koch WJ, Schwartz RJ, Dorn GW., 2nd Cardiac-specific ablation of G-protein receptor kinase 2 redefines its roles in heart development and beta-adrenergic signaling. Circ Res. 2006;99(9):996–1003. doi: 10.1161/01.RES.0000247932.71270.2c. [DOI] [PubMed] [Google Scholar]
- Nijboer CH, Heijnen CJ, Willemen HL, Groenendaal F, Dorn GW, 2nd, Bel FV, Kavelaars A. Cell-specific roles of GRK2 in onset and severity of hypoxic-ischemic brain damage in neonatal mice. Brain Behav Immun. 2009 doi: 10.1016/j.bbi.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijboer CH, Kavelaars A, Vroon A, Groenendaal F, van Bel F, Heijnen CJ. Low endogenous G-protein-coupled receptor kinase 2 sensitizes the immature brain to hypoxia-ischemia-induced gray and white matter damage. J Neurosci. 2008;28(13):3324–3332. doi: 10.1523/JNEUROSCI.4769-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7(5):353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- Parameswaran N, Pao CS, Leonhard KS, Kang DS, Kratz M, Ley SC, Benovic JL. Arrestin-2 and G protein-coupled receptor kinase 5 interact with NFkappaB1 p105 and negatively regulate lipopolysaccharide-stimulated ERK1/2 activation in macrophages. J Biol Chem. 2006;281(45):34159–34170. doi: 10.1074/jbc.M605376200. [DOI] [PubMed] [Google Scholar]
- Patial S, Luo J, Porter KJ, Benovic JL, Parameswaran N. G-protein coupled receptor kinases mediate TNFalpha-induced NFkappaB signaling via direct interaction with and phosphorylation of IkappaBalpha. Biochem J. 2009 doi: 10.1042/BJ20090908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peregrin S, Jurado-Pueyo M, Campos PM, Sanz-Moreno V, Ruiz-Gomez A, Crespo P, Mayor F, Jr., Murga C. Phosphorylation of p38 by GRK2 at the docking groove unveils a novel mechanism for inactivating p38MAPK. Curr Biol. 2006;16(20):2042–2047. doi: 10.1016/j.cub.2006.08.083. [DOI] [PubMed] [Google Scholar]
- Ribas C, Penela P, Murga C, Salcedo A, Garcia-Hoz C, Jurado-Pueyo M, Aymerich I, Mayor F., Jr. The G protein-coupled receptor kinase (GRK) interactome: role of GRKs in GPCR regulation and signaling. Biochim Biophys Acta. 2007;1768(4):913–922. doi: 10.1016/j.bbamem.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Rittirsch D, Hoesel LM, Ward PA. The disconnect between animal models of sepsis and human sepsis. J Leukoc Biol. 2007;81(1):137–143. doi: 10.1189/jlb.0806542. [DOI] [PubMed] [Google Scholar]
- Salomao R, Martins PS, Brunialti MK, Fernandes Mda L, Martos LS, Mendes ME, Gomes NE, Rigato O. TLR signaling pathway in patients with sepsis. Shock. 2008;30(Suppl 1):73–77. doi: 10.1097/SHK.0b013e318181af2a. [DOI] [PubMed] [Google Scholar]
- Standiford TJ, Kunkel SL, Lukacs NW, Greenberger MJ, Danforth JM, Kunkel RG, Strieter RM. Macrophage inflammatory protein-1 alpha mediates lung leukocyte recruitment, lung capillary leak, and early mortality in murine endotoxemia. J Immunol. 1995;155(3):1515–1524. [PubMed] [Google Scholar]
- Standiford TJ, Rolfe MW, Kunkel SL, Lynch JP, 3rd, Burdick MD, Gilbert AR, Orringer MB, Whyte RI, Strieter RM. Macrophage inflammatory protein-1 alpha expression in interstitial lung disease. J Immunol. 1993;151(5):2852–2863. [PubMed] [Google Scholar]
- Symons A, Beinke S, Ley SC. MAP kinase kinase kinases and innate immunity. Trends Immunol. 2006;27(1):40–48. doi: 10.1016/j.it.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Tarrant TK, Rampersad RR, Esserman D, Rothlein LR, Liu P, Premont RT, Lefkowitz RJ, Lee DM, Patel DD. Granulocyte chemotaxis and disease expression are differentially regulated by GRK subtype in an acute inflammatory arthritis model (K/BxN). Clin Immunol. 2008;129(1):115–122. doi: 10.1016/j.clim.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Berghe G, Wilmer A, Hermans G, Meersseman W, Wouters PJ, Milants I, Van Wijngaerden E, Bobbaers H, Bouillon R. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354(5):449–461. doi: 10.1056/NEJMoa052521. [DOI] [PubMed] [Google Scholar]
- Vroon A, Kavelaars A, Limmroth V, Lombardi MS, Goebel MU, Van Dam AM, Caron MG, Schedlowski M, Heijnen CJ. G protein-coupled receptor kinase 2 in multiple sclerosis and experimental autoimmune encephalomyelitis. J Immunol. 2005;174(7):4400–4406. doi: 10.4049/jimmunol.174.7.4400. [DOI] [PubMed] [Google Scholar]
- Vroon A, Lombardi MS, Kavelaars A, Heijnen CJ. Changes in the G-protein-coupled receptor desensitization machinery during relapsing-progressive experimental allergic encephalomyelitis. J Neuroimmunol. 2003;137(1-2):79–86. doi: 10.1016/s0165-5728(03)00050-x. [DOI] [PubMed] [Google Scholar]
- Waterfield M, Jin W, Reiley W, Zhang M, Sun SC. IkappaB kinase is an essential component of the Tpl2 signaling pathway. Mol Cell Biol. 2004;24(13):6040–6048. doi: 10.1128/MCB.24.13.6040-6048.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong ET, Tergaonkar V. Roles of NF-kappaB in health and disease: mechanisms and therapeutic potential. Clin Sci (Lond) 2009;116(6):451–465. doi: 10.1042/CS20080502. [DOI] [PubMed] [Google Scholar]