

Abstract
Mnt, a tetrameric repressor encoded by bacteriophage P22, uses N-domain dimers to contact each half of its operator site. Experiments with a double mutant and structural homology with the P22 Arc repressor suggest that contacts made by Arg-28 and stabilized by Glu-33 are largely responsible for dimer–dimer cooperativity in Mnt. These dimer–dimer contacts are energetically more important for operator binding than solution tetramerization, which is mediated by an independent C-terminal coiled-coil domain. Indeed, once one dimer of the Mnt tetramer contacts an operator half site, binding of the second dimer occurs with an effective concentration much lower than that expected if both dimers were flexibly tethered. These results suggest that binding of the second dimer introduces some strain into the protein–DNA complex, a mechanism that could serve to limit the affinity of operator binding and to prevent strong binding of the Mnt tetramer to nonoperator sites.
Keywords: DNA binding, dimer–dimer interactions, effective concentration, tetramerization domain, ribbon-helix-helix family
The Mnt repressor of bacteriophage P22 is a modular ribbon-helix-helix transcription factor that consists of a dimeric N-terminal domain that binds operator DNA and a C-terminal coiled-coil domain that mediates protein tetramerization (1–2). All members of the ribbon-helix-helix family possess DNA-binding domains with a common interwoven fold composed of a two-stranded antiparallel β-sheet (the DNA-binding motif) and four α-helices (3). Some family members contain just this DNA-binding domain, whereas others, like Mnt, have additional C-terminal domains that are subfamily specific. The structures of protein–DNA complexes are known for three ribbon-helix-helix proteins—MetJ, Arc, and CopG (4–6). For Mnt, solution structures of the isolated N- and C-terminal domains have been determined (2, 7), but structures of the intact protein or DNA complexes are not known.
Cooperative interactions mediated by protein–protein contacts are required for high levels of binding affinity and specificity for many DNA-binding proteins, including the ribbon-helix-helix family. For example, contacts observed between tandemly bound dimers in the Arc-operator structure are essential for strong and cooperative operator binding (5, 8, 9). Dimer–dimer contacts are also seen in the cocrystal structures of MetJ and CopG, but very different parts of the dimer surfaces interact in each case. Fig. 1 shows a model for the Mnt-operator complex based on homology with Arc, its closest relative. In this model, the side chains of two Arg-28 side chains, stabilized by interactions with Glu-33, are positioned to make cooperative contacts with the adjacent dimer near the center of the DNA site. In this study, we show that Arg-28 and Glu-33 are required for strong binding of Mnt to its operator, quantify the contribution of these interactions to binding affinity and cooperativity, and determine the energetic contributions of dimer–dimer cooperativity and Mnt tetramer formation to overall operator binding.
Figure 1.

Model of Mnt repressor bound to operator DNA. Each N-terminal domain dimer has been positioned on the operator on the basis of the structure of the homologous Arc repressor–operator complex (5). The model shows residues 1–43 of the N-terminal DNA-binding domain (7) and residues 52–82 of the C-terminal tetramerization domain (2). The positions of the Arg-28 (R28) and Glu-33 (E33) side chains, which are replaced by Met-28 and Tyr-33 in the Mnt-MYI mutant, are also marked. The linkers that connect the C terminus (C) of each N-terminal segment in the intact protein to the N terminus (N) of each C-terminal subunit are not shown because their conformations are unknown. Nooren et al. (14) have suggested a model in which the C-terminal domain is rotated 90° around a vertical axis from the orientation shown here. Figure prepared by using molscript (23).
Materials and Methods
Construction, Purification, and Characterization.
Genes encoding wild-type or mutant (Arg-28→Met/Glu-33→Tyr) variants of Mnt or its N-terminal domain (residues 1–52) with C-terminal His6 tails were generated by PCR and cloned into plasmid pET-24-d (+) (Novagen). Proteins were expressed in Escherichia coli strain X90 (λ DE3) and purified under denaturing conditions by chromatography on Ni2+-NTA agarose (Qiagen, Chatsworth, CA) with 10 mM imidazole in the column loading and washing steps. The purified proteins were dialyzed against four changes of 4 liters of S-buffer [50 mM Tris⋅Cl (pH 7.5)/100 mM KCl/0.1 mM EDTA/5% glycerol] and were stored at −80°C. Proteins were greater than 95% pure as assayed by PAGE followed by staining with Coomassie brilliant blue, and concentrations were determined by UV absorbance by using extinction coefficients at 280 nm of 1,197 M−1 cm−1 for Mnt-N-H6, 2,394 M−1 cm−1 for Mnt-H6 and the Mnt-N-MYI mutant, and 3,591 M−1 cm−1 for Mnt-MYI-H6.
Circular-dichroism spectra were taken on an Aviv Model 60DS spectrometer (Aviv Associates, Lakewood, NJ), and urea denaturation at 25°C was monitored by changes in ellipticity at 234 nm. Both experiments were performed in S-buffer. Forward and reverse urea melts were similar, as expected for reversible reactions. Gel-filtration chromatography was performed in S-buffer on a 1 × 30 cm Superdex 75 column (Pharmacia).
DNA-Binding Assays.
A DNA fragment containing the mnt operator with a silent G:C → A:T mutation at position 20 was generated by digestion of the plasmid pIO101 (10) with EcoRI and EcoRV. A DNA fragment containing an mnt operator half site was generated by digestion of the plasmid pBluescript SK+ (mnt LI) (1) with SacI and Asp718. The fragments were labeled by end-filling the restriction overhangs with the Klenow fragment of DNA polymerase I (New England Biolabs) by using [α-32P] dATP and nonradioactive dCTP, dGTP, and dTTP, and were purified by electrophoresis on a native 5% polyacrylamide 1× TBE gel (90 mM Tris/64.6 mM boric acid/2.5 mM EDTA, pH 8.3).
DNase I footprinting reactions contained ≈10,000 cpm of DNA and were performed in 30 mM Hepes KOH (pH 7.5)/100 mM potassium glutamate/10 mM MgCl2/1.5 mM CaCl2/0.1 mM EDTA/100 μg/ml BSA/0.02% Nonidet P-40. Proteins were diluted in buffer, and 20 μl was mixed with 2.2 μl of buffer containing DNA. Reactions were preincubated for 2 h at 25°C, and 5.6 μl of buffer containing DNase I (Worthington) was added to give a final enzyme concentration of 0.1 μg/ml. Reactions were allowed to proceed for 1 min before quenching with an equal volume of 2.5 M ammonium acetate/20 mM EDTA/10 μg/ml salmon sperm DNA. The reaction mixture was extracted with phenol/chloroform/isoamyl alcohol (25:24:1) (Sigma), precipitated with ethanol, washed with 80% ethanol, dried, and resuspended in 85% formamide/10 mM NaOH/1 mM EDTA/0.1% xylene cyanol/0.1% bromophenol blue. Samples were electrophoresed on 8% polyacrylamide/8.3 M urea gels in 1× TBE. Cleavage data were quantified by PhosphorImager analysis. To calculate fractional protection (FP), the intensities of bands within the operator were normalized by dividing by the intensities of nonoperator regions and then dividing by the same ratio for a lane containing no protein (11). Half-site binding data were fitted to the equation:
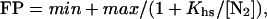 |
1 |
where FP is the observed fractional protection, min and max represent the minimal and maximal fractional protection, Khs is the equilibrium constant for dissociation for the dimer from the operator half site, and [N2] is the free concentration of N-terminal domain dimer calculated by using the dimerization constants obtained from urea-denaturation experiments and the total protein concentration. Protection of the intact mnt operator by cooperative binding of two N-terminal domain dimers was fit by using the equation:
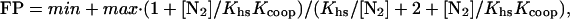 |
2 |
where Kcoop is the cooperativity constant, and Khs is the microscopic equilibrium dissociation constant for half-site binding. All fitting was performed by using a nonlinear least squares subroutine in the program kaleidagraph (Synergy Software, Reading, PA).
For intact Mnt-H6 and Mnt-MYI-H6, binding to end-labeled operator was performed for 2 h at 25°C in the same buffer used for DNase protection, and bound and free DNA were separated by electrophoresis on a 6% polyacrylamide gel in 0.5× TBE buffer (12). The intensities of the bound and free bands were determined by PhosphorImager analysis and were fit to the model
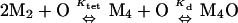 |
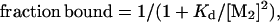 |
3 |
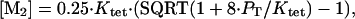 |
4 |
where PT is the total Mnt concentration in dimer equivalents.
Results
Mutant Design and Characterization.
Using a cooperativity mutant of Arc repressor as a guide, we designed an analogous mutant of Mnt. In Arc, Arg-31 in one dimer hydrogen bonds to the backbone of the adjacent dimer to stabilize the tetrameric DNA complex and is also part of a salt-bridge/hydrogen-bond network with Glu-36 and Arg-40 in the same subunit (5, 8, 9). Individual substitutions at positions 31, 36, or 40 destabilize Arc, but these residues can be replaced en masse by Met-31, Tyr-36, and Leu-40 to give a hyperstable mutant (Arc-MYL), in which hydrophobic contacts replace the polar interactions (13). Arc-MYL is defective in DNA-binding cooperativity. The homologous residues in Mnt are Arg-28, Glu-33, and Ile-37, with the charged side chains being close enough in the solution structure to form a salt bridge (7). By analogy with Arc-MYL, we replaced Arg-28 and Glu-33 in Mnt with the hydrophobic residues Met-28 and Tyr-33, leaving Ile-37 unchanged. We refer to this Mnt mutant as MYI.
The Arg-28→Met/Glu-33→Tyr double substitution was constructed by site-directed mutagenesis in a protein fragment corresponding to the N-terminal domain of Mnt with a C-terminal His6 tail. The purified mutant fragment (Mnt-N-MYI-H6) and an otherwise identical wild-type fragment (Mnt-N-H6) had essentially the same far-UV circular dichroism (CD) spectra (Fig. 2a), indicating similar secondary structures, and eluted at the same position (apparent MR 11.3 kDa) in gel-filtration experiments, indicating that both formed dimers. Dimerization and folding are tightly coupled reactions for these fragments, and urea-denaturation experiments, monitored by changes in CD ellipticity, were used to determine the equilibrium dissociation constant for dimer dissociation and unfolding (Fig. 2b). The mutant MYI dimer was significantly more stable (Ku = 86 pM) than the wild-type dimer (Ku = 5.3 nM).
Figure 2.

(a) Far UV circular-dichroism spectra of the Mnt-N-H6 (triangles) and Mnt-N-MYI-H6 (squares) fragments. Spectra were taken at 25°C in a cuvette with a path length of 1 mm with a protein concentration of 50 μM in S buffer. (b) Urea denaturation of Mnt-N-H6 and Mnt-N-MYI-H6. Experiments were performed at a protein concentration of 10 μM in S buffer in a cuvette with a path length of 1 cm. Fitting gave m-values of −1.43 and −1.51 kcal/mol⋅M and ΔGu values of 11.2 and 13.7 kcal/mol for Mnt-N-H6 and Mnt-N-MYI-H6, respectively.
Half-Operator and Operator Binding.
The Mnt-N-H6 and Mnt-N-MYI-H6 dimers showed essentially identical binding to a DNA fragment bearing a single mnt operator half site in DNase I footprinting assays (Fig. 3 Top). As expected, both reactions were first order with respect to dimer concentration. Values of Khs—the equilibrium constant for dissociation of a dimer⋅half-site complex to free dimer and DNA—were calculated from fitting to be 4.8 (±0.9) × 10−8 M for Mnt-N-H6 and 3.4 (±0.7) × 10−8 M for Mnt-N-MYI-H6. These data show that the mutant and wild-type N-domain dimers bind to operator half sites with very similar affinities.
Figure 3.

DNA-binding assays. Top and Middle show binding of N-domain homodimers of Mnt-N-H6 and Mnt-N-MYI-H6 to a half operator site (Top) and to the full length mnt operator (Middle), as assayed by DNase I footprinting. Bottom shows binding of the tetrameric Mnt-H6 and Mnt-MYI-H6 proteins to the intact mnt operator as determined by gel mobility-shift assays. The solid lines in Top, Middle, and Bottom are theoretical fits of the experimental data calculated as described in Materials and Methods.
Footprinting was also used to assay binding of Mnt-N-H6 and Mnt-N-MYI-H6 to a DNA fragment containing the intact mnt operator, which consists of two identical subsites related by 2-fold symmetry (Fig. 3 Middle). With both half sites of the operator present, the wild-type N-domain dimer bound significantly better than the mutant, as would be expected if the mutations reduced dimer–dimer cooperativity. These data were fit to a model in which a single dimer binds to either half of the operator with the equilibrium constant (Khs) determined above for half-site binding, and two dimers bind with an equilibrium constant of Khs2⋅Kcoop, where the latter value represents the cooperativity constant. For the wild-type fragment, Kcoop was 3.3 (±1.7) × 10−4, indicating strong positive cooperativity. For the MYI fragment, Kcoop was only 0.18 (±0.6), exhibiting weak cooperativity. These Kcoop values correspond to favorable free energies of dimer–dimer cooperativity (ΔGcoop) of −4.7 (±0.5) kcal/mol for the wild-type N-terminal domain but only −1.0 (±0.2) kcal/mol for the MYI domain. These experiments show that the Arg-28→Met/Glu-33→Tyr double mutation disrupts most of the cooperative interactions that stabilize binding of adjacently bound N-terminal domain dimers to the intact mnt operator.
Effects of Cooperativity Mutations in Intact Mnt.
The experiments described above were performed by using fragments corresponding to the N-terminal domain of Mnt. To test whether the Arg-28→Met/Glu-33→Tyr mutations also affected operator binding in intact, tetrameric Mnt, we constructed and purified a variant (Mnt-MYI-H6) containing both mutations and a His6 tail. In gel mobility-shift experiments, intact Mnt-MYI-H6 bound substantially more poorly than the Mnt-H6 protein to a DNA fragment containing the mnt operator (Fig. 3 Bottom). Addition of the C-terminal coiled-coil increased overall binding affinity relative to the constructs lacking this tetramerization domain but did not suppress the binding defect caused by the Arg-28→Met/Glu-33→Tyr mutations (Fig. 3 Middle and Bottom). Hence, dimer–dimer cooperativity mediated by the latter residues is important both in stabilizing the binding of N-domain dimers and the intact Mnt tetramer to operator DNA.
Operator binding by wild-type Mnt-H6 was fit well by using an equilibrium constant of 1.9 (±1.1) ×10−12 M for the dissociation of tetramers to dimers (Ktet) and a constant of 4.6 (±1.8) × 10−13 M for dissociation of the tetramer from operator DNA (Kd) (Fig. 3 Bottom). Operator binding by Mnt-MYI-H6 was first-order in total protein concentration, as expected if this mutant were largely tetrameric at concentrations above 10−10 M. Fitting of these data gave a Kd of 7.8 (±3.1) × 10−11 M for dissociation of the mutant MYI tetramer from the operator.
As shown in Fig. 4, the overall binding of the Mnt tetramer to its operator can be subdivided into one bimolecular and two unimolecular reactions. In this model, Kd equals 0.25⋅Khs⋅Kdock⋅Kcoop, the product of a statistical factor and three equilibrium constants. The 0.25⋅Khs term accounts for binding of one N-terminal domain dimer in the tetramer to either operator half site in a bimolecular reaction. The Kdock term corresponds to docking of the other N-terminal domain dimer with the second operator half site in a unimolecular reaction. The final term (Kcoop) corresponds to unimolecular formation of the cooperative interactions between adjacently bound N-terminal domain dimers. Equilibrium constants and free energies can be assigned to each of the steps in the Fig. 4 model by assuming that the half-site binding and cooperativity reactions have the same Khs and Kcoop values determined experimentally for the N-terminal domain dimers. We believe that this assumption is justified because the C-terminal domain makes no detectable DNA contacts (1) and does not alter the structure of the N-terminal domain in any discernible fashion (14). Using Kd values for the intact proteins and Khs and Kcoop values for the N-terminal dimers gave Kdock values within error for wild-type Mnt (0.12 ± 0.07) and the MYI mutant (0.049 ± 0.027). Kdock is the ratio of operators with one bound dimer to operators with two bound dimers in the absence of the dimer–dimer cooperativity contacts. Hence, without dimer–dimer cooperativity, a significant fraction of operators (5–10%) have just one dimer in contact with the DNA even though the tetramerization domain constrains the other dimer to be nearby.
Figure 4.

Models for operator binding of the wild-type and MYI Mnt tetramers divided into discrete steps: (i) bimolecular binding of one dimeric DNA-binding domain to a single operator half-site; (ii) intramolecular docking of the second dimeric DNA-binding domain to the adjacent operator half site; and (iii) formation of cooperative contacts between DNA-binding domains. Equilibrium constants were calculated from the data in Fig. 3, as described in the text. ΔG values are calculated for a standard state of 1 μM for the bimolecular reaction and are within error for steps (i), and (ii) for the wild-type and mutant proteins. ΔGcoop for step (iii) is dramatically reduced for the MYI mutant.
Discussion
We have shown that the Arg-28→Met/Glu-33→Tyr double mutation causes a significant defect in the cooperative binding of dimers of the N-terminal domain of Mnt to adjacent operator half sites. Quantifying dimer–dimer cooperativity gave a ΔGcoop value of −4.7 kcal/mol for the wild-type N-domain but only −1.0 kcal/mol for the MYI mutant. The mutant domain had a circular dichroism spectrum similar to wild type, formed dimers with the same chromatographic properties as wild type, and showed wild-type affinity for operator half sites. These results suggest that the MYI cooperativity defect is not produced by global structural changes but rather is caused by the loss of contacts between adjacently bound dimers mediated by Arg-28 and/or Glu-33. Arg-28 corresponds to the residue that makes cooperative contacts between operator-bound Arc dimers (5, 8–9), making it likely that this side chain also makes critical dimer–dimer cooperativity contacts in Mnt.
In the context of Mnt's N-terminal domain, the MYI mutant is more stable than the wild-type domain by 2.5 kcal/mol. We presume that this hyperstability arises because hydrophobic interactions between the mutant side chains are more favorable than the wild-type salt-bridge interactions. This, in turn, probably results from the high cost of desolvating the charged residues and forming the salt bridge in a partially buried environment (15). Interestingly, the increased stability of the MYI mutant, in which two salt-bridged residues are replaced with nonpolar side chains, is almost exactly two-thirds of the increased stability of Arc-MYL, in which three salt-bridged residues are replaced (13). Salt-bridge and hydrogen-bonding interactions between Arg-28 and Glu-33 in Mnt would serve to structurally support cooperative interactions mediated by Arg-28.
Mnt uses two distinct types of quaternary contacts to stabilize operator binding (Fig. 4). First, cooperative interactions between DNA-bound N-terminal domain dimers contribute 4–5 kcal/mol of favorable free energy to operator binding in the wild-type case. These contacts are largely disrupted by the Arg-28/Glu-33 mutations. Second, tetramerization mediated by the C-terminal domain also stabilizes operator binding, because once one N-domain dimer in the tetramer binds to a half site, intramolecular docking of the second N-domain dimer to the other half site occurs with a favorable free energy change of 1–2 kcal/mol. At a standard state of 1 μM—corresponding to roughly 1,000 free Mnt tetramers per cell—the half-site binding reaction, intramolecular docking reaction, and dimer–dimer cooperativity reaction contribute 30, 15, and 55%, respectively, of the total free energy of stabilization (−8.5 kcal/mol; Fig. 4). Under these conditions, the dimer–dimer contacts are clearly a major source of complex stabilization, even though they add just a few extra bonds to the operator complex.
The dimer–dimer contacts are likely to be so favorable because half-site operator binding orients the two DNA-binding domains so precisely that a relatively negligible entropic cost accompanies formation of these cooperative interactions. By contrast, bimolecular half-site binding involves a much larger set of protein–DNA contacts but contributes less to the overall free energy of stabilization because a much larger entropic cost is required to bring two molecules together at a standard-state concentration of 1 μM. Intramolecular half-site docking results in formation of a similarly large number of protein-DNA contacts but contributes even less to complex stabilization than the bimolecular reaction. One way of comparing otherwise comparable bimolecular and unimolecular interactions is to calculate an effective concentration (Ceff) by dividing the equilibrium dissociation constant for the bimolecular reaction (here Khs) by the corresponding equilibrium constant for the unimolecular reaction (here Kdock). The Ceff values for wild-type Mnt (0.41 ± 0.29 μM) and Mnt-MYI (0.69 ± 0.4 μM) are within error. Thus, following binding of one dimeric DNA-binding domain to an operator half-site, the tetramerization domain of Mnt holds the other DNA-binding domain at an effective concentration of approximately 0.5 μM. As discussed below, this Ceff value is significantly lower than would be expected from entropic considerations alone.
NMR studies show that the linkers between the C- and N-terminal domains of Mnt are highly flexible (14). The linker in each monomer is 8–10 residues, and thus there are a total of 36–40 linker residues in the Mnt tetramer. In other flexible-linker systems, effective concentrations vary in rough proportion to (linker length)−3/2, as expected from the entropic considerations of a simple random-walk model (16). In these systems, flexible linkers of 8–40 residues have been shown to have Ceff values ranging from a high of roughly 100 mM to a low of roughly 0.5 mM (16–21). Because the linkers between the C- and N-terminal domains of Mnt tether these domains at an effective concentration of 0.5 μM, this value is at least 1,000-fold lower than would be expected on the basis of linker length and entropic considerations alone. This discrepancy could be explained if the linkage between the tetramerization and DNA-binding domains of Mnt results in a modest degree of enthalpic strain (≈3 kcal/mol) when both DNA-binding domains contact the operator. Such strain could be caused by an orientation of the N-terminal domains in the free tetramer that needs to be distorted on DNA binding. For example, in solution the tetramerization domain might hold the N-terminal DNA-binding domains too far apart to contact both operator half sites without bending of the C-domain helical bundle or unwinding of several residues at the N terminus of each C-domain helix. One prediction of this model is that Mnt variants with longer linkers between domains would bind operator DNA more strongly. Alternatively, bending or distortions of the operator DNA required to accommodate binding of both N-terminal domains in the tetramer could be the source of strain.
The Ceff values calculated for the Mnt system depend on the assumptions inherent in calculating Kdock, namely that the half-site binding and cooperativity portions of the operator-binding reaction for intact Mnt have the same Khs and Kcoop values determined experimentally for the N-terminal domain dimers. What if this assumption were incorrect? The enthalpic strain could no longer be assigned to the Kdock step but would now have to be assigned to the Khs and/or Kcoop steps. As a result, it would still be true that operator binding of both dimeric DNA-binding domains in the Mnt tetramer was less favorable than that expected based on the binding of the isolated domains and the flexible linkage between the tetramerization and DNA-binding domains.
There are several ways in which strain associated with simultaneous DNA binding of both N-terminal domains in the Mnt tetramer might serve a useful biological purpose. One would be to reduce operator affinity, thereby making it easier to lift Mnt repression of the arc and ant genes during the switch from lysogenic to lytic growth of phage P22 (22). Strain could also be useful in reducing the affinity or half-life of Mnt for nonoperator DNA. In principle, it should be even more difficult for both DNA-binding domains in the Mnt tetramer to interact simultaneously with nonoperator than with operator DNA, because both the Kdock and Kcoop steps should be less favorable in the former case. Kdock should be less favorable for nonspecific DNA, because there would be fewer specific contacts between the protein and a half-site to counteract the unfavorable intramolecular entropy and possibly strain of docking. Kcoop should also be less favorable for nonspecific DNA because half-site binding would not orient both DNA-binding domains properly and thus formation of the cooperativity contacts would be associated with a much larger entropic penalty. It seems reasonable to predict, therefore, that nonoperator binding of the Mnt tetramer would chiefly involve just one of the two DNA-binding domains. In the operator complex, by contrast, both the docking and cooperative reactions would be more favorable, allowing both DNA-binding domains to contact the operator and each other. This, in turn, would allow formation of a complex that would be sufficiently stable to mediate repression but one that would not be too stable to allow induction.
Acknowledgments
We thank members of the lab for advice and helpful discussions and Mike Nohaile, Matt Cordes, and Jeff Miller for assistance. This work was supported in part by National Institutes of Health Grants AI-16892, AI-15706 and by a predoctoral fellowship from the National Science Foundation (A.B.).
References
- 1.Waldburger C D, Sauer R T. Biochemistry. 1995;34:13109–13116. doi: 10.1021/bi00040a023. [DOI] [PubMed] [Google Scholar]
- 2.Nooren I M, Kaptein R, Sauer R T, Boelens R. Nat Struct Biol. 1999;6:755–759. doi: 10.1038/11531. [DOI] [PubMed] [Google Scholar]
- 3.Phillips S E. Annu Rev Biophys Biomol Struct. 1994;23:671–701. doi: 10.1146/annurev.bb.23.060194.003323. [DOI] [PubMed] [Google Scholar]
- 4.Somers W S, Phillips S E. Nature (London) 1992;359:387–393. doi: 10.1038/359387a0. [DOI] [PubMed] [Google Scholar]
- 5.Raumann B E, Rould M A, Pabo C O, Sauer R T. Nature (London) 1994;367:754–757. doi: 10.1038/367754a0. [DOI] [PubMed] [Google Scholar]
- 6.Gomis-Ruth F X, Sola M, Acebo P, Parraga A, Guasch A, Eritja R, Gonzalez A, Espinosa M, del Solar G, Coll M. EMBO J. 1998;17:7404–7415. doi: 10.1093/emboj/17.24.7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burgering M J, Boelens R, Gilbert D E, Breg J N, Knight K L, Sauer R T, Kaptein R. Biochemistry. 1994;33:15036–15045. doi: 10.1021/bi00254a012. [DOI] [PubMed] [Google Scholar]
- 8.Brown B M, Milla M E, Smith T L, Sauer R T. Nat Struct Biol. 1994;1:164–168. doi: 10.1038/nsb0394-164. [DOI] [PubMed] [Google Scholar]
- 9.Smith T L, Sauer R T. J Mol Biol. 1995;249:729–742. doi: 10.1006/jmbi.1995.0332. [DOI] [PubMed] [Google Scholar]
- 10.Knight K L, Sauer R T. EMBO J. 1992;11:215–223. doi: 10.1002/j.1460-2075.1992.tb05044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brenowitz M, Senear D F, Shea M A, Ackers G K. Methods Enzymol. 1986;130:132–181. doi: 10.1016/0076-6879(86)30011-9. [DOI] [PubMed] [Google Scholar]
- 12.Brown B M, Sauer R T. Biochemistry. 1993;32:1354–1363. doi: 10.1021/bi00056a022. [DOI] [PubMed] [Google Scholar]
- 13.Waldburger C D, Schildbach J F, Sauer R T. Nat Struct Biol. 1995;2:122–128. doi: 10.1038/nsb0295-122. [DOI] [PubMed] [Google Scholar]
- 14.Nooren I M A, Folkers G E, Kaptein R, Sauer R T, Boelens R. J Biomol Struct Dyn. 2000;11:113–122. doi: 10.1080/07391102.2000.10506611. [DOI] [PubMed] [Google Scholar]
- 15.Hendsch Z S, Tidor B. Protein Sci. 1994;3:211–226. doi: 10.1002/pro.5560030206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng Z Y, Wu L, Kim P S. Biochemistry. 1995;34:3248–3252. doi: 10.1021/bi00010a014. [DOI] [PubMed] [Google Scholar]
- 17.Creighton T E, Goldenberg D P. J Mol Biol. 1984;179:497–526. doi: 10.1016/0022-2836(84)90077-9. [DOI] [PubMed] [Google Scholar]
- 18.Lin T-Y, Kim P S. Biochemistry. 1989;28:5282–5287. doi: 10.1021/bi00438a054. [DOI] [PubMed] [Google Scholar]
- 19.Lin T-Y, Kim P S. Proc Natl Acad Sci USA. 1991;88:10573–10577. doi: 10.1073/pnas.88.23.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Klemm J D, Pabo C O. Genes Dev. 1996;10:27–36. doi: 10.1101/gad.10.1.27. [DOI] [PubMed] [Google Scholar]
- 21.Robinson C R, Sauer R T. Proc Natl Acad Sci USA. 1998;95:5929–5934. doi: 10.1073/pnas.95.11.5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Susskind M M, Youderian P. In: Lambda II. Hendrix R W, Roberts J W, Stahl F W, Weisberg R, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1983. pp. 347–366. [Google Scholar]
- 23.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]