

Abstract
Two models for speciation via selection have been proposed. In the well-known model of ‘ecological speciation’, divergent natural selection between environments drives the evolution of reproductive isolation. In a second ‘mutation-order’ model, different, incompatible mutations (alleles) fix in different populations adapting to the same selective pressure. How to demonstrate mutation-order speciation has been unclear, although it has been argued that it can be ruled out when gene flow occurs because the same, most advantageous allele will fix in all populations. However, quantitative examination of the interaction of factors influencing the likelihood of mutation-order speciation is lacking. We used simulation models to study how gene flow, hybrid incompatibility, selective advantage, timing of origination of new mutations and an initial period of allopatric differentiation affect population divergence via the mutation-order process. We find that at least some population divergence can occur under a reasonably wide range of conditions, even with moderate gene flow. However, strong divergence (e.g. fixation of different alleles in different populations) requires very low gene flow, and is promoted when (i) incompatible mutations have similar fitness advantages, (ii) less fit mutations arise slightly earlier in evolutionary time than more fit alternatives, and (iii) allopatric divergence occurs prior to secondary contact.
Keywords: Dobzhansky–Muller incompatibilities, ecological speciation, gene flow, migration, reproductive isolation, uniform selection
1. Introduction
Natural selection commonly promotes speciation [1–5]. This statement is supported by widespread correlations between levels of ecological divergence and reproductive isolation [3,6], cases of divergence with levels of gene flow too high to allow genetic drift [7], examples of traits under selection that also cause reproductive isolation [8], and theoretical studies [1,2,9]. More specifically, reproductive isolation can arise if the different alleles favoured by selection within each population are incompatible with one another when brought together in the genome of a hybrid [1,2]. Indeed, negative interactions between alleles at different loci have been shown to result in hybrid inviability and sterility (Dobzhansky–Muller incompatibilities denoted as DMIs hereafter; [2,10,11]) and molecular studies have demonstrated that selection can drive the evolution of the genes involved [1,12].
However, different types of selection might drive the evolution of reproductive isolation. Consequently, models of speciation via selection have recently been classified into two main categories: ecological speciation and mutation-order speciation [4,13]. In ‘ecological speciation’, divergent natural selection between different environments drives the evolution of reproductive isolation. This model has received substantial support from laboratory experimental evolution studies ([14] for review), case studies in nature [15–18] and comparative analyses [3].
In ‘mutation-order speciation’, different and incompatible mutations (alleles) fix in different populations adapting to the same selective pressure (i.e. uniform selection, following Mani & Clarke [19]; reviewed in Schluter [4]). The order in which new mutations arise can affect which mutations fix, making ‘mutation-order’ a potentially important stochastic process in evolution. This mechanism has also been referred to within the more inclusive term ‘non-ecological speciation’, but as noted by Sobel et al. [5], uniform selection can still involve ecology. While laboratory experimental evolution studies have demonstrated that uniform selection on isolated populations can lead to divergence (e.g. [20,21]), mutation-order speciation is less substantiated than ecological speciation, in part because how to demonstrate it in nature is unclear [4]. Thus, it is important to develop predictions for when mutation-order speciation is likely and tests that can distinguish it from ecological speciation. This is the aim of the current article. We focus on how five factors impact mutation-order speciation: (i) rates of gene flow, (ii) the degree of hybrid incompatibility, (iii) the relative fitness of different new mutations, (iv) the timing of origination of different mutations, and (v) an initial period of allopatric divergence preceding secondary contact and gene flow. We consider reproductive isolation (i.e. low hybrid fitness) arising from both within-locus and between-locus incompatibilities. Population differentiation in genes reducing hybrid fitness is our measure of divergence via the mutation-order process, with divergence of more strongly incompatible alleles representing the evolution of stronger reproductive isolation.
It has been argued verbally that mutation-order speciation is difficult, and can perhaps be ruled out, when gene flow occurs [4,13]. The logic is as follows. Suppose that two new adaptive but incompatible alleles arise in two different populations. The alleles will move between populations via gene flow, and because one allele will almost always have at least a slightly higher selective advantage than others, a likely outcome is that the same, most advantageous allele will fix in both populations. Additionally, with gene flow, incompatible genotypic combinations will be created (i.e. ‘hybrids’), and alleles that are part of this combination will be removed by selection [1]. These processes should prevent different incompatible alleles from fixing in different populations which experience gene flow. However, based on verbal arguments alone, it is unclear what level of gene flow would prohibit divergence. It is also unclear how evolutionary factors other than gene flow affect mutation-order speciation, and how different factors interact. In summary, clearer predictions are required about mutation-order speciation.
Previous theoretical work has examined DMIs and mutation-order divergence in a few important ways. Seminal studies demonstrated that pairwise interactions between loci accumulate in a nonlinear fashion, leading to a ‘snowball’ of DMI accumulation [10,11]. However, these studies did not consider gene flow or specify the type of selection driving divergence. Some considered the importance of mutation-order when speciation is driven solely via drift [22–25]. The effect of mutation-order during divergence via selection on new, advantageous alleles in a uniform environment has also been examined in some contexts. Cohan [26] showed that uniform selection in isolated populations accelerates divergence relative to that observed via drift alone. An insightful paper by Mani & Clarke [19] used simulations to establish mutation-order as an important stochastic process in evolution, but one distinct from genetic drift, because mutations fix via selection. Similarly, Unckless & Orr [27] derived analytical expressions for probabilities of the evolution of a DMI when two populations experience identical environments. However, all these studies examined populations that experience no gene flow.
Other models examined the mutation-order process with gene flow [2,28–31]. For example, Gavrilets presents a simple model of mutation-order speciation ([2], pp. 139–140) and reviews more complex spatial models of the process ([2], pp. 221–228). The main conclusion to date is that if there is gene flow, then ‘Once established in a local population, an advantageous allele will tend to spread across the whole system’ ([2], p. 225). Previous models, however, have not ‘competed’ new mutations against one another in the face of gene flow, varying their selective advantage and timing or origination, to test the factors promoting and constraining mutation-order speciation.
The current study extends useful past models in at least three general ways. First, we vary the fitness of new alleles and hybrid genotypes relative to each other and relative to ancestral alleles and genotypes. Second, we vary the timing of origination of selectively advantageous alleles relative to each other. Third, we consider the effects of initial periods of allopatric divergence preceding the onset of gene flow. We use the results to develop predictions about the conditions that affect mutation-order speciation.
2. General methods
We used simulation models to study populations of diploid, sexually reproducing organisms with discrete, non-overlapping generations. We assume that individuals are divided between two identical patches, and we refer to each patch as a population. Within populations, we assume random mating with respect to genotype. Our first model considered a single locus (model 1: ‘single locus model’). The second model considered epistasis between two completely unlinked loci (i.e. a DMI; model 2: ‘epistasis model’). Thus, both models study the origin of an incompatibility, whether it leads to divergence, and if so, what degree of divergence. Although speciation probably often involves divergence at more than two loci [1], studying a two-locus model is the most logical starting point for a multi-locus model because: (i) the first alleles to diverge are those that initiate the speciation process, and (ii) once one incompatibility is established, the establishment of future incompatibilities via a ‘snowball’ process [10,11] becomes possible. Because qualitatively similar results were observed between models, we present here only the results for the more generally accepted scenario of a DMI (our ‘epistasis model’). Results for the single-locus model are presented in the electronic supplementary material. Unless otherwise noted, descriptions below apply to the epistasis model.
Both models considered the fate of two new mutations that were selectively advantageous, as might be expected for speciation with gene flow [2,32,33]. Thus, we assume no genetic drift. The possible genotypes and their fitnesses are given in table 1. Within a patch, we assume Hardy–Weinberg genotype frequencies after random mating has occurred. Given these assumptions, we can use very straightforward equations (described below) to model allele frequency change from one generation to the next.
Table 1.
Alleles, genotypes and fitnesses in the two models.
| model | possible alleles | possible genotypes | fitnesses |
|---|---|---|---|
| model 1: single-locus (within-locus incompatibilities) | b, a, A | ancestral: bb derived: ab, aa, Ab, AA, Aa | wbb = 1 wab = waa > 1 wAb = wAA > waa, wAa ≤ 1 |
| model 2: epistasis (between-locus incompatibilities) | locus 1: a, A locus 2: b, B | ancestral: aabb derived: Aabb, AAbb, aaBb, aaBB, AaBb, AaBB, AABb, AABB | waabb = 1 waaBb = waaBB > 1 wAabb = wAAbb > waaBbwAaBb = wAaBB = wAABb = wAABB ≤ 1 |
The general scheme is as follows (figure 1). Populations are initialized with a single recessive allele initially fixed at each locus of interest. The genotype with only the ancestral, recessive allele(s) is assigned a fitness of 1. We study the spread of two new mutations (alleles), each of which has higher fitness than the ancestral allele but is also incompatible, to some degree, with the other new allele (table 1). When a new allele arises, it is assumed to be dominant to the ancestral allele at its locus, and initially arises with a starting frequency of 0.001 in one of the patches (and 0 in the other patch). Each simulation run then proceeds with the iteration of a two-step cycle for 107 generations or until allele frequencies have stopped changing in both patches (whichever comes first).
Figure 1.

A schematic representation of the epistasis model. There are two patches, 1 and 2, that experience uniform selection for the same optimum. Symmetrical migration between patches occurs at rate m. Each patch is initially fixed for the ancestral alleles a and b. During the course of the simulations, two new mutations arise, denoted as A and B. Both new mutations confer a selective advantage over the ancestral genotype. A is the most favoured allele, and B is more favoured than b, but less favoured than A (table 1). ‘Hybrid’ individuals carrying both A and B exhibit reduced fitness because these two alleles are incompatible. Mutations may arise simultaneously or in staggered fashion such that mutation B arises first in patch 1, reaches a frequency qthresh in at least one of the patches, following which mutation A arises in patch 2. Scenarios of initial allopatric divergence were also examined by setting m initially to zero for tcontact generations.
In the first step of the cycle, natural selection adjusts allele frequencies according to the selection coefficients for each genotype. In the second step of the cycle, migration between patches occurs. We assume that migration is random with respect to patch and genotype, and we vary the migration rate between simulation runs. Thus, a fraction m of each genotype was moved from each patch to the other patch. Fixation of an allele in a patch was defined to have occurred if the allele reached a frequency greater than 0.9999 in that patch. To determine if ending simulations at 107 generations affected our results, we examined the final 100 timesteps of thousands of cases that reached 107 generations, and in every case the allele-frequency changes were extremely small in magnitude (less than 10−15). That is, the system had essentially reached equilibrium before the simulation was ended, and only infinitesimal changes (near the limit of computational precision for double-precision floating-point decimal numbers) were occurring. All simulations were programmed in C and run on a 64-bit Intel Macintosh computer. Source code and spreadsheets with full results are available upon request.
3. Between-locus incompatibilities: the epistasis model
Here we consider two incompatible alleles that arise at separate loci and interact to reduce hybrid fitness. The ancestral alleles at the two loci are designated a and b, and the two new adaptive but incompatible alleles are A (arises at the locus where a is ancestral) and B (arises at the locus where b is ancestral). Since we are interested in the divergence of populations, we assume that the new alleles always arise in different patches. The patch in which B arises is designated patch 1, and the patch in which A arises is designated patch 2. We let pi represent the frequency of allele A in patch i (i = 1 or 2) and qi represent the frequency of allele B in patch i. Thus, the frequencies of alleles a and b in patch i are, respectively, ri = 1 − pi and si = 1 − qi. We are interested in scenarios in which both new alleles are advantageous when compared with ancestral alleles, but one new allele is more advantageous than the other. Thus, AAbb (or Aabb) was the most favoured genotype and aaBB (or aaBb) was more favoured than aabb, but less favoured than AAbb (table 1 for full fitness scheme).
Allele frequencies changed from the current generation to the next as follows. With the assumptions stated above, we calculate the frequency of A in patch i in the next generation as its proportional weighted fitness (e.g. [34], p. 42):
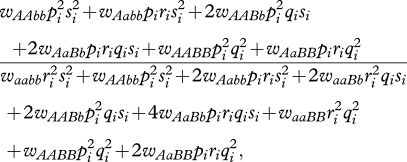 |
3.1 |
where wAAbb is the fitness of an individual of genotype AAbb, wAabb is the fitness of an individual of genotype Aabb, and so on. The frequency of a in patch i in the next generation was simply 1 − [equation (3.1)]. The frequencies of B and b in the next generation were calculated in an analogous manner.
To explore the effects of the relative timing of origination of the two alleles, we allowed allele B (the less fit of the two new alleles) to arise in patch 1 (with initial frequency q1 = 0.001) and spread to a frequency of qthresh in at least one of the patches, while holding p1 = p2 = 0. In essence, we gave the less advantageous mutation a ‘leg-up’, such that it could spread to various frequencies before the second, more advantageous mutation originated. Following this time-lag period set by qthresh, we then allowed allele A to arise in patch 2 with a frequency p2 = 0.001. In addition to adding biological realism, this feature addresses questions about whether newer alleles that are better adapted can displace incompatible alleles that already have a foothold in a population. We varied qthresh over several values (0, 0.01, 0.1, 0.333333, 0.5 and 0.9), with qthresh = 0 corresponding to simultaneous origination of both mutations.
To explore the effects of an initial period of allopatric divergence preceding gene flow, we ran simulations as above, but set m to zero for the first tcontact number of generations. We varied tcontact over a wide range of values (0, 10, 25, 30, 40, 75, 100, 1000, 10 000). We focused on analysing the role of allopatry using the parameter combinations of qthresh and hybrid incompatibility that yielded the most divergent predictions about evolutionary outcomes (qthresh = 0, wAABB = 0.9 and qthresh = 0.333, wAABB = 0.001, see §4). Exploration of a wider range of parameter values revealed that these two sets of parameter combinations accurately encapsulated the range of effects of allopatric divergence (electronic supplementary material, figures S4 and S5).
Two main sets of simulation runs were conducted that we present here. The first set was used to explore large, biologically realistic areas of parameter space. Simulations in this set were run with all combinations of the following ranges of parameter values (with the constraint that only combinations with wAAbb > waaBB were used): wAAbb = [1.005, 1.01, 1.02, 1.03, 1.04, 1.05, 1.1, 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.5]; waaBB = [1.001, 1.005, 1.01, 1.02, 1.03, 1.04, 1.05, 1.1, 1.15, 1.2, 1.25, 1.3, 1.35, 1.4, 1.45, 1.49]; wAABB = [1, 0.999, 0.995, 0.99, 0.98, 0.97, 0.96, 0.95, 0.9, 0.8, 0.7, 0.6, 0.5, 0.4, 0.3, 0.2, 0.1, 0.05, 0.01, 0.005, 0.001, 0]; m = [0, 10−5, 10−4, 10−3, 10−2, 0.0125, 0.025, 0.05, 0.1]; qthresh = [0, 0.01, 0.1, 0.333, 0.5 and 0.9]; and tcontact = 0. For the most part, changing parameter values had moderate, quantitative effects on the results, with the migration rate and ratio of mutant fitnesses having the largest effects. As such, we show a subset of data that illustrate the main trends.
We then performed a second set of simulations that were used to gain higher resolution in regions of parameter space that illustrate the effects of each parameter. For this second set, we fixed waaBB = 1.2 and then varied wAAbb over 100 evenly spaced values between 1.206 and 1.8. This varied the ratio of fitnesses, wAAbb/waaBB, between 1.005 and 1.5 in increments of 0.005. Simultaneously, we varied m over 100 logarithmically spaced values between 10−5 and 10−1. This resulted in a set of 10 000 simulation runs which we display together in a single phase diagram. Different phase diagrams show the results for different combinations of the values of qthresh (0, 0.01 or 0.333), tcontact (0, 10, 25, 40 or 75 generations) and wAABB (0.001 or 0.9).
Using the following classification scheme, we show the locations of boundaries between different evolutionary outcomes. We categorized our results into five outcomes, which correspond to the differently shaded regions shown in phase diagrams (M.O. = mutation-order divergence): (i) ‘extreme M.O.’ = when the two alternative, incompatible alleles each reached and maintained a frequency greater than 0.99 in their respective patches, (ii) ‘moderate M.O.’ = when the alternative, incompatible alleles reached and maintained frequencies greater than 0.9 (but one or both alleles had a frequency less than 0.99) in their respective patches, (iii) ‘weak M.O’ = all cases when A and B were both maintained but divergence was weaker than for ‘moderate M.O.’, (iv) ‘A fixed’ when allele A was fixed in both patches and allele B was eliminated, and (v) ‘B fixed’ when allele B was fixed in both patches and allele A was eliminated. We stress that outcomes (i)–(iii) are not qualitatively different: they all represent population divergence via the mutation-order process. Nonetheless, categorization into three levels is useful for visualizing and discussing the magnitude of population divergence observed, in each case occurring in the absence of ecologically based divergent selection. By contrast, outcomes (iv) and (v) represent cases in which there is no divergence.
4. Epistasis model: results
At least some population divergence via the mutation-order process was observed under a relatively wide range of conditions (figures 2–4; electronic supplementary material, figures S4 and S5). However, strong divergence was unlikely unless gene flow was absent or extremely low. Specifically, moderate M.O. and extreme M.O. were never observed at migration rates above 0.01 and in general consistently occurred only when m < 0.001.
Figure 2.

The effects of migration rate and relative fitnesses on mutation-order divergence when the novel alleles arise at different loci (epistasis model). Squares with continuous line show the frequency of allele A in patch 1 (P1); circles with dashed lines show the frequency of allele A in patch 2 (P2). In panels (a,b), the mutations (alleles A and B) arise simultaneously. In panels (c–l), allele B reaches a frequency qthresh in at least one patch before allele A arises in patch 2. In all panels, there is no allopatric divergence period (i.e. tcontact = 0). Strong population divergence (i.e. p1 ≈ 0 and p2 ≈ 1) via the mutation-order process occurs when there is no migration (all panels) or when the migration rate is low and fitnesses are relatively similar (right-hand panels). Note that while the frequencies of allele B are not shown, by the end of a simulation, we have qi ≈ 1 − pi (i.e. either A or B becomes common in a patch by the end).
Figure 4.

The role of a period of tcontact generations of allopatric divergence on the outcome of mutation-order divergence in the epistasis model. Shading as for figure 3; top row: parameter values as in figure 3a; bottom row: parameters values as in figure 3f. (a,e) tcontact = 10 generations; (b,f) 25 generations; (c,g) 40 generations; (d,h) 75 generations.
Three factors other than gene flow strongly affected the probability of divergence via the mutation-order process. First, as the difference in selective advantage of the two new mutations decreased, mutation-order divergence became more likely (figures 2–4). This was because small differences in relative fitness caused the most fit allele, A, to have a smaller chance of fixation in both patches.
Second, staggering the timing of origination of mutations affected the outcome, but its effect depended upon other parameters. When the first, less-fit mutation (allele B) originated slightly before the second, mutation-order divergence was promoted; for example, being observed when differences in the selective advantage of new mutations were relatively large. By contrast, when the first mutation, B, originated long before the second mutation (i.e. was given a large ‘leg-up’), the less advantageous allele could actually spread between and even fix in both patches, preventing mutation-order divergence. In part, the latter was owing to the second mutation, A, often finding itself in a low fitness hybrid (AaBb, AaBB, AABb or AABB). The latter effect could only be ameliorated by extremely low migration rates that prevented allele B from rising in frequency in patch 2.
Third, an initial period of allopatric divergence tended to promote mutation-order divergence, which can be seen by noting that all three M.O. regions expand as one moves left to right across the panels in figure 4. Just 25–75 generations of allopatric divergence promoted mutation-order divergence. Periods of allopatry longer than 75 generations has little additional effect (e.g. results for 100–10 000 generations of allopatry are very similar to those for 75 generations; electronic supplementary material, figures S4 and S5).
In contrast to the effects of the other factors, hybrid fitness had relatively weak effects: greater incompatibility caused only slight increases in the conditions favouring mutation-order divergence. Nonetheless, as wAABB decreased from 0.9 (moderate incompatibility; figure 3a,b) to 0.001 (nearly complete incompatibility; figure 3d,e), the M.O. regions all expanded somewhat (figure 3). Overall, these results were qualitatively very similar to those observed for the single locus model (for details see electronic supplementary material, figures S1–S3).
Figure 3.

Outcomes of simulations from the epistasis model as influenced by migration rate, the ratio (wAAbb/waaBB) of fitnesses of the two favoured alleles, and the fitness wAABB (=wAaBB = wAABb = wAaBb) of genotypes having at least one copy of each of the two incompatible alleles. Each panel represents outcomes from 10 000 simulations for varying combinations of the fitness ratio (wAAbb/waaBB) and the migration rate. In all panels, there is no allopatric divergence period (i.e. tcontact = 0). Outcome classifications are according to the shading as detailed in (e) and as explained in the text. (a) no lag, wAABB = 0.9; (b) qthresh = 0.01, wAABB = 0.9; (c) qthresh = 0.333, wAABB = 0.9; (d) no lag, wAABB = 0.001; (e) qthresh = 0.01, wAABB = 0.001; (f) qthresh = 0.333, wAABB = 0.001.
5. Discussion
Previous considerations of mutation-order speciation have primarily focused on a single prediction: divergence should be unlikely if there is gene flow [4]. However, there has been a lack of specificity about what level of gene flow would prevent mutation-order divergence, and a lack of predictions about the influence of factors other than gene flow. While our models partly confirm intuitive effects of gene flow (i.e. more gene flow = less divergence), we also show that even with moderate gene flow, the mutation-order process can cause DMIs to become established, thereby beginning the process of population divergence, and potentially sowing the seeds for a ‘snowball’ [10,11] of DMI accumulation. Furthermore, our model offers straightforward predictions about how four other factors affect the likelihood of mutation-order speciation. Here, we discuss the predictions our model generates, how these can be tested empirically, and extensions for future work.
(a). Predictions
(i). Gene flow
The mutation-order process can lead to at least some divergence of populations even when there is gene flow, but strong population divergence requires little or no gene flow (figures 2–4). There is an upper limit of gene flow beyond which any divergence at all via a mutation-order process is unlikely. This is seen by noting the presence of a boundary on the right-hand sides of the different M.O. regions of figures 3 and 4. Finally, previous verbal arguments about mutation-order speciation (discussed above) suggested that high gene flow would lead to fixation of the most fit allele. Our model shows that this is sometimes, but not always, true. While high gene flow will lead to fixation of a derived allele (rather than divergence of populations), it is not necessarily the most fit allele that fixes in such cases; which allele fixes depends upon other factors (predictions below).
(ii). Relative fitness of incompatible alleles
Not surprisingly, increasing the ratio of fitnesses (wAAbb/waaBB) favoured allele A, causing (i) a transition from mutation-order divergence to complete fixation of A (figure 3a,d; qthresh = 0, i.e. no lag), and (ii) a transition from fixation of allele B to mutation-order divergence (figure 3c,f; qthresh = 0.333). Thus, mutation-order divergence will be more likely when new, incompatible alleles have more similar fitnesses (see also Unckless & Orr [27]).
(iii). Staggered emergence of new alleles
An intermediate length of time between mutations is the most likely to produce mutation-order divergence (figure 3b,e). When there was no delay, much of the M.O. regions were replaced by the complete fixation of the most favoured allele, A. On the other hand, when the delay was much longer, the M.O. regions were eroded by complete fixation of B. In general, by arising somewhat earlier a less-fit allele could often exclude another, better adapted allele.
(iv). Period of allopatry
Coyne & Orr [1] and Barton [28] suggested that allopatric divergence followed by secondary contact could facilitate mutation-order divergence. Our models support this suggestion. In fact, if the allopatric period lasted enough generations, the influences of all parameters except migration rate were nearly eliminated. Intuitively, this is explained by the fact that when tcontact is large, each of the derived alleles has (nearly or completely) gone to fixation in its original patch before any migration occurred; the outcome after secondary contact is then a consequence of migration-selection balance. While this might be expected via intuition alone, a less obvious finding is that relatively short periods of allopatry could promote mutation-order divergence, with longer periods having little additional effect on promoting divergence. However, a long enough period of allopatric divergence eliminated the chance that the less fit of the two new alleles could go to fixation. Periods of allopatric divergence are thus predicted to counter the effects of long time lags between mutations. Some examples of ecological speciation between currently sympatric taxa involved an allopatric element [35,36], but data from cases of mutation-order divergence are required to test the role of allopatry.
(v). Incompatibility between alleles
Somewhat surprisingly, the degree of incompatibility of the two new mutations had effects that were quite small in magnitude, as seen by noting that the top and bottom rows of panels in figure 3 have widely different hybrid fitness values, yet the sizes of the different regions are changed only slightly. Thus, divergence in genes causing weak or strong reproductive isolation occurred, so long as gene flow was low. Intuitively, greater incompatibility did (slightly) increase mutation-order divergence.
(b). Strong inference tests of ecological and mutation-order speciation
A powerful way to test for mutation-order speciation would be to use experimental evolution in controlled laboratory experiments. Some experiments have found divergence or premating isolation to evolve between different replicate populations evolved in the same environment ([14,21]; for review [20]). However, most such experiments were designed to test ecological speciation, or did not manipulate the variables our model predicts are important for mutation-order divergence (e.g. Cohan & Hoffmann [21] evolved populations in similar environments without gene flow). Thus, experimental evolution studies that test the mutation-order process and manipulate the factors we have highlighted are warranted.
How might mutation-order speciation versus ecological speciation be supported in nature? Our predictions above generate strong inference (sensu [37]) tests of these two speciation processes. Namely, one could conduct reciprocal transplant experiments using populations that are known to exhibit some reproductive isolation (e.g. a DMI). Such experiments would rigorously establish (i) whether a given genotype has the same fitness across different environments (i.e. whether selection is nearly uniform or not), and (ii) the relative fitnesses of different genotypes in the same environment. If selection is uniform across environments, as observed in some experiments (Leimu & Fischer [38] for review), the hypothesis of ecological speciation is eliminated. On the other hand, evidence for divergent selection would falsify a basic assumption of mutation-order speciation.
If transplant experiments eliminated ecological speciation as a viable explanation, further information might add support for mutation-order divergence, by being consistent with the mutation-order process being at work in a specific set of populations. For example, mutation-order speciation would be more strongly supported if the populations under study have low rates of gene flow. Additionally, small fitness differences between genotypes would make mutation-order more likely. While estimating the selective advantage of specific alternative alleles might be difficult, specific mutations underlying adaptation (Hubbard et al. [39] for review) and speciation (Coyne & Orr [1] for review) have been detected, and at least one experiment has estimated the strength of selection acting on a specific mutation [40]. A third factor of interest would be to determine the strength of hybrid incompatibility, with a high degree of incompatibility making mutation-order speciation a more likely explanation. Finally, coalescent-based techniques might allow tests of whether a period of allopatric divergence preceded gene flow (Hey [7] for review), although the efficacy of such techniques requires consideration [41].
As for the overall prevalence of mutation-order divergence as a driver of speciation, comparative studies across many sets of populations are required. In such studies, the predictions above could be examined by looking at whether the various factors correlate with the incidence of mutation-order divergence.
(c). Future work: extensions to the model
Two obvious extensions are to consider the effects of: (i) many alleles at many loci and (ii) stochastic changes via drift. With regard to the first point: how many loci and alleles tend to cause incompatibility? Empirical evidence is mixed. Strong hybrid incompatibility can be caused by just a few loci [1,42–44], but many different sets of a few loci can potentially generate incompatibility [45]. Rapid evolution (i.e. fixation) of advantageous mutations is possible, once they emerge [1,42,43,46], arguing that numerous advantageous mutations will rarely be simultaneously segregating within a population. At the very least, our scenario of two alleles at two loci is a necessary biological precursor to any greater number of new alleles/loci. Nonetheless, we expect that interesting dynamics could emerge if more alleles/loci are considered, as might occur when gene flow maintains polymorphism. For example, an increased number of alleles could create more possible ways of achieving mutation-order speciation, but could also create more possible incompatibilities, which would inhibit the spread of newer, rare alleles. Analysis of this issue is beyond the scope of this article, but future work could modify past models of the accumulation of DMIs [10,11,27].
Another extension would be stochastic changes in allele frequencies. Our deterministic treatment is justified because we considered the rise and fixation of selectively advantageous mutations [2,32,33]. Adding stochasticity to the dynamics of beneficial allele replacements should not affect the conclusions qualitatively, especially with large population sizes [11,47]. This argument is bolstered by the fact that drift is expected to affect the results in the same direction as the documented trends. That is, when differences in the selective advantage of different mutations are large, the less selectively advantageous mutation should be lost by drift more readily than the more advantageous mutation, preventing mutation-order speciation. This is the exact outcome observed in our deterministic model when differences in selective advantages were great. When such differences are slighter, each mutation would be similarly affected by drift. Nonetheless, stochastic simulations could address how divergent selection, mutation-order and genetic drift might interact. Another avenue would be to examine how divergence of selected loci via the mutation-order process might promote a general barrier to gene flow at neutral loci (cf. [2,48]).
These represent interesting avenues for further research, although they should not alter our main conclusions: some mutation-order divergence can occur under relatively broad conditions, but fixation of alternative alleles via the mutation-order process speciation is extremely difficult unless gene flow is very low, and in such cases where gene flow is low, a number of other factors can promote or inhibit divergence.
Acknowledgements
We thank R. Safran and D. Schluter for discussion. S. Gavrilets, H. Rundle and two anonymous reviewers provided useful comments on earlier drafts of the manuscript.
References
- 1.Coyne J. A., Orr H. A. 2004. Speciation. Sunderland, MA: Sinauer Associates, Inc [Google Scholar]
- 2.Gavrilets S. 2004. Fitness landscapes and the origin of species. Princeton, NJ: Princeton University Press [Google Scholar]
- 3.Funk D. J., Nosil P., Etges B. 2006. Ecological divergence exhibits consistently positive associations with reproductive isolation across disparate taxa. Proc. Natl Acad. Sci. USA 103, 3209–3213 10.1073/pnas.0508653103 (doi:10.1073/pnas.0508653103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schluter D. 2009. Evidence for ecological speciation and its alternative. Science 323, 737–741 10.1126/science.1160006 (doi:10.1126/science.1160006) [DOI] [PubMed] [Google Scholar]
- 5.Sobel J. M., Chen G. F., Watt L. R., Schemske D. W. 2010. The biology of speciation. Evolution 64, 295–315 10.1111/j.1558-5646.2009.00877.x (doi:10.1111/j.1558-5646.2009.00877.x) [DOI] [PubMed] [Google Scholar]
- 6.Rundle H. D., Nosil P. 2005. Ecological speciation. Ecol. Lett. 8, 336–352 10.1111/j.1461-0248.2004.00715.x (doi:10.1111/j.1461-0248.2004.00715.x) [DOI] [Google Scholar]
- 7.Hey J. 2006. Recent advances in assessing gene flow between diverging populations and species. Curr. Opin. Genet. Dev. 16, 592–596 10.1016/j.gde.2006.10.005 (doi:10.1016/j.gde.2006.10.005) [DOI] [PubMed] [Google Scholar]
- 8.Jiggins C. D., Naisbit R. E., Coe R. L., Mallet J. 2001. Reproductive isolation caused by colour pattern mimicry. Nature 411, 302–305 10.1038/35077075 (doi:10.1038/35077075) [DOI] [PubMed] [Google Scholar]
- 9.Kirkpatrick M., Ravigné V. 2002. Speciation by natural and sexual selection: models and experiments. Am. Nat. 159, S22–S35 10.1086/338370 (doi:10.1086/338370) [DOI] [PubMed] [Google Scholar]
- 10.Orr H. A. 1995. The population genetics of speciation: the evolution of hybrid incompatibilities. Genetics 139, 1805–1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orr H. A., Turelli M. 2001. The evolution of postzygotic isolation: accumulating Dobzhansky–Muller incompatibilities. Evolution 55, 1085–1094 [DOI] [PubMed] [Google Scholar]
- 12.Tang S. W., Presgraves D. C. 2009. Evolution of the Drosophila nuclear pore complex results in multiple hybrid incompatibilities. Science 323, 779–782 10.1126/science.1169123 (doi:10.1126/science.1169123) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Price T. D. 2007. Speciation in birds. Greenwood Village, CO: Roberts and Company [Google Scholar]
- 14.Rice W. R., Hostert E. E. 1993. Laboratory experiments on speciation: what have we learned in 40 years. Evolution 47, 1637–1653 10.2307/2410209 (doi:10.2307/2410209) [DOI] [PubMed] [Google Scholar]
- 15.Funk D. J. 1998. Isolating a role for natural selection in speciation: host adaptation and sexual isolation in Neochlamisus bebbianae leaf beetles. Evolution 52, 1744–1759 10.2307/2411347 (doi:10.2307/2411347) [DOI] [PubMed] [Google Scholar]
- 16.Rundle H. D., Nagel L., Boughman J. W., Schluter D. 2000. Natural selection and parallel speciation in sympatric sticklebacks. Science 287, 306–308 10.1126/science.287.5451.306 (doi:10.1126/science.287.5451.306) [DOI] [PubMed] [Google Scholar]
- 17.Langerhans R. B., Gifford M. E., Joseph E. O. 2007. Ecological speciation in Gambusia fishes. Evolution 61, 2056–2074 10.1111/j.1558-5646.2007.00171.x (doi:10.1111/j.1558-5646.2007.00171.x) [DOI] [PubMed] [Google Scholar]
- 18.Nosil P. 2007. Divergent host-plant adaptation and reproductive isolation between ecotypes of Timema cristinae walking-sticks. Am. Nat. 169, 151–162 10.1086/510634 (doi:10.1086/510634) [DOI] [PubMed] [Google Scholar]
- 19.Mani G. S., Clarke B. C. 1990. Mutational order: a major stochastic process in evolution. Proc. R. Soc. Lond. B 240, 29–37 10.1098/rspb.1990.0025 (doi:10.1098/rspb.1990.0025) [DOI] [PubMed] [Google Scholar]
- 20.Travisano M., Vasi F., Lenski R. E. 1995. Long-term experimental evolution in Escherichia coli. III. Variation among replicate populations in correlated responses to novel environments. Evolution 49, 189–200 10.2307/2410304 (doi:10.2307/2410304) [DOI] [PubMed] [Google Scholar]
- 21.Cohan F. M., Hoffmann A. A. 1989. Uniform selection as a diversifying force in evolution: evidence from Drosophila. Am. Nat. 134, 613–637 [Google Scholar]
- 22.Gavrilets S., Li H., Vose M. D. 1998. Rapid parapatric speciation on holey adaptive landscapes. Proc. R. Soc. Lond. B 265, 1483–1489 10.1098/rspb.1998.0461 (doi:10.1098/rspb.1998.0461) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gavrilets S., Li H., Vose M. D. 2000. Patterns of parapatric speciation. Evolution 54, 1126–1134 [DOI] [PubMed] [Google Scholar]
- 24.Gavrilets S. 2000. Waiting time to parapatric speciation. Proc. R. Soc. Lond. B 256, 2483–2492 10.1098/rspb.2000.1309 (doi:10.1098/rspb.2000.1309) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoelzer G. A., Drewes R., Meier J., Doursat R. 2008. Isolation-by-distance and outbreeding depression are sufficient to drive parapatric speciation in the absence of environmental influences. PLoS Comput. Biol. 4, e1000126. 10.1371/journal.pcbi.1000126 (doi:10.1371/journal.pcbi.1000126) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohan F. M. 1984. Can uniform selection retard random genetic divergence between isolated conspecific populations? Evolution 38, 495–504 10.2307/2408699 (doi:10.2307/2408699) [DOI] [PubMed] [Google Scholar]
- 27.Unckless R. L., Orr H. A. 2009. Dobzhansky–Muller incompatibilities and adaptation to a shared environment. Heredity 102, 214–217 10.1038/hdy.2008.129 (doi:10.1038/hdy.2008.129) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barton N. H. 2001. The role of hybridization in evolution. Mol. Ecol. 10, 551–568 10.1046/j.1365-294x.2001.01216.x (doi:10.1046/j.1365-294x.2001.01216.x) [DOI] [PubMed] [Google Scholar]
- 29.Church S. A., Taylor D. A. 2002. The evolution of reproductive isolation in spatially structured populations. Evolution 56, 1859–1862 [DOI] [PubMed] [Google Scholar]
- 30.Kondrashov A. S. 2003. Accumulation of Dobzhansky–Muller incompatabilities within a spatially structured population. Evolution 57, 151–153 [DOI] [PubMed] [Google Scholar]
- 31.Aguiar M. A. M., Baranger M., Baptestini E. M., Kaufman L., Bar-Yam Y. 2009. Global patterns of speciation and diversity. Nature 460, 384–387 10.1038/nature08168 (doi:10.1038/nature08168) [DOI] [PubMed] [Google Scholar]
- 32.Mallet J. 2006. What has Drosophila genetics revealed about speciation? Trends Ecol. Evol. 21, 186–193 10.1016/j.tree.2006.05.004 (doi:10.1016/j.tree.2006.05.004) [DOI] [PubMed] [Google Scholar]
- 33.Wu C.-I., Ting C.-T. 2004. Genes and speciation. Nat. R. Genet. 5, 114–122 10.1038/nrg1269 (doi:10.1038/nrg1269) [DOI] [PubMed] [Google Scholar]
- 34.Graur D., Li W.-H. 2000. Fundamentals of molecular evolution, 2nd ed. Sunderland, MA: Sinauer Associates [Google Scholar]
- 35.Feder J. L., et al. 2003. Allopatric genetic origins for sympatric host-plant shifts and race formation in Rhagoletis. Proc. Natl Acad. Sci. USA 100, 10 314–10 319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rundle H. D., Schluter D. 2004. Natural selection and ecological speciation in sticklebacks.In Adaptive speciation (eds Dieckmann U., Doebeli M., Metz J. A. J., Tautz D.), pp. 192–209 Cambridge, UK: Cambridge University Press [Google Scholar]
- 37.Platt J. R. 1964. Strong inference. Science 146, 347–353 10.1126/science.146.3642.347 (doi:10.1126/science.146.3642.347) [DOI] [PubMed] [Google Scholar]
- 38.Leimu R., Fischer M. 2008. A meta-analysis of local adaptation in plants. PLoS ONE 3, e4010. 10.1371/journal.pone.0004010 (doi:10.1371/journal.pone.0004010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hubbard J., Uy J. A. C., Hauber M. E., Hoekstra H. E., Safran R. J. 2010. Vertebrate pigmentation: from genes to adaptive function. Trends Genet. 26, 231–239 10.1016/j.tig.2010.02.002 (doi:10.1016/j.tig.2010.02.002) [DOI] [PubMed] [Google Scholar]
- 40.Barrett R. D. H., Rogers S. M., Schluter D. 2008. Natural selection on a major armor gene in threespine stickleback. Science 322, 255–257 10.1126/science.1159978 (doi:10.1126/science.1159978) [DOI] [PubMed] [Google Scholar]
- 41.Strasburg J. L., Rieseberg L. H. 2010. How robust are ‘Isolation with Migration’ analyses to violations of the IM model? A simulation study. Mol. Biol. Evol. 27, 297–310 10.1093/molbev/msp233 (doi:10.1093/molbev/msp233) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ting C. T., Tsaur S. C., Wu M. L., Wu C. I. 1998. A rapidly evolving homeobox at the site of a hybrid sterility gene. Science 282, 1501–1504 10.1126/science.282.5393.1501 (doi:10.1126/science.282.5393.1501) [DOI] [PubMed] [Google Scholar]
- 43.Presgraves D. C., Balagopalan L., Abmayr S. M., Orr H. A. 2003. Adaptive evolution drives divergence of a hybrid inviability gene between two species of Drosophila. Nature 423, 715–719 10.1038/nature01679 (doi:10.1038/nature01679) [DOI] [PubMed] [Google Scholar]
- 44.Sweigart A., Fishman L., Willis J. H. 2006. A simple genetic incompatibility causes hybrid male sterility in Mimulus. Genetics 172, 2465–2479 10.1534/genetics.105.053686 (doi:10.1534/genetics.105.053686) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Presgraves D. C. 2003. A fine-scale genetic analysis of hybrid incompatibilities in Drosophila. Genetics 163, 955–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hendry A. P., Nosil P., Rieseberg L. H. 2007. The speed of ecological speciation. Funct. Ecol. 21, 455–464 10.1111/j.1365-2435.2007.01240.x (doi:10.1111/j.1365-2435.2007.01240.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barton N. H. 2000. Genetic hitchhiking. Phil. Trans. R. Soc. Lond. B 355, 1553–1562 10.1098/rstb.2000.0716 (doi:10.1098/rstb.2000.0716) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barton N., Bengtsson B. O. 1986. The barrier to genetic exchange between hybridizing populations. Heredity 57, 357–376 10.1038/hdy.1986.135 (doi:10.1038/hdy.1986.135) [DOI] [PubMed] [Google Scholar]