


Abstract
Objective:
Retinal nerve fiber layer (RNFL) abnormalities detected by optical coherence tomography (OCT) are useful markers for axonal loss and visual dysfunction in multiple sclerosis (MS), but their role in routine clinical management is not well-studied.
Methods:
Clinical and OCT examinations were performed on 240 patients attending a neurology clinic. Using OCT 5th percentile to define abnormal RNFL thickness, we compared eyes classified by neurologists as having optic atrophy to RNFL thickness, and afferent pupillary defect (APD) to RNFL thickness ratios of eye pairs.
Results:
Mean RNFL thickness was less in eyes classified by neurologists as having optic atrophy (79.4 ± 21 μm; n = 63) vs those without (97.0 ± 15 μm; n = 417; p < 0.001, t test) and in eyes with an APD (84.1 ± 16 μm; n = 44) than without an APD (95.8 ± 17 μm; n = 436; p < 0.001). Physicians' diagnostic accuracy for detecting pallor in eyes with an abnormal RNFL thickness was 79% (sensitivity = 0.56; specificity = 0.82). Accuracy for detecting a RAPD in patients with mean RNFL ratio (affected eye to unaffected eye) <0.90 was 73% (sensitivity = 0.30; specificity = 0.86). Ability to detect visual pathway injury via assessment of atrophy and APD differed between neurologists.
Conclusions:
OCT reveals RNFL abnormality in many patients in whom eyes are not classified by neurologic examiners as having optic atrophy. Further study is needed to define the role of OCT measures in the context of examinations for optic atrophy and APD by neuroophthalmologists. OCT-measured RNFL thickness is likely to have an important future role in the clinical setting.
GLOSSARY
- APD
= afferent pupillary defect;
- MS
= multiple sclerosis;
- OCT
= optical coherence tomography;
- RNFL
= retinal nerve fiber layer.
Multiple sclerosis (MS) is the primary cause of nontraumatic disability among young adults, and visual dysfunction is a major cause of disability in MS.1–3 Examination of the peripapillary retinal nerve fiber layer (RNFL) provides a unique opportunity to directly visualize unmyelinated CNS axons and detect evidence of CNS pathology.4–6 Traditionally, physicians have used the detection of optic disc pallor and afferent pupillary defects (APD) during clinical examination as signs of optic neuropathy.7,8
Optical coherence tomography (OCT) is a high-resolution, noninvasive imaging technique that uses near infrared light to produce images used to generate quantitative measurements of the RNFL.9–11 Studies have demonstrated that RNFL thickness correlates with disease subtype, visual acuity, disability status, disease activity, and MRI measures of optic nerve atrophy in MS.4,12–17 As such, use of OCT as an endpoint in clinical trials of neuroprotective drugs and as a structural marker for disease activity has been suggested.15,18
However, OCT use in everyday clinical management of patients with MS has not been well-elucidated. Because both optic atrophy and APD are subjective measures that may be influenced by a physician's clinical training and experience, an objective measure of RNFL thickness, such as OCT, may provide an additional assessment of underlying RNFL thinning. If so, OCT may provide additional information important for the routine management of patients with CNS demyelinating disorders. The purpose of our study was to compare neurologists' assessment of optic atrophy and APD to RNFL thickness as measured by OCT-3.
METHODS
Standard protocol approvals, registrations, and patient consent.
This study was approved by the Johns Hopkins Hospital Institutional Review Board and performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from all patients participating in this study.
Study procedures.
A convenience sample of 240 patients who attended the Neurology Outpatient Clinic at the Johns Hopkins Hospital between December 2007 and August 2008 were enrolled. Patients with a previous diagnosis of glaucoma and any moderate or severe additional ocular pathology or surgery were excluded. One patient was excluded because of inability to independently maintain visual fixation during OCT evaluation.
Each patient first underwent a routine neurologic examination after which neurologists were asked to note categorically whether optic atrophy and APD were present or absent. Funduscopic evaluations were performed using Welch Allen hand-held direct ophthalmoscopes provided in clinic rooms. In order to approximate usual clinical practice, physicians did not receive any specialized training, and no attempt was made to develop a standardized protocol for funduscopic examination or a consensus assessment of examinations. The presence vs absence of optic atrophy was therefore based on the neurologists' prior understanding of the clinical features underlying this finding.
Twelve neurologists each examined at least 1 patient (mean = 20; range 1-45). Seven senior faculty members, 1 junior faculty member, 3 fellows, and 1 resident participated. None was a neuroophthalmologist. Among the subcohort who examined at least 15 patients were 5 senior faculty members, 1 junior faculty member, and 1 fellow.
Each participant then underwent OCT evaluation with the Stratus OCT-3 (Carl Zeiss Meditec, Inc.; Dublin, CA) using the OCT-3 fast RNFL thickness protocol. OCT software (OCT 4.0, version A2; Carl Zeiss Meditec) was used to calculate RNFL thickness measurements and percentiles of RNFL thickness distributions based on the reference database included in this software. Clinical and OCT examination were performed on nondilated eyes. The examining physicians were blinded to the OCT results but did have access to a patient's clinical history and results of investigations performed prior to study enrollment.
Statistical analysis.
Statistical analyses were performed using Intercooled Stata 10.0 (StataCorp, College Station, TX). Demographic characteristics were calculated, and comparisons were then made between eyes with and without an abnormality as designated categorically by the neurologists on clinical examination. Univariate logistic regression models including a random effect of participant were used to compare clinical and OCT characteristics between groups. One study participant was excluded from all paired analyses because an APD was documented in both eyes.
When comparing clinical examination to OCT evaluation, the neurologists' assessments were analyzed as the index tests, and OCT results were analyzed as the reference test. Classification of optic atrophy was compared to the population distribution percentile of mean RNFL thickness as determined by OCT software. RNFL thicknesses of <5th percentile or the 1st percentile of age-matched healthy control eyes were used as abnormal thresholds. Detection of an APD in eye pairs was compared to RNFL ratios (RNFL thickness in affected eye to unaffected eye). While a clinically significant reduction in RNFL ratios has not been previously identified, prior studies have demonstrated that an APD is not clinically detectable until the RNFL ratio is between 0.7 and 0.8.19–21 Therefore, we prospectively decided to investigate ratios of <0.90, <0.80, <0.70, and <0.60 as thresholds for abnormal values. For each abnormal threshold, several measures of test accuracy were determined. Finally, neurologists who examined 15 or more study participants were included in an analysis of individual examiner results using the same analyses described above.
RESULTS
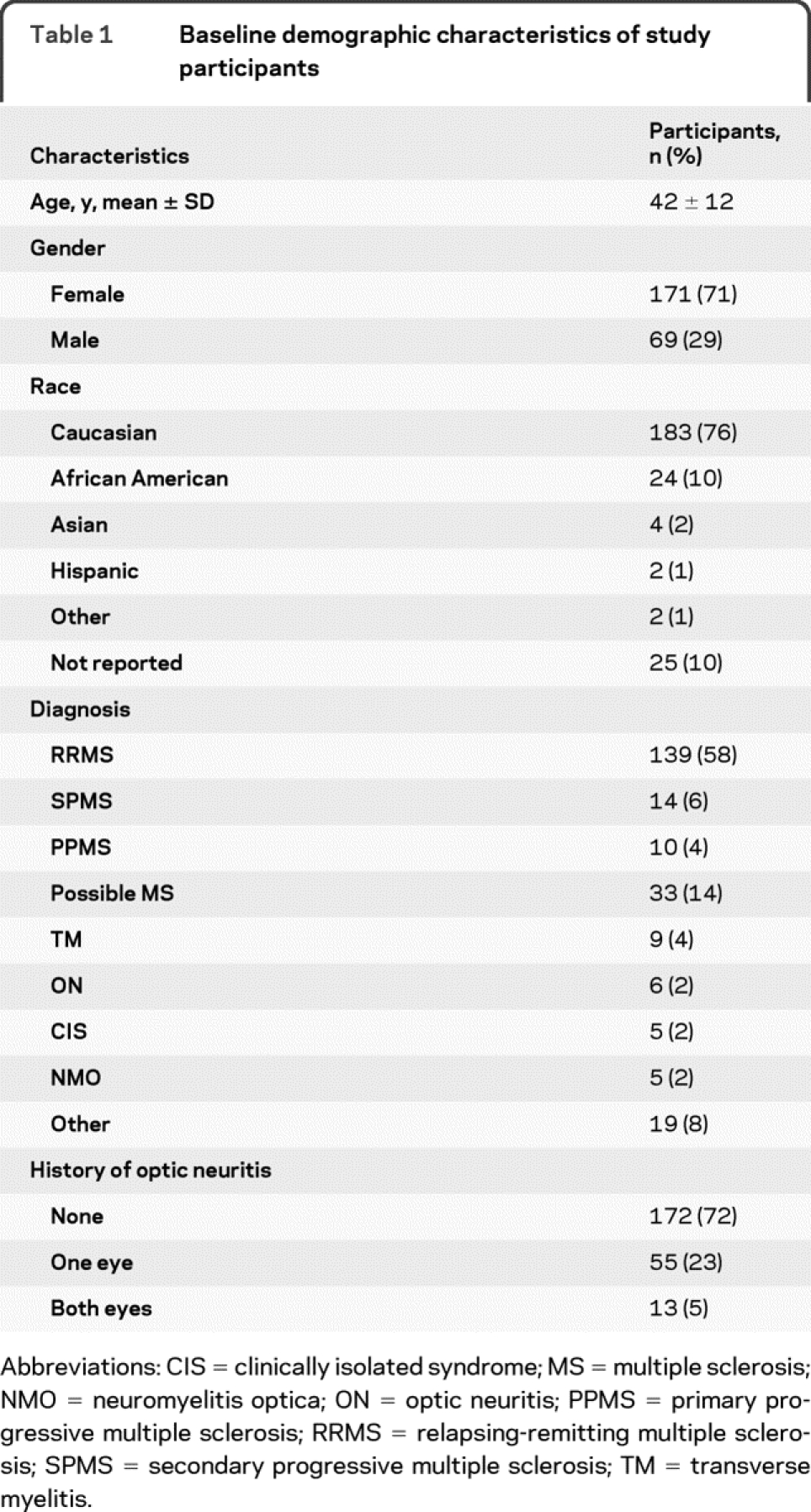
Demographic characteristics of study participants are listed in table 1. The majority of participants were women (71.2%) and white (76.2%). More than two-thirds (67.9%) of participants had a definite diagnosis of MS (57.9% relapsing-remitting MS), and 13.7% of participants had possible MS, which included individuals who have had >1 clinical attack but do not fulfill McDonald diagnostic criteria or who have had paroxysmal symptoms beyond a presenting clinical attack (table 1). Overall, 28.3% (n = 68) of study participants had a history of at least 1 clinical episode of optic neuritis and 5% (n = 13) had a bilateral history of optic neuritis.
Table 1 Baseline demographic characteristics of study participants
Compared to eyes not classified by neurologists as having optic atrophy, eyes with suggestive findings were more likely to have a history of optic neuritis (44% vs 13%; p < 0.001) and to have an APD detected on clinical examination (32% vs 6%; p < 0.001) (table 2). Mean RNFL thickness was less in eyes classified as having optic atrophy (79.4 ± 21 μm) than in eyes without (97.0 ± 15 μm; p < 0.001). When eyes with a known history of ON were excluded from the analysis, all of these results retained significance (data not shown).
Table 2 Comparison of clinical characteristics of eyes classified by neurologists as having optic atrophy and APD
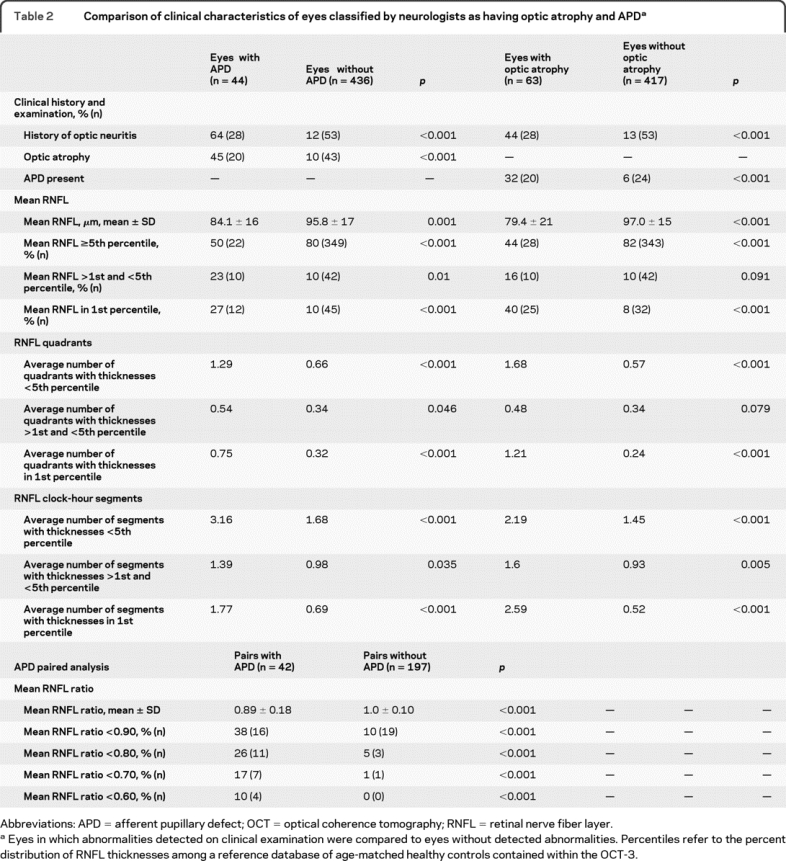
Compared to eyes in which no APD was detected, eyes with an APD were more likely to have a history of optic neuritis (64% vs 12%; p < 0.001) and to have optic atrophy detected on clinical examination (45% vs 10%; p < 0.001) (table 2). Eyes in which an APD was detected had mean RNFL thicknesses which were thinner (84.1 ± 16 μm) than eyes without an APD (95.8 ± 17 μm; p = 0.001). Eyes classified as having optic atrophy and eyes with an APD were also significantly more likely to have an abnormal mean RNFL and a higher number of retinal quadrants and clock-hour segments with abnormal RNFL thicknesses (table 2). However, none of these differences was significant when eyes with a known history of ON were excluded from the analysis.
In paired analyses, eye pairs with an APD had lower mean RNFL thickness ratios than pairs without an APD (0.89 ± 0.18 vs 1.0 ± 0.10; p < 0.001, t test) (table 2). However, the proportion of eye pairs with an APD that had abnormal RNFL ratios was significantly greater than the proportion of eye pairs with no APD, regardless of the threshold used.
When comparing classification of optic atrophy to mean RNFL thickness, both abnormal threshold values were characterized by similarly high specificities (<5th percentile = 92%; 1st percentile = 91%) and negative predictive values (<5th percentile = 82%; 1st percentile = 92%) but low sensitivities (<5th percentile = 44%; 1st percentile = 32%) and positive predictive values (<5th percentile = 56%; 1st percentile = 40%) (table 3). Overall agreement was higher for the 1st percentile cutoff. When physicians were considered independently, the ability to detect visual pathway injury by assessing pallor differed (table 4). Sensitivity ranged from 14% to 50% for the <5th percentile cutoff and from 0% to 100% for the 1st percentile cutoff. Specificity was more stable, ranging between 85% and 97% for both cutoffs.
Table 3 Measures of test agreement for clinical examination vs OCT
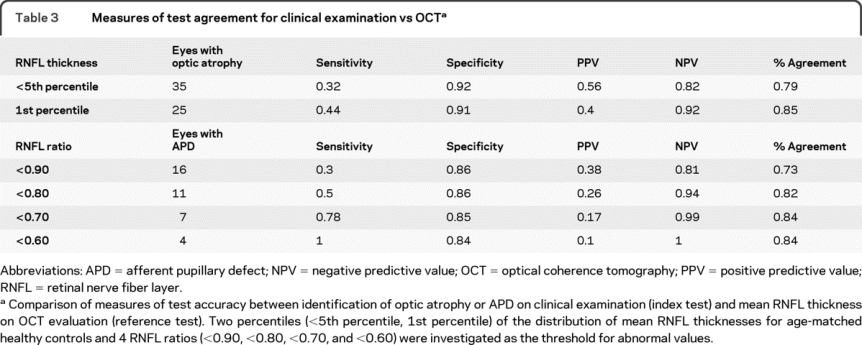
Table 4 Measures of test agreement for clinical examination vs OCT by neurologist
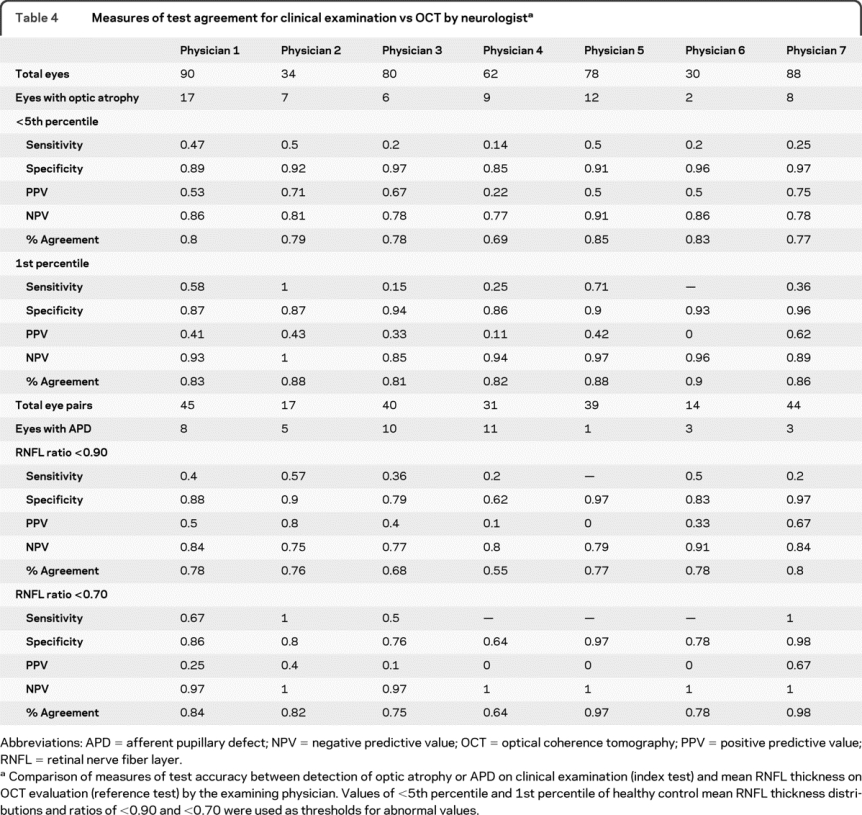
Clinical detection of an APD was compared to mean RNFL ratios (table 3). A mean RNFL ratio of <0.7 demonstrated 78% sensitivity and 85% specificity, while a ratio of <0.6 was 100% sensitive and 84% specific. At the lowest threshold ratio (<0.6), overall agreement was 84%, which was 11% higher than at the highest threshold ratio (<0.9). As with optic disc pallor, test accuracy measures for each individual physician's detection of APDs varied (table 4). Sensitivities ranged from 0% to 57% for a threshold ratio <0.9 and from 50% to 100% for a threshold ratio <0.7. Specificities fluctuated between 62% and 97% at both thresholds. Overall percent agreement varied between 55% and 80% for a threshold ratio of <0.90 and between 64% and 98% for a threshold ratio of <0.70.
DISCUSSION
Our findings corroborate the results of prior studies that have found RNFL thickness reduction is more pronounced in MS eyes with a history of acute optic neuritis.12,14,22,23 In addition, we have shown an association between abnormal mean RNFL thicknesses and classification of eyes by neurologists as having optic atrophy and between abnormal mean RNFL ratios and detection of APDs. This finding provides evidence that clinical signs of optic atrophy and RNFL thicknesses determined by OCT evaluation are evaluating similar clinical phenomena.
As expected, in this cohort the probability of classifying eyes as having optic atrophy or as having an APD during clinical examination increased as the severity of RNFL defects increased. Using the OCT software 5th percentile cutoff for abnormal, our data suggest that OCT can detect more retinal abnormalities than are observed by clinical examination of optic atrophy or APD, at least among a cohort of academic neurologists. Furthermore, these results indicate that a substantial degree of axonal loss in the RNFL and optic nerve occurs before eyes are classified as having optic atrophy or an APD.
While the sensitivity of detecting clinical abnormalities increased as the severity of RNFL abnormalities increased in both the overall study cohort and within the subcohorts examined by each physician, neurologists' likelihood to classify eyes as having optic atrophy and APDs at a given severity of RNFL defects varied substantially. Whether this difference was due to a physician's clinical experience and training, the assortment of patients encountered by each physician, or both is unclear. If clinical experience was a factor, additional training in the examination of the optic nerve and retina may be beneficial during neurology residency and subspecialty training. In addition, OCT could be useful as a training tool to correlate with clinical findings. However, these data must also be interpreted with caution, as the measures of sensitivity and positive predictive value are estimated with poor precision within each physician due to the small number of patients examined by each physician.
Few studies have assessed the association between clinical examination and OCT findings. One recent study compared RNFL defects in patients with retinitis pigmentosa to detailed funduscopic examinations.24 All patients with moderate to severe optic atrophy and retinal abnormalities on funduscopic examination had at least 1 quadrant or 2 clock-hour segments with abnormal RNFL thicknesses. Only 7% of individuals with normal optic discs or mild disc pallor had an abnormal quadrant thickness, and only 13% had an abnormal clock-hour thickness measure. The investigators concluded that RNFL thinning could be present in patients with normal-appearing optic discs during the clinical examination and in patients without significant loss of visual acuity. However, the probability of RNFL thinning increased with the severity of optic atrophy. The results of our study are remarkably similar. For example, eyes classified as having optic atrophy in our cohort averaged more than 1 quadrant and more than 2 clock-hour segments with abnormal RNFL thickness measures (table 2).
Two human studies and 1 animal study investigating the correlation between APDs and mean RNFL ratios have been reported.19–21 In both human studies, APDs were quantified using log-scaled neutral density filters over the unaffected eye during the swinging flashlight test. In a study of 20 patients with unilateral optic atrophy of various etiologies, the mean RNFL thickness of the affected eye was significantly less than the unaffected eye, and the correlation coefficient between a clinically detectable APD and the mean RNFL ratio was 0.48.20 In addition, the investigators found that an APD was clinically undetectable until the mean RNFL ratio was ≤0.75. In a similar study in 29 patients with glaucoma, the correlation coefficient between an APD and mean RNFL ratio was 0.49, and an APD was clinically undetectable until the mean RNFL ratio was ≤0.73.21 In this study, too, the mean RNFL thickness in the affected eye was significantly less than in the unaffected eye. A study in monkeys investigating the correlation between an APD and RNFL thickness also found that a 25% to 50% decrease in RNFL thickness was necessary to produce a clinically detectable APD.19
Despite the more quantitative methods used to detect APDs in the previously reported studies, our results were similar. In our cohort, mean RNFL thickness was also significantly less in eyes in which an APD was detected than in eyes without an APD (table 2). While a significant increase in sensitivity was seen when the threshold for an abnormal RNFL ratio was decreased from 0.80 to 0.70 (table 3)—a level consistent with the findings of the previously reported studies—most APDs identified in this study were detected at RNFL ratios >0.7. This may be the result of noise from the poor resolution and high coefficient of variance of OCT, which is one limitation to its clinical utility.
Despite the consistency of these results with previously published studies, our study has several limitations. While each neurologist included in the analyses examined at least 15 patients, none of the patients was examined by more than 1 physician. Therefore, we cannot directly compare agreement between neurologists' classifications of optic atrophy and APDs among the same cohort of patients. In addition, only 1 OCT evaluation was performed on each eye in study participants. Using average RNFL measurements obtained from repeated scans would help reduce random noise and variability associated with OCT RNFL measurements. Another important limitation is the multiple etiologies of optic nerve disease among our study cohort. It is possible that each disease etiology could have a different relationship between detection of optic atrophy or an APD and RNFL thickness reduction. However, the majority of patients in our cohort had demyelinating disorders and most had a diagnosis of MS, making significant pathophysiologic heterogeneity unlikely.
Clinical and OCT examinations were also performed during routine neurologic clinic visits. While physicians were blinded to the results of the OCT examination, they did have knowledge of a patient's clinical history, including whether a history of optic neuritis was present and results of previous investigations that had been performed. This is appropriate in the clinical setting, as it cannot be overemphasized that the identification of true optic atrophy does depend upon the presence of suggestive clinical findings, including visual dysfunction, color desaturation, and the APD.
However, due to concern that knowledge of ON history would bias the clinical assessment, we also analyzed the data after excluding eyes with a known history of ON. While the differences in OCT characteristics between eyes with and without optic atrophy remained significant, the differences between eyes with and without APD were no longer significant. Whether the loss of significance was due to eliminating bias obtained from clinical history or to the small number of eyes with APD remaining in this analysis (n = 44 eyes with APD when ON eyes are included; n = 16 eyes with APD when ON eyes are excluded) is difficult to determine.
In addition, OCT and clinical examinations were performed by non-ophthalmologists on undilated eyes and did not incorporate equipment such as red-free filters for ophthalmoscopy, neutral density filters, infrared pupillography, or stereo fundus photographs. Optic atrophy was identified in only 34% of patients with a history of optic neuritis, which is a lower proportion than expected. However, we purposed to examine whether OCT may provide useful information to supplement clinical examination in everyday outpatient practices. Therefore, we did not provide specialized training in the identification of atrophy or APD. It is therefore not surprising that clinical assessments were heterogenous among participating neurologists, since they relied upon the physicians' prior knowledge and skills. However, despite the nonstandardized examination conditions, our results are remarkably similar to previously published results in study cohorts with different diagnoses and are potentially applicable to routine clinical practice.
Finally, the lack of a detailed ophthalmologic assessment of study participants limited our ability to adjust for factors that may have influenced RNFL thickness or funduscopic findings suggestive of optic atrophy. Previous studies have found that increased ophthalmic axial length, decreased optic disc area, and myopia are associated with thinner RNFL measurements in normal and abnormal eyes independent of disease processes.25–27 However, these variables were not measured for study participants, so RNFL measurements could not be adjusted for these factors in our statistical analyses. In addition, a recent report demonstrated that visual evoked potentials were more sensitive than OCT in detecting optic neuritis. However, because demyelination alone does not cause optic atrophy, we believe OCT was the best method with which to associate atrophy as they represent axonal loss.28
Despite these limitations, our results add to the growing literature suggesting that OCT may allow for earlier detection of MS axonal loss in the visual pathway as measured by RNFL defects. Our study also supports the utility of OCT as a structural marker of axonal loss in clinical trials of neuroprotective drugs, and suggests a future clinical role. Our results demonstrate that OCT evaluation may be a useful adjunct to routine clinical management of patients with demyelinating and axonal degenerative disorders and may well provide complementary and confirmatory information to that which can be obtained from clinical examination. Earlier and more accurate detection of RNFL defects using OCT may be indicative of subclinical disease activity and could allow for earlier and more aggressive disease management.
DISCLOSURE
D. Cettomai has received research/salary support from the NIH/Johns Hopkins Pre-Doctoral Clinical Research Training Program (T32), the University of California, San Francisco, and the Doris Duke Foundation. Dr. Hiremath reports no disclosures. Dr. Ratchford serves as a consultant for Sun Pharmaceutical Industries Ltd. and receives research support from Novartis. Dr. Venkatesan serves on a scientific advisory board for GlaxoSmithKline and receives research support from the NIH (NIDA K08 DA022946 [PI]) and the Howard Hughes Medical Institute. Dr. Greenberg has received speaker honoraria from Biogen Idec, Teva Pharmaceutical Industries Ltd., and EMD Serono, Inc.; serves as a consultant for and holds equity in DioGenix, Inc.; receives research support from the University of Texas Southwestern, the Guthy Jackson Foundation, and the Accelerated Cure Project; and has given expert testimony in medico-legal cases. Dr. McGready and Dr. Pardo report no disclosures. Dr. Kerr has received funding for travel and speaker honoraria from Teva Pharmaceutical Industries Ltd. and Biogen Idec; served as an Associate Editor for the Annals of Neurology; is an author on patents re: Strategies to create immune tolerance of CNS allografts and cell-based compositions and Methods for treating conditions of the nervous system; and is a founder and equity holder and served as Chief Scientific Consultant for Nerveda, Inc. Dr. Frohman has served on speakers' bureaus for and received funding for travel and speaker honoraria from Biogen Idec, Teva Pharmaceutical Industries Ltd., and Bayer Schering Pharma; and has served as a consultant for Biogen Idec, Teva Pharmaceutical Industries Ltd., Athena Diagnostics, Inc., and Abbott. Dr. Balcer serves on a scientific advisory board for and has received funding for travel and speaker honoraria from Biogen Idec and receives research support from the NIH (NEI K24 EY 018136 [PI]) and the National Multiple Sclerosis Society. Dr. McArthur serves as an unpaid consultant to Allergan, Inc., Pfizer Inc., and Biogen Idec; receives royalties from the publication of Current Therapy in Neurologic Disease, 7th edition (Mosby, 2006); is an author on patents re: Device for thermal stimulation of small neural fibers and Immunophilin ligand treatment of antiretroviral toxic neuropathy; and receives research support from Biogen Idec, the NIH (R01 MH075673 [PI], RO1 NS44807 [PI], RO1 NS49465 [PI], and R01 MH067831 [Co-I]), the National Multiple Sclerosis Society, and the Foundation for Peripheral Neuropathy. Dr. Calabresi is an author on a patent re: Role of Kv1.3 as neuroprotective; has served as a consultant for Novartis, EMD Serono, Inc., Teva Pharmaceutical Industries Ltd., Biogen Idec, Vertex Pharmaceuticals, Amplimmune, Inc., Centocor Ortho Biotech Inc., Genentech, Inc., and Novo Nordisk; and has received research support from EMD Serono, Inc., Teva Pharmaceutical Industries Ltd., Biogen Idec, Genentech, Inc., Bayer Schering Pharma, Vertex Pharmaceuticals, the NIH (NINDS NS RO-1 041435 [PI]), and the National Multiple Sclerosis Society.
Address correspondence and reprint requests to Dr. Peter A. Calabresi, Pathology Building, Suite 627, Johns Hopkins Hospital, 600 N. Wolfe St., Baltimore, MD 21287 calabresi@jhmi.edu
Editorial, page 1312
e-Pub ahead of print on September 1, 2010, at www.neurology.org.
Study funding: Supported by the Johns Hopkins School of Medicine/National Institutes of Health T-32 Predoctoral Clinical Research Training Program, the National Multiple Sclerosis Society TR grant, and the Nancy Davis Foundation.
Disclosure: Author disclosures are provided at the end of the article.
Received October 16, 2009. Accepted in final form May 14, 2010.
REFERENCES
- 1.Confavreux C, Vukusic S. The clinical epidemiology of multiple sclerosis. Neuroimaging Clin N Am 2008;18:589–622. [DOI] [PubMed] [Google Scholar]
- 2.McDonald WI, Barnes D. The ocular manifestations of multiple sclerosis: 1: abnormalities of the afferent visual system. J Neurol Neurosurg Psychiatry 1992;55:747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Holmes BA, Bates D. The cost of MS. Br J Med Econ 1995;8:181–193. [Google Scholar]
- 4.Trip SA, Schlottmann PG, Jones SJ, et al. Optic nerve atrophy and retinal nerve fibre layer thinning following optic neuritis: evidence that axonal loss is a substrate of MRI-detected atrophy. Neuroimage 2006;31:286–293. [DOI] [PubMed] [Google Scholar]
- 5.Frohman E, Costello F, Zivadinov R, et al. Optical coherence tomography in multiple sclerosis. Lancet Neurol 2006;5:853–863. [DOI] [PubMed] [Google Scholar]
- 6.Laterza A, Nappo A. Optic nerve: a concise review of the anatomy, pathophysiology and principal acquired disorders. Ital J Neurol Sci 1987;8:529–535. [DOI] [PubMed] [Google Scholar]
- 7.Wirtschafter JD. Optic nerve axons and acquired alterations in the appearance of the optic disc. Trans Am Ophthalmol Soc 1983;81:1034–1091. [PMC free article] [PubMed] [Google Scholar]
- 8.Wilhelm H. Neuro-ophthalmology of pupillary function: practical guidelines. J Neurol 1998;245:573–583. [DOI] [PubMed] [Google Scholar]
- 9.Sharma P, Sample PA, Zangwill LM, Schuman JS. Diagnostic tools for glaucoma detection and management. Surv Ophthalmol 2008;53(suppl 1):S17–S32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pagliara MM, Lepore D, Balestrazzi E. The role of OCT in glaucoma management. Prog Brain Res 2008;173:139–148. [DOI] [PubMed] [Google Scholar]
- 11.Huang D, Swanson EA, Lin CP, et al. Optical coherence tomography. Science 1991;254:1178–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pulicken M, Gordon-Lipkin E, Balcer LJ, et al. Optical coherence tomography and disease subtype in multiple sclerosis. Neurology 2007;69:2085–2092. [DOI] [PubMed] [Google Scholar]
- 13.Henderson AP, Trip SA, Schlottmann PG, et al. An investigation of the retinal nerve fibre layer in progressive multiple sclerosis using optical coherence tomography. Brain 2008;131:277–287. [DOI] [PubMed] [Google Scholar]
- 14.Fisher JB, Jacobs DA, Markowitz CE, et al. Relation of visual function to retinal nerve fiber layer thickness in multiple sclerosis. Ophthalmology 2006;113:324–332. [DOI] [PubMed] [Google Scholar]
- 15.Sepulcre J, Murie-Fernandez M, Salinas-Alaman A, et al. Diagnostic accuracy of retinal abnormalities in predicting disease activity in MS. Neurology 2007;68:1488–1494. [DOI] [PubMed] [Google Scholar]
- 16.Siger M, Dziegielewski K, Jasek L, et al. Optical coherence tomography in multiple sclerosis: thickness of the retinal nerve fiber layer as a potential measure of axonal loss and brain atrophy. J Neurol 2008;255:1555–1560. [DOI] [PubMed] [Google Scholar]
- 17.Gordon-Lipkin E, Chodkowski B, Reich DS, et al. Retinal nerve fiber layer is associated with brain atrophy in multiple sclerosis. Neurology 2007;69:1603–1609. [DOI] [PubMed] [Google Scholar]
- 18.Frohman EM, Fujimoto JG, Frohman TC, et al. Optical coherence tomography: a window into the mechanisms of multiple sclerosis. Nat Clin Pract Neurol 2008;4:664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kerrison JB, Buchanan K, Rosenberg ML, et al. Quantification of optic nerve axon loss associated with a relative afferent pupillary defect in the monkey. Arch Ophthalmol 2001;119:1333–1341. [DOI] [PubMed] [Google Scholar]
- 20.Nakanishi Y, Nakamura M, Tatsumi Y, et al. Quantification of retinal nerve fiber layer thickness reduction associated with a relative afferent pupillary defect. Graefes Arch Clin Exp Ophthalmol 2006;244:1480–1484. [DOI] [PubMed] [Google Scholar]
- 21.Tatsumi Y, Nakamura M, Fujioka M, et al. Quantification of retinal nerve fiber layer thickness reduction associated with a relative afferent pupillary defect in asymmetric glaucoma. Br J Ophthalmol 2007;91:633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trip SA, Schlottmann PG, Jones SJ, et al. Retinal nerve fiber layer axonal loss and visual dysfunction in optic neuritis. Ann Neurol 2005;58:383–391. [DOI] [PubMed] [Google Scholar]
- 23.Kallenbach K, Frederiksen J. Optical coherence tomography in optic neuritis and multiple sclerosis: a review. Eur J Neurol 2007;14:841–849. [DOI] [PubMed] [Google Scholar]
- 24.Walia S, Fishman GA, Edward DP, Lindeman M. Retinal nerve fiber layer defects in RP patients. Invest Ophthalmol Vis Sci 2007;48:4748–4752. [DOI] [PubMed] [Google Scholar]
- 25.Budenz DL, Anderson DR, Varma R, et al. Determinants of normal retinal nerve fiber layer thickness measured by Stratus OCT. Ophthalmology 2007;114:1046–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jun JH, Lee SY. The effects of optic disc factors on retinal nerve fiber layer thickness measurement in children. Korean J Ophthalmol 2008;22:115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medeiros FA, Zangwill LM, Bowd C, et al. Influence of disease severity and optic disc size on the diagnostic performance of imaging instruments in glaucoma. Invest Ophthalmol Vis Sci 2006;47:1008–1015. [DOI] [PubMed] [Google Scholar]
- 28.Naismith R, Tutlam N, Xu J, et al. Optical coherence tomography is less sensitive than visual evoked potentials in optic neuritis. Neurology 2009;73:46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]