


Abstract
Objectives:
In Alzheimer disease (AD), mounting evidence points to a greater role for synaptic loss than neuronal loss. Supporting this notion, multiple postmortem studies have demonstrated that the hippocampal CA1 apical neuropil is one of the earliest sites of pathology, exhibiting tau aggregates and then atrophy before there is substantial loss of the CA1 pyramidal neurons themselves. In this cross-sectional study, we tested whether tissue loss in the CA1 apical neuropil layer can be observed in vivo in patients with mild AD.
Methods:
We performed ultra-high-field 7-T MRI on subjects with mild AD (n = 14) and age-matched normal controls (n = 16). With a 2-dimensional T2*-weighted gradient-recalled echo sequence that was easily tolerated by subjects, we obtained cross-sectional slices of the hippocampus at an in-plane resolution of 195 μm.
Results:
On images revealing the anatomic landmarks of hippocampal subfields and strata, we observed thinning of the CA1 apical neuropil in subjects with mild AD compared to controls. By contrast, the 2 groups exhibited no difference in the thickness of the CA1 cell body layer or of the entire CA1 subfield. Hippocampal volume, measured on a conventional T1-weighted sequence obtained at 3T, also did not differentiate these patients with mild AD from controls.
Conclusions:
CA1 apical neuropil atrophy is apparent in patients with mild AD. With its superior spatial resolution, 7-T MRI permits in vivo analysis of a very focal, early site of AD pathology.
GLOSSARY
- AD
= Alzheimer disease;
- CDR
= Clinical Dementia Rating;
- DG
= dentate gyrus;
- GRE
= gradient-recalled echo;
- NC
= normal control;
- PiB
= Pittsburgh Compound B;
- SP
= stratum pyramidale;
- SRLM
= stratum radiatum and stratum lacunosum-moleculare;
- TIV
= total intracranial volume.
Neuronal death is a late manifestation of Alzheimer disease (AD), with synaptic dysfunction and loss occurring much earlier.1 Within the hippocampal CA1 subfield, one of the earliest sites of pathologic change in AD,2 numerous lines of evidence point to disproportionate synaptic loss in the apical neuropil layer, where CA1 pyramidal neurons receive synapses both from the Schaffer collateral axons originating from hippocampal CA3 neurons, and from the perforant pathway axons projecting from the entorhinal cortex. In a rodent model of AD, electrophysiologic evidence of synaptic loss within the CA1 apical neuropil preceded both neuronal death and the appearance of histopathologic changes.3 In humans, hyperphosphorylated tau aggregates appeared inside the distal apical dendritic branches of hippocampal CA1 pyramidal neurons at Braak stage II (clinically silent), prior to any loss of the neurons themselves.4–6 These dendritic segments became dilated and dystrophic, then disappeared by Braak stages IV-V (as clinical signs of AD become fully developed), apparently by amputation and resorption.4 In postmortem studies of AD, other groups found focal loss of both synapses and overall tissue volume within the hippocampal apical neuropil.7,8
We sought to investigate whether focal thinning of the hippocampal CA1 apical neuropil may be observed in vivo in patients with mild AD. To test this hypothesis, we turned to ultra-high-field 7-T MRI, which can depict detailed human hippocampal subfield anatomy with unprecedented spatial resolution.9,10
METHODS
Subject recruitment and characterization.
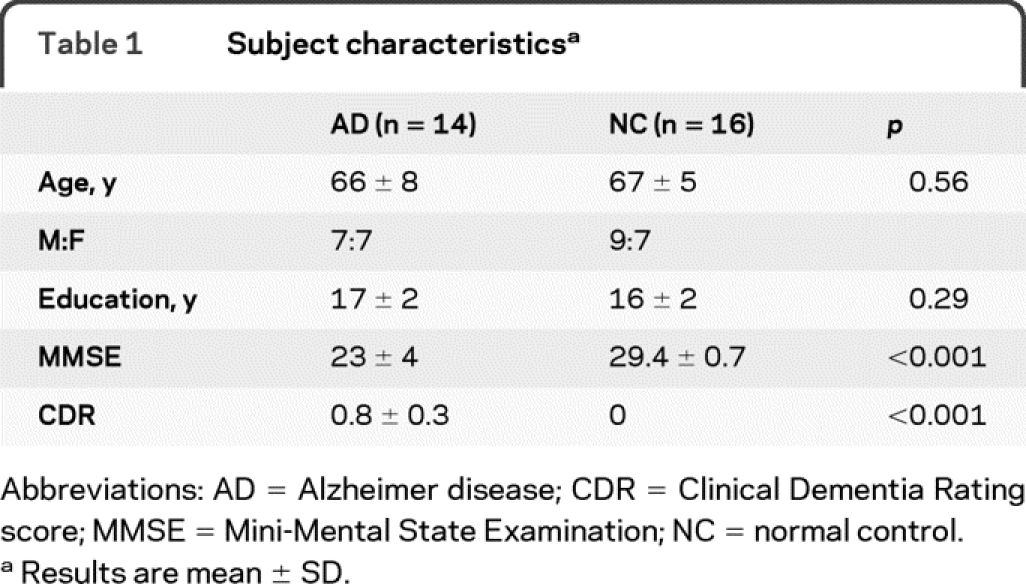
For this cross-sectional study, all subjects were recruited from existing research cohorts at the UCSF Memory and Aging Center, and all underwent a clinical evaluation that included a history and physical examination by a neurologist, a structured caregiver interview by a nurse, and a battery of neuropsychological tests, followed by a multidisciplinary team conference to reach a consensus diagnosis. We recruited 16 subjects with probable AD, according to National Institute of Neurological and Communicative Disorders and Stroke-Alzheimer's Disease and Related Disorders Association research criteria, and a low Clinical Dementia Rating (CDR) score of 0.5-1. The data from 2 subjects were excluded because of motion artifacts. In the course of participating in another study at our center, 6 of the 14 subjects with AD included in this study also underwent PET with the β-amyloid ligand Pittsburgh Compound B (PiB)11 using a previously described protocol.12 All 6 subjects were judged to have elevated cortical PiB signal by 2 visual raters blinded to clinical diagnosis, and high PiB binding was confirmed in all subjects using empirically derived quantitative thresholds.13 We also recruited 16 age-matched normal controls (NCs) who exhibited neither subjective nor objective cognitive problems. Table 1 details the characteristics of the groups.
Table 1 Subject characteristics
Standard protocol approvals, registrations, and patient consents.
Each participant provided written, informed consent according to a protocol approved by the UCSF Committee on Human Research.
Image acquisition.
All subjects were scanned on a 7-T GE Excite MRI system (General Electric, Waukesha, WI) using a volume transmit coil and a custom-constructed 8-channel phased-array receive head coil (Nova Medical, Inc., Wilmington, MA). All but 2 subjects were scanned on the same day on a 3-T GE Excite MRI scanner equipped with an 8-channel phased-array head coil (MRI Devices, Waukesha, WI) for comparison; the other 2 subjects (both with AD) underwent imaging on a 3-T Siemens TIM Trio scanner (Siemens Medical Solutions, Malvern, PA) with a 12-channel head coil less than 5 months before their 7-T scan.
Imaging at 7 T was targeted to the medial temporal lobes, with acquisition of oblique coronal images oriented perpendicular to the longitudinal axis of the hippocampus, as visualized in parasagittal scout films (figure 1). Soft pads were placed between a subject's head and the receive coil to minimize motion during the scan. All images were acquired using a conventional T2*-weighted gradient-recalled echo (GRE) sequence with a low flip angle of 20°, a repetition time of 250 msec, and an echo time of 15 msec. Scan time was 9.6 minutes, using a 1,024 × 768 matrix with 3 repetitions over a 20-cm field of view using a slice thickness of 2 mm. Some subjects were scanned using a slice gap of 3 mm (8 AD and 7 NCs), and the others had no gap between slices.

Figure 1 7-T gradient-recalled echo hippocampal imaging
Oblique coronal slices were obtained orthogonal to the longitudinal axis of the hippocampal body, as illustrated. Slices through the mid-hippocampal body at the level of the red nucleus afforded the best and most consistent views of hippocampal cross-sectional anatomy. These images were obtained from a normal control, and are displayed in radiologic orientation.
Image analysis.
Manual measurements of hippocampal subfield dimensions were performed on raw 7-T GRE images using Dicom Viewer 2.0 software (Rubo Medical Imaging, the Netherlands). On each side of the brain, we made 6 linear measurements of the thickness of CA1-SRLM at different points along the CA1 subfield, 3 each on 2 adjacent slices at the level of the red nucleus. Thickness was defined as the distance from 1 tissue plane interface to another using an orthogonal line. These values were then averaged to generate 1 value per side per subject. The thickness of the CA1-SP was measured in the same manner. All manual measurements were performed by a neurologist (G.A.K.), and the locations of the measurements were checked and confirmed by a neuroradiologist (C.P.H.), both blinded to diagnosis.
3-T T1 image processing was performed with FSL 4.1 (www.fmrib.ox.ac.uk/fsl/). Total intracranial volume was measured after brain extraction14 and segmentation15 by adding the volumes of CSF, gray matter, and white matter. Automated hippocampal segmentation was performed with the FIRST tool in FSL, which uses mesh models trained with a large amount of rich hand-segmented training data to segment cerebral structures.
Statistics.
All data are expressed as mean ± SD. Single-metric comparisons between AD and NC subjects were performed with the Mann-Whitney nonparametric U test, using p < 0.05 as the threshold for significance. Correlations were tested by linear regression. All statistical testing was performed with the SPSS 17.0 software package.
RESULTS
We performed 7-T MRI on subjects with mild AD and age-matched normal controls (table 1), as described in Methods. All subjects tolerated the imaging well, reporting no adverse reactions, except for 1 subject who reported tinnitus 1 day after the examination. No sedation of any kind was used.
Hippocampal cross-sectional anatomy was best appreciated on oblique coronal slices through the mid-hippocampal body at the level of the red nucleus (figure 1). Qualitatively, the hippocampi of participants with normal cognition or mild AD appeared similar on 7-T GRE imaging (figure 2). Cell body layers exhibited more intense signal than intervening neuropil, permitting direct visualization of hippocampal subfields and strata using classic anatomic landmarks16 and MRI studies of autopsy specimens17–19 as guides. As illustrated in figure 2, the dentate gyrus (DG, or fascia dentata) cups the CA4 subfield. The CA3 subfield emerges out of the hilus, ending where the pyramidal cell body layer, or stratum pyramidale (SP), reaches its narrowest dimension. No border between DG, CA4, or CA3 was consistently visible by this technique. CA2, signified by a thin SP, begins at the end of CA3 and terminates where SP widens, marking the beginning of CA1. These landmarks were easily identified on scans for each subject. The gradual transition between CA1 and the subiculum was not visible and was defined as the point at which the SP passes the most medial point of the DG. We considered the stratum radiatum and stratum lacunosum-moleculare as a single unit (SRLM), because there was no identifiable boundary between these layers (figure 3A). We use CA1-SP and CA1-SRLM to indicate the portions of SP and SRLM contained within the CA1 subfield.
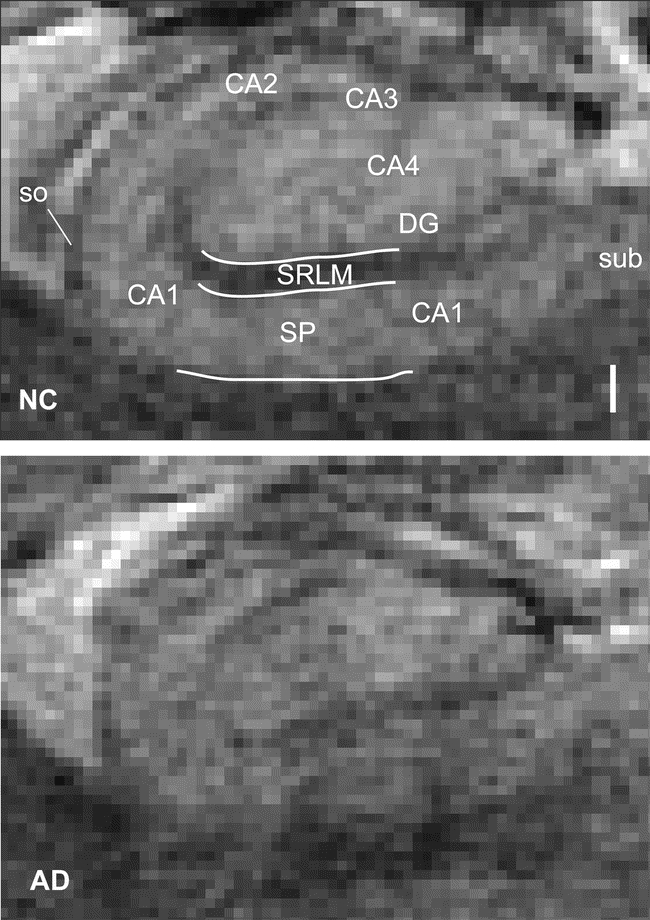
Figure 2 Hippocampus in normal aging and Alzheimer disease (AD)
Illustrated for comparison are cross-sections through the right hippocampus at the level of the red nucleus in a normal control (NC) and a subject with mild AD, obtained using a 7-T gradient-recalled echo sequence. The scale bar is 1 mm. DG = dentate gyrus; SP = stratum pyramidale; so = stratum oriens; SRLM = stratum radiatum/stratum lacunosum-moleculare; sub = subiculum.

Figure 3 Hippocampal subfield metrics
The thickness of the CA1 stratum radiatum/stratum lacunosum-moleculare (SLRM) (A) and the thickness of the CA1 stratum pyramidale (SP) (B) were measured for each individual. At left are examples of how measurements were performed (see Methods), drawn on the right hippocampus of a normal control (NC). The scale bar is 1 mm. Normalized data (see table 2) are summarized in the graphs, with error bars representing the SD. AD = Alzheimer disease.
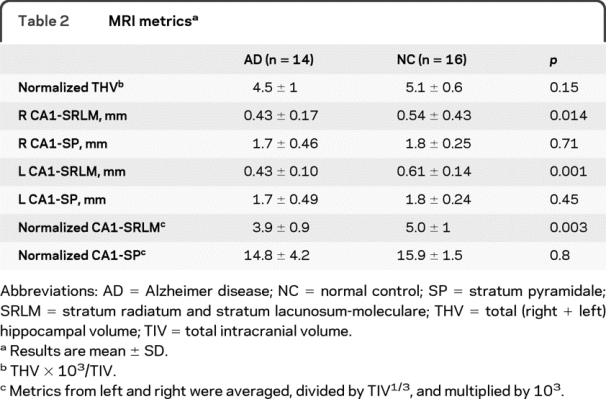
The CA1-SRLM was significantly thinner in subjects with AD compared to NCs (table 2 and figure 3A). By contrast, there was no significant difference in the thickness of CA1-SP in subjects with AD compared to NCs (table 2 and figure 3B). Metrics from the left and right hippocampi were symmetric (table 2). This finding—thinning of CA1-SRLM but not CA1-SP in subjects with mild AD—was maintained with or without normalizing the measurements to total intracranial volume (TIV) (table 2), which is intended to correct for premorbid brain size. The thickness of the overall CA1 subfield (CA1-SP + CA1-SRLM) was no different between AD and NC subjects (p = 0.29), probably reflecting the greater contribution of CA1-SP to the overall thickness. Although the men in this study had a larger TIV than the women (p = 0.009), CA1-SRLM and CA1-SP thicknesses had no relationship with gender, whether using normalized or raw values (p > 0.3). By linear regression analysis, there was also no significant relationship between CA1-SP or CA1-SRLM thickness and age (p = 0.26 and 0.37). Furthermore, there was at most a weak trend toward reduced hippocampal volume in the subjects with AD compared to controls (table 2). Finally, among the subjects with AD, there was a trend relating CA1-SRLM thinning to worse verbal memory performance (total number of correct items freely recalled during 4 trials of a 9-item word list, calculated as a scaled score relative to age-matched controls) (R2 = 0.32; p = 0.09); by contrast, there was no such trend for CA1-SP thickness (p = 0.36) or hippocampal volume (p = 0.51).
Table 2 MRI metrics
DISCUSSION
We report on the selective thinning of the CA1 apical neuropil layer relative to the CA1 cell body layer in subjects with mild AD, visualized in vivo using 7-T MRI. By permitting analysis of the microscopic hippocampal structures that are most sensitive to early pathology in AD, ultra-high-field 7-T MRI may be able to contribute to AD diagnosis and tracking.
The CA1 apical neuropil is an early target of AD pathology. Neurofibrillary tangles appear first outside the hippocampus, in the entorhinal cortex.2 The perforant pathway axons, projecting from the entorhinal cortex to the hippocampus, target the outer molecular layer of the dentate gyrus and the distal apical dendrites of CA1 neurons in the stratum lacunosum-moleculare. It is of interest, then, that these 2 neuropil areas of the hippocampus are among the next sites for tau pathology to appear, still quite early in AD pathogenesis.4,6 Some have hypothesized that tau pathology spreads in a network-dependent fashion,4,5,20 perhaps passively by deafferentation,21 or more actively by a prion-based mechanism.22 In the stratum lacunosum-moleculare, the varicose, tau-laden CA1 dendrites go on to disappear as clinical signs of AD become more pronounced, possibly corresponding to the thinning of the CA1-SRLM observed in vivo in this study and in prior studies of postmortem tissue.7,8
It was not surprising that CA1-SRLM thickness was better than overall hippocampal volume at distinguishing subjects with AD from NCs. While global hippocampal atrophy in AD is now a well-accepted phenomenon,23 cross-sectional studies often use larger sample sizes or more severely affected subjects with AD to detect group differences. In our small study of subjects with mild AD, there was no significant loss of hippocampal volume. Our work supports the notion that areas of the hippocampus less affected by AD may dilute changes in heavily, but focally affected areas. Importantly, as sensitive as CA1-SRLM thickness was at distinguishing subjects with AD from NCs at the group level in this dataset, there was still considerable overlap between the groups (figure 3). Therefore, like many other neuroimaging markers investigated to date, it is not clear whether measuring CA1-SRLM thickness alone will be able to contribute to a diagnosis in an individual patient.
A focal, early hit to the CA1-SRLM is consistent with the fact that short-term episodic memory impairment is the most frequent first symptom in AD. The synapses in the CA1 apical neuropil are well-known for their robust propensity to undergo NMDA receptor-dependent long-term potentiation, a form of synaptic plasticity associated with synapse unsilencing and memory formation,24 and it is conceivable that loss of those synapses may contribute to short-term memory loss in AD. A prior study revealed a tight correlation between neuropsychological measures and synaptic density in this region at autopsy,25 supporting the notion that there may be phenotypic relevance to atrophy of the apical neuropil in AD. Besides AD, there are other disease processes that cause damage to CA1 and result in memory impairment. As an example, CA1 pyramidal neurons are selectively vulnerable to global cerebral ischemia, as experienced during cardiac arrest,26 and it would be of interest to investigate whether any changes observed in the CA1-SP and CA1-SRLM at 7 T correspond to the degree of episodic memory deficits.
Our work adds to growing efforts to visualize the microscopic features of the human hippocampus using MRI technology. At lower field strength, several groups have traced approximate hippocampal subfield boundaries onto 3-dimensional hippocampal projection maps and shown that CA1 and subiculum atrophy occurs in AD and predicts cognitive decline in normal or mildly impaired individuals.27–30 This technique, while powerful in using advanced computational methods to extract detailed information from low-resolution raw data, does not permit analysis of the hippocampal strata visible only in cross-section.
Several other groups have reported AD-associated changes in hippocampal cross-sectional anatomy using MRI.31–34 One group noted thinning of CA1 but not other subfields in subjects with dementia relative to controls using 1.5-T MRI.31 Also noting CA1 thinning in AD, another group used 4-T MRI to produce better visualization of anatomic landmarks, and thus improved reliability in marking the dimensions of individual subfields.32,33 Both groups considered the CA1 subfield as a whole unit, without differentiating CA1-SP from CA1-SRLM. In this study of patients with mild AD, no difference in overall CA1 thickness was observed. Aside from one 7-T MRI study of young subjects,9 the use of MRI to visualize hippocampal strata has been limited to autopsy specimens.17–19
We reliably distinguished CA1-SRLM as the hypointense band between the more intense DG and CA1-SP cell layers.9 While 7-T GRE MRI afforded excellent in-plane resolution and gray-white differentiation, it was only good enough to separate neuropil from cell body layers, and significantly better resolution (∼60 μm) would be necessary for differentiation of stratum radiatum from stratum lacunosum-moleculare.35 Likewise, borders between DG, CA4, and CA3 remained obscure in our study, and the stratum oriens was only barely visible as a hypointense band superficial to the SP measuring about 1 pixel (200 μm) in thickness (figure 2). In this study, the SRLM was only approximately 2-3 voxels (400-600 μm) wide, and the values for subjects with AD and NC differed on average by only about 1 voxel. Thus, the thinning of the SRLM associated with mild AD in this study was already at the detection limit of our 7-T GRE protocol, and we could not detect any difference in SRLM thickness between the groups on our 3-T images (not shown). This point highlights an important limitation of our technique and of our study, that the absolute measurements that we recorded, as interpolations on raw, pixelated images, are only rough approximations of the true tissue dimensions. Although there is no reason for the presence of AD to bias the measurements one way or the other (raters were blinded to the diagnosis), and thus no reason to discount the presence of some difference between the groups, it is not possible to draw any firm conclusion about the absolute magnitude of this difference. None of the subjects in this study has come to autopsy, and it was not possible to validate our imaging measurements with postmortem histopathology. Amyloid plaques may also be at or beyond the limit of ultra-high-field MRI resolution. Others have observed amyloid plaques as areas of increased susceptibility in mice36 and in human brains in vitro,37 possibly even in vivo.38 Although we did not explicitly attempt strong susceptibility weighting, we observed no plaque-like formations in our images.
This study was powered to demonstrate the feasibility of performing 7-T MRI in elderly cognitively impaired patients and to detect differences in hippocampal metrics between 2 groups. To determine the sensitivity and specificity of CA1-SRLM measurements for detecting mild AD, more work would be required on larger numbers of subjects, including subjects with other clinical diagnoses. To establish CA1-SRLM thinning as an objective imaging biomarker of AD, it would additionally be important to establish intrarater and interrater reliability of the manual measurements used in this study. Importantly, the use of 7-T MRI as a clinical tool is limited by its current availability in only a few academic centers, and by its greater sensitivity to motion and other artifacts compared to lower field MRI.
Our data support the use of 7-T MRI as a tool for detecting submillimeter changes in hippocampal structure, as well as the growing notion that AD is more a disease of synaptic loss than of neuronal loss.1 Use of CA1-SRLM thickness as a potential biomarker of AD is a rational extension from pathology and is now feasible in vivo, using a 7-T MRI sequence easily tolerated by patients. In addition to addressing the limitations discussed above, longitudinal analysis of NCs and patients with mild cognitive impairment will also be important to determine whether CA1-SRLM thinning predicts future cognitive decline.
ACKNOWLEDGMENT
The authors thank Reva Wilheim for coordinating subjects' visits; Niles Bruce, Bert Jimenez, and Mary McPolin for technical and nursing assistance in performing the scans; Ken Edwards, Alefiyah Pishori, Eric Sullivan, and Sri Veeraraghavan for logistical and technical support; and William J. Jagust and his laboratory members for assistance with amyloid imaging.
DISCLOSURE
Dr. Kerchner receives research support from the Larry L. Hillblom Foundation. Dr. Hess and Dr. Hammond-Rosenbluth report no disclosures. Dr. Xu receives research support from the NIH (1R01EB009756 [Co-PI]). Dr. Rabinovici serves on scientific advisory boards for Novartis and GE Healthcare and receives research support from the NIH (NIA K23-AG031861 [PI]), the Alzheimer's Association, and the John Douglas French Alzheimer's Foundation. Dr. Kelley is a full-time employee of and holds stock in GE Healthcare. Dr. Vigneron receives research support from GE Healthcare. Dr. Nelson receives research support from GE Healthcare and the NIH (NCI, SPORE 2P50CA097257 [Project leader], RO1 CA127612-01A1 [PI], PO1CA118816-01A2 [Project lease and Core Director], and R01 CA59880 [PI]). Dr. Miller serves on a scientific advisory board for the Alzheimer's Disease Clinical Study; serves as an Editor for Neurocase and as an Associate Editor of ADAD; receives royalties from the publication of Behavioral Neurology of Dementia (Cambridge, 2009), Handbook of Neurology (Elsevier, 2009), and The Human Frontal Lobes (Guilford, 2008); serves as a consultant for Lundbeck Inc., Elan Corporation, and Allon Therapeutics, Inc.; serves on speakers' bureaus for Novartis and Pfizer Inc.; and receives research support from Novartis and the NIH (NIA P50 AG23501 [PI] and NIA P01 AG19724 [PI]) and the State of California Alzheimer's Center.
Address correspondence and reprint requests to Dr. Geoffrey A. Kerchner, Stanford Center for Memory Disorders, Department of Neurology and Neurological Sciences, Stanford University School of Medicine, 300 Pasteur Drive, Room A343, Stanford, CA 94305-5235 kerchner@stanford.edu
Study funding: Supported by the Larry L. Hillblom Foundation (to G.A.K.), the National Institutes of Health, and the Industry-University Cooperative Research Program of California in conjunction with GE Healthcare (ITL-BIO04-10148).
Disclosure: Author disclosures are provided at the end of the article.
Received April 13, 2010. Accepted in final form June 25, 2010.
REFERENCES
- 1.Selkoe DJ. Alzheimer's disease is a synaptic failure. Science 2002;298:789–791. [DOI] [PubMed] [Google Scholar]
- 2.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006;112:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsia AY, Masliah E, McConlogue L, et al. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA 1999;96:3228–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braak E, Braak H. Alzheimer's disease: transiently developing dendritic changes in pyramidal cells of sector CA1 of the Ammon's horn. Acta Neuropathol 1997;93:323–325. [DOI] [PubMed] [Google Scholar]
- 5.Lace G, Savva GM, Forster G, et al. Hippocampal tau pathology is related to neuroanatomical connections: an ageing population-based study. Brain 2009;132:1324–1334. [DOI] [PubMed] [Google Scholar]
- 6.Thal DR, Holzer M, Rub U, et al. Alzheimer-related tau-pathology in the perforant path target zone and in the hippocampal stratum oriens and radiatum correlates with onset and degree of dementia. Exp Neurol 2000;163:98–110. [DOI] [PubMed] [Google Scholar]
- 7.Scheff SW, Price DA, Schmitt FA, DeKosky ST, Mufson EJ. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007;68:1501–1508. [DOI] [PubMed] [Google Scholar]
- 8.Mizutani T, Kasahara M. Hippocampal atrophy secondary to entorhinal cortical degeneration in Alzheimer-type dementia. Neurosci Lett 1997;222:119–122. [DOI] [PubMed] [Google Scholar]
- 9.Thomas BP, Welch EB, Niederhauser BD, et al. High-resolution 7T MRI of the human hippocampus in vivo. J Magn Reson Imaging 2008;28:1266–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Theysohn JM, Kraff O, Maderwald S, et al. The human hippocampus at 7 T in vivo MRI. Hippocampus 2009;19:1–7. [DOI] [PubMed] [Google Scholar]
- 11.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol 2004;55:306–319. [DOI] [PubMed] [Google Scholar]
- 12.Rabinovici GD, Furst AJ, Alkalay A, et al. Increased metabolic vulnerability in early-onset Alzheimer's disease is not related to amyloid burden. Brain 2010;133:512–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Aizenstein HJ, Nebes RD, Saxton JA, et al. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol 2008;65:1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith SM. Fast robust automated brain extraction. Hum Brain Mapp 2002;17:143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Y, Brady M, Smith S. Segmentation of brain MR images through a hidden Markov random field model and the expectation-maximization algorithm. IEEE Trans Med Imaging 2001;20:45–57. [DOI] [PubMed] [Google Scholar]
- 16.Amaral D, Lavenex P. Hippocampal neuroanatomy. In: Andersen P, Morris R, Amaral D, Bliss T, O'Keefe J, eds. The Hippocampus Book. New York: Oxford University Press; 2007:37–114. [Google Scholar]
- 17.Chakeres DW, Whitaker CDS, Dashner RA, et al. High-resolution 8 Tesla imaging of the formalin-fixed normal human hippocampus. Clin Anat 2005;18:88–91. [DOI] [PubMed] [Google Scholar]
- 18.Miller M, Mark L, Ho K, Haughton V. MR appearance of the internal architecture of Ammon's horn. AJNR Am J Neuroradiol 1996;17:23–26. [PMC free article] [PubMed] [Google Scholar]
- 19.Yushkevich PA, Avants BB, Pluta J, et al. A high-resolution computational atlas of the human hippocampus from postmortem magnetic resonance imaging at 9.4 T. Neuroimage 2009;44:385–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seeley WW, Crawford RK, Zhou J, Miller BL, Greicius MD. Neurodegenerative diseases target large-scale human brain networks. Neuron 2009;62:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohm TG, Münch S, Schönheit B, et al. Transneuronally altered dendritic processing of tangle-free neurons in Alzheimer's disease. Acta Neuropathol 2002;103:437–443. [DOI] [PubMed] [Google Scholar]
- 22.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 2009;284:12845–12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jack CR Jr, Petersen RC, O'Brien PC, Tangalos EG. MR-based hippocampal volumetry in the diagnosis of Alzheimer's disease. Neurology 1992;42:183–188. [DOI] [PubMed] [Google Scholar]
- 24.Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat Rev Neurosci 2008;9:813–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Samuel W, Masliah E, Hill LR, Butters N, Terry R. Hippocampal connectivity and Alzheimer's dementia: effects of synapse loss and tangle frequency in a two-component model. Neurology 1994;44:2081–2088. [DOI] [PubMed] [Google Scholar]
- 26.Kerchner G, Kim A, Choi D. Glutamate-mediated excitotoxicity. In: Jonas P, Monyer H, eds. Ionotropic Glutamate Receptors in the CNS. Berlin: Springer-Verlag; 1999:443–469. [Google Scholar]
- 27.Chételat G, Fouquet M, Kalpouzos G, et al. Three-dimensional surface mapping of hippocampal atrophy progression from MCI to AD and over normal aging as assessed using voxel-based morphometry. Neuropsychologia 2008;46:1721–1731. [DOI] [PubMed] [Google Scholar]
- 28.Csernansky JG, Wang L, Swank J, et al. Preclinical detection of Alzheimer's disease: hippocampal shape and volume predict dementia onset in the elderly. Neuroimage 2005;25:783–792. [DOI] [PubMed] [Google Scholar]
- 29.Frisoni GB, Ganzola R, Canu E, et al. Mapping local hippocampal changes in Alzheimer's disease and normal ageing with MRI at 3 Tesla. Brain 2008;131:3266–3276. [DOI] [PubMed] [Google Scholar]
- 30.Miller MI, Priebe CE, Qiu A, et al. Collaborative computational anatomy: An MRI morphometry study of the human brain via diffeomorphic metric mapping. Hum Brain Mapp 2009;30:2132–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adachi M, Kawakatsu S, Hosoya T, et al. Morphology of the inner structure of the hippocampal formation in Alzheimer disease. AJNR Am J Neuroradiol 2003;24:1575–1581. [PMC free article] [PubMed] [Google Scholar]
- 32.Mueller SG, Schuff N, Raptentsetsang S, Elman J, Weiner MW. Selective effect of Apo e4 on CA3 and dentate in normal aging and Alzheimer's disease using high resolution MRI at 4 T. Neuroimage 2008;42:42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mueller SG, Stables L, Du AT, et al. Measurement of hippocampal subfields and age-related changes with high resolution MRI at 4T. Neurobiol Aging 2007;28:719–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reitz C, Brickman AM, Brown TR, et al. Linking hippocampal structure and function to memory performance in an aging population. Arch Neurol 2009;66:1385–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shepherd TM, Ozarslan E, Yachnis AT, King MA, Blackband SJ. Diffusion tensor microscopy indicates the cytoarchitectural basis for diffusion anisotropy in the human hippocampus. AJNR Am J Neuroradiol 2007;28:958–964. [PMC free article] [PubMed] [Google Scholar]
- 36.Jack CR Jr, Marjanska M, Wengenack TM, et al. Magnetic resonance imaging of Alzheimer's pathology in the brains of living transgenic mice: a new tool in Alzheimer's disease research. Neuroscientist 2007;13:38–48. [DOI] [PubMed] [Google Scholar]
- 37.Benveniste H, Einstein G, Kim KR, Hulette C, Johnson GA. Detection of neuritic plaques in Alzheimer's disease by magnetic resonance microscopy. Proc Natl Acad Sci USA 1999;96:14079–14084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nakada T, Matsuzawa H, Igarashi H, Kwee YFIL. In vivo visualization of senile-plaque-like pathology in Alzheimer's disease patients by MR microscopy on a 7T system. J Neuroimaging 2008;18:125–129. [DOI] [PubMed] [Google Scholar]